

Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
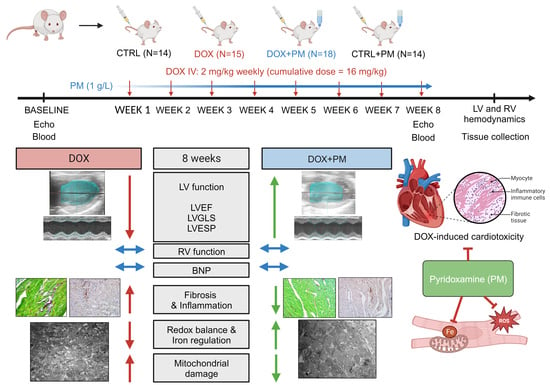
2.1. Animal Model
2.2. Study Design
2.3. Echocardiography
2.3.1. Conventional Echocardiography and 4D Imaging
2.3.2. Strain Imaging
2.4. Hemodynamic Measurements
2.5. Quantification of Plasma BNP Levels
2.6. Histology
2.7. Immunohistochemistry
2.8. Assessment of Cardiomyocyte Organization and Mitochondrial Structure
2.9. Real-Time PCR
2.10. Cluster Analysis
2.11. Statistical Analysis
3. Results
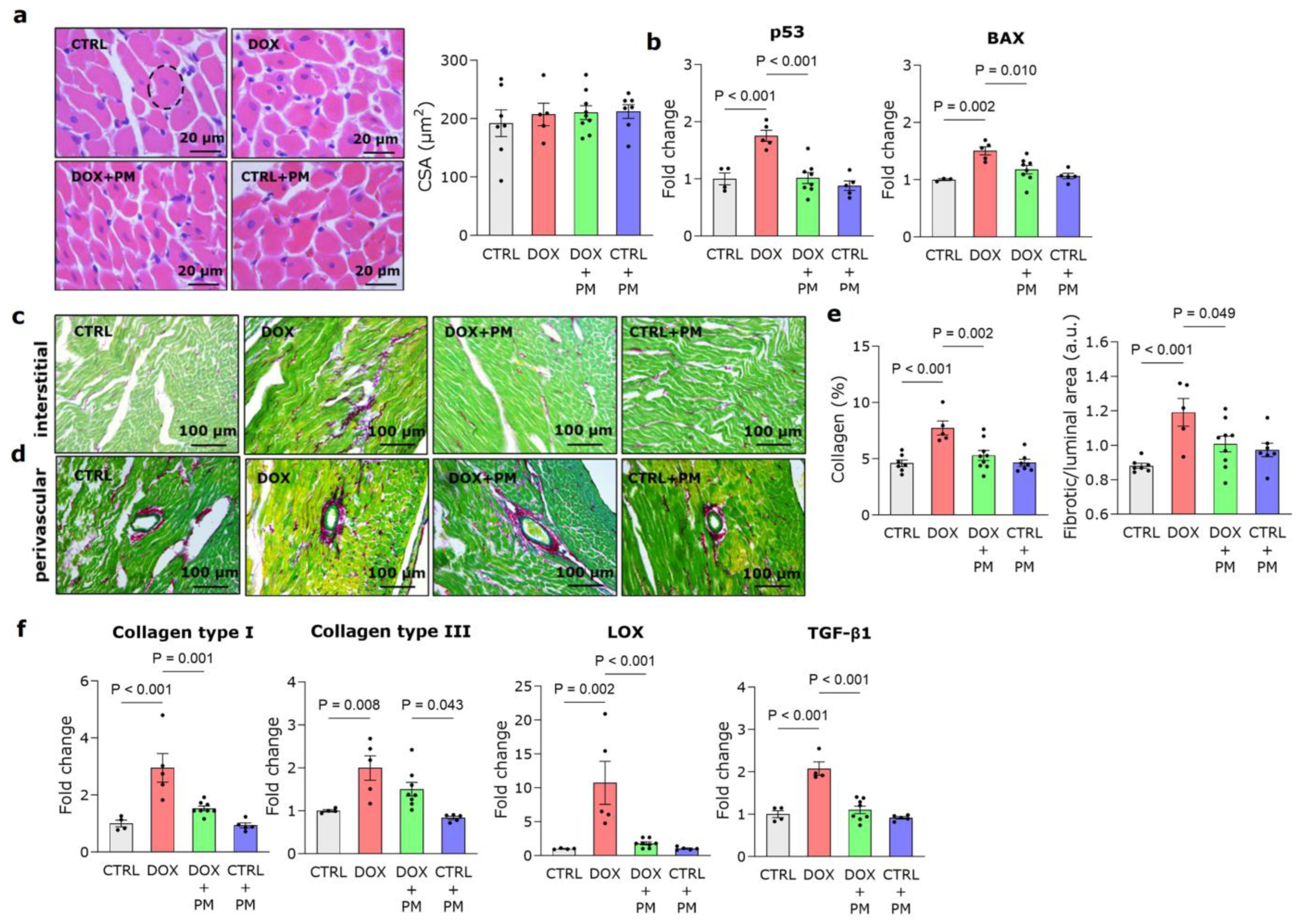
3.1. PM Protects against DOX-Induced LV Cardiomyopathy
3.2. PM Reduces DOX-Induced LV Fibrotic Remodeling
3.3. PM Alleviates DOX-Induced Macrophage-Driven Myocardial Inflammation
3.4. PM Restores DOX-Induced Disturbance of Redox and Iron Regulation
3.5. PM Attenuates DOX-Induced Mitochondrial Damage
3.6. PM Improves Adverse Gene Expression Patterns Involved in DOX-Induced Cardiotoxicity
4. Discussion
4.1. PM as a Cardioprotective Strategy during DOX Treatment
4.2. Underlying Mechanisms of PM Cardioprotection
4.3. Current Limitations and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021. online ahead of print. [Google Scholar] [CrossRef]
- Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 9 March 2023).
- Miller, K.D.; Nogueira, L.; Devasia, T.; Mariotto, A.B.; Yabroff, K.R.; Jemal, A.; Kramer, J.; Siegel, R.L. Cancer treatment and survivorship statistics, 2022. CA Cancer J. Clin. 2022, 72, 409–436. [Google Scholar] [CrossRef] [PubMed]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef]
- Lopez-Sendon, J.; Alvarez-Ortega, C.; Zamora Aunon, P.; Buno Soto, A.; Lyon, A.R.; Farmakis, D.; Cardinale, D.; Canales Albendea, M.; Feliu Batlle, J.; Rodriguez Rodriguez, I.; et al. Classification, prevalence, and outcomes of anticancer therapy-induced cardiotoxicity: The CARDIOTOX registry. Eur. Heart J. 2020, 41, 1720–1729. [Google Scholar] [CrossRef]
- Sturgeon, K.M.; Deng, L.; Bluethmann, S.M.; Zhou, S.; Trifiletti, D.M.; Jiang, C.; Kelly, S.P.; Zaorsky, N.G. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur. Heart J. 2019, 40, 3889–3897. [Google Scholar] [CrossRef] [PubMed]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Simunek, T.; Sterba, M.; Popelova, O.; Adamcova, M.; Hrdina, R.; Gersl, V. Anthracycline-induced cardiotoxicity: Overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacol. Rep. 2009, 61, 154–171. [Google Scholar] [CrossRef]
- Sawicki, K.T.; De Jesus, A.; Ardehali, H. Iron Metabolism in Cardiovascular Disease: Physiology, Mechanisms, and Therapeutic Targets. Circ. Res. 2023, 132, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Lyon, A.R.; Lopez-Fernandez, T.; Couch, L.S.; Asteggiano, R.; Aznar, M.C.; Bergler-Klein, J.; Boriani, G.; Cardinale, D.; Cordoba, R.; Cosyns, B.; et al. 2022 ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur. Heart J. 2022, 43, 4229–4361. [Google Scholar] [CrossRef]
- Gulati, G.; Heck, S.L.; Ree, A.H.; Hoffmann, P.; Schulz-Menger, J.; Fagerland, M.W.; Gravdehaug, B.; von Knobelsdorff-Brenkenhoff, F.; Bratland, A.; Storas, T.H.; et al. Prevention of cardiac dysfunction during adjuvant breast cancer therapy (PRADA): A 2 × 2 factorial, randomized, placebo-controlled, double-blind clinical trial of candesartan and metoprolol. Eur. Heart J. 2016, 37, 1671–1680. [Google Scholar] [CrossRef]
- Livi, L.; Barletta, G.; Martella, F.; Saieva, C.; Desideri, I.; Bacci, C.; Del Bene, M.R.; Airoldi, M.; Amoroso, D.; Coltelli, L.; et al. Cardioprotective Strategy for Patients with Nonmetastatic Breast Cancer Who Are Receiving an Anthracycline-Based Chemotherapy: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1544–1549. [Google Scholar] [CrossRef]
- Vaduganathan, M.; Hirji, S.A.; Qamar, A.; Bajaj, N.; Gupta, A.; Zaha, V.; Chandra, A.; Haykowsky, M.; Ky, B.; Moslehi, J.; et al. Efficacy of Neurohormonal Therapies in Preventing Cardiotoxicity in Patients with Cancer Undergoing Chemotherapy. JACC CardioOncology 2019, 1, 54–65. [Google Scholar] [CrossRef]
- Macedo, A.V.S.; Hajjar, L.A.; Lyon, A.R.; Nascimento, B.R.; Putzu, A.; Rossi, L.; Costa, R.B.; Landoni, G.; Nogueira-Rodrigues, A.; Ribeiro, A.L.P. Efficacy of Dexrazoxane in Preventing Anthracycline Cardiotoxicity in Breast Cancer. JACC CardioOncology 2019, 1, 68–79. [Google Scholar] [CrossRef] [PubMed]
- van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C. Cardioprotective interventions for cancer patients receiving anthracyclines. Cochrane Database Syst. Rev. 2011, 2011, CD003917. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Gerber, M.C.; Weisberg, S.; York, M.; Spicer, D.; Jones, S.E.; Wadler, S.; Desai, A.; Vogel, C.; et al. Cardioprotection with dexrazoxane for doxorubicin-containing therapy in advanced breast cancer. J. Clin. Oncol. 1997, 15, 1318–1332. [Google Scholar] [CrossRef]
- Deluyker, D.; Ferferieva, V.; Driesen, R.B.; Verboven, M.; Lambrichts, I.; Bito, V. Pyridoxamine improves survival and limits cardiac dysfunction after MI. Sci. Rep. 2017, 7, 16010. [Google Scholar] [CrossRef]
- Watson, A.M.; Soro-Paavonen, A.; Sheehy, K.; Li, J.; Calkin, A.C.; Koitka, A.; Rajan, S.N.; Brasacchio, D.; Allen, T.J.; Cooper, M.E.; et al. Delayed intervention with AGE inhibitors attenuates the progression of diabetes-accelerated atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 2011, 54, 681–689. [Google Scholar] [CrossRef]
- Kumrungsee, T.; Peipei, Z.; Yanaka, N.; Suda, T.; Kato, N. Emerging cardioprotective mechanisms of vitamin B6: A narrative review. Eur. J. Nutr. 2022, 61, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Stach, K.; Stach, W.; Augoff, K. Vitamin B6 in Health and Disease. Nutrients 2021, 13, 3229. [Google Scholar] [CrossRef] [PubMed]
- Asnani, A.; Moslehi, J.J.; Adhikari, B.B.; Baik, A.H.; Beyer, A.M.; de Boer, R.A.; Ghigo, A.; Grumbach, I.M.; Jain, S.; Zhu, H.; et al. Preclinical Models of Cancer Therapy-Associated Cardiovascular Toxicity: A Scientific Statement from the American Heart Association. Circ. Res. 2021, 129, e21–e34. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Herman, E.H. Anthracycline cardiotoxicity: The importance of horizontally integrating pre-clinical and clinical research. Cardiovasc. Res. 2018, 114, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Pilz, P.M.; Ward, J.E.; Chang, W.T.; Kiss, A.; Bateh, E.; Jha, A.; Fisch, S.; Podesser, B.K.; Liao, R. Large and Small Animal Models of Heart Failure with Reduced Ejection Fraction. Circ. Res. 2022, 130, 1888–1905. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Wilcox, N.S.; Rotz, S.J.; Mullen, M.; Song, E.J.; Ky Hamilton, B.; Moslehi, J.; Armenian, S.H.; Wu, J.C.; Rhee, J.W.; Ky, B. Sex-Specific Cardiovascular Risks of Cancer and Its Therapies. Circ. Res. 2022, 130, 632–651. [Google Scholar] [CrossRef] [PubMed]
- Evens, L.; Belien, H.; D’Haese, S.; Haesen, S.; Verboven, M.; Rummens, J.L.; Bronckaers, A.; Hendrikx, M.; Deluyker, D.; Bito, V. Combinational Therapy of Cardiac Atrial Appendage Stem Cells and Pyridoxamine: The Road to Cardiac Repair? Int. J. Mol. Sci. 2021, 22, 9266. [Google Scholar] [CrossRef] [PubMed]
- Zacchigna, S.; Paldino, A.; Falcao-Pires, I.; Daskalopoulos, E.P.; Dal Ferro, M.; Vodret, S.; Lesizza, P.; Cannata, A.; Miranda-Silva, D.; Lourenco, A.P.; et al. Towards standardization of echocardiography for the evaluation of left ventricular function in adult rodents: A position paper of the ESC Working Group on Myocardial Function. Cardiovasc. Res. 2021, 117, 43–59. [Google Scholar] [CrossRef]
- Haesen, S.; Col, U.; Schurgers, W.; Evens, L.; Verboven, M.; Driesen, R.B.; Bronckaers, A.; Lambrichts, I.; Deluyker, D.; Bito, V. Glycolaldehyde-modified proteins cause adverse functional and structural aortic remodeling leading to cardiac pressure overload. Sci. Rep. 2020, 10, 12220. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Deluyker, D.; Ferferieva, V.; Noben, J.P.; Swennen, Q.; Bronckaers, A.; Lambrichts, I.; Rigo, J.M.; Bito, V. Cross-linking versus RAGE: How do high molecular weight advanced glycation products induce cardiac dysfunction? Int. J. Cardiol. 2016, 210, 100–108. [Google Scholar] [CrossRef]
- Derveaux, S.; Vandesompele, J.; Hellemans, J. How to do successful gene expression analysis using real-time PCR. Methods 2010, 50, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- Celutkiene, J.; Plymen, C.M.; Flachskampf, F.A.; de Boer, R.A.; Grapsa, J.; Manka, R.; Anderson, L.; Garbi, M.; Barberis, V.; Filardi, P.P.; et al. Innovative imaging methods in heart failure: A shifting paradigm in cardiac assessment. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2018, 20, 1615–1633. [Google Scholar] [CrossRef]
- Chan, B.Y.H.; Roczkowsky, A.; Cho, W.J.; Poirier, M.; Sergi, C.; Keschrumrus, V.; Churko, J.M.; Granzier, H.; Schulz, R. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2021, 117, 188–200. [Google Scholar] [CrossRef]
- McLean, B.A.; Patel, V.B.; Zhabyeyev, P.; Chen, X.; Basu, R.; Wang, F.; Shah, S.; Vanhaesebroeck, B.; Oudit, G.Y. PI3Kalpha Pathway Inhibition with Doxorubicin Treatment Results in Distinct Biventricular Atrophy and Remodeling with Right Ventricular Dysfunction. J. Am. Heart Assoc. 2019, 8, e010961. [Google Scholar] [CrossRef]
- Garcia-Diez, G.; Monreal-Corona, R.; Mora-Diez, N. Complexes of Copper and Iron with Pyridoxamine, Ascorbic Acid, and a Model Amadori Compound: Exploring Pyridoxamine’s Secondary Antioxidant Activity. Antioxidants 2021, 10, 208. [Google Scholar] [CrossRef] [PubMed]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- El-Agamy, D.S.; El-Harbi, K.M.; Khoshhal, S.; Ahmed, N.; Elkablawy, M.A.; Shaaban, A.A.; Abo-Haded, H.M. Pristimerin protects against doxorubicin-induced cardiotoxicity and fibrosis through modulation of Nrf2 and MAPK/NF-kB signaling pathways. Cancer Manag. Res. 2019, 11, 47–61. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, A.; Sun, X.; Yang, Y.; Zhang, L.; Bai, H.; Ben, J.; Zhu, X.; Li, X.; Yang, Q.; et al. Self-Maintenance of Cardiac Resident Reparative Macrophages Attenuates Doxorubicin-Induced Cardiomyopathy through the SR-A1-c-Myc Axis. Circ. Res. 2020, 127, 610–627. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Li, D.; Liu, X.; Pi, W.; Zhang, Y.; Yu, L.; Xu, C.; Sun, Z.; Jiang, J. Fisetin Attenuates Doxorubicin-Induced Cardiomyopathy In Vivo and In Vitro by Inhibiting Ferroptosis Through SIRT1/Nrf2 Signaling Pathway Activation. Front. Pharmacol. 2021, 12, 808480. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol. 2004, 382, 115–144. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.G.; Leisenring, W.M.; Gonzalez-Covarrubias, V.M.; Kawashima, T.I.; Davies, S.M.; Relling, M.V.; Robison, L.L.; Sklar, C.A.; Stovall, M.; Bhatia, S. Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer 2008, 112, 2789–2795. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Persson, H.L.; Richardson, D.R. Molecular pharmacology of the interaction of anthracyclines with iron. Mol. Pharmacol. 2005, 68, 261–271. [Google Scholar] [CrossRef]
- Panjrath, G.S.; Patel, V.; Valdiviezo, C.I.; Narula, N.; Narula, J.; Jain, D. Potentiation of Doxorubicin cardiotoxicity by iron loading in a rodent model. J. Am. Coll. Cardiol. 2007, 49, 2457–2464. [Google Scholar] [CrossRef]
- Huo, M.; Tang, Z.; Wang, L.; Zhang, L.; Guo, H.; Chen, Y.; Gu, P.; Shi, J. Magnesium hexacyanoferrate nanocatalysts attenuate chemodrug-induced cardiotoxicity through an anti-apoptosis mechanism driven by modulation of ferrous iron. Nat. Commun. 2022, 13, 7778. [Google Scholar] [CrossRef]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef]
- Tadokoro, T.; Ikeda, M.; Ide, T.; Deguchi, H.; Ikeda, S.; Okabe, K.; Ishikita, A.; Matsushima, S.; Koumura, T.; Yamada, K.I.; et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight 2020, 5, e132747. [Google Scholar] [CrossRef]
- Chen, X.; Comish, P.B.; Tang, D.; Kang, R. Characteristics and Biomarkers of Ferroptosis. Front. Cell Dev. Biol. 2021, 9, 637162. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.P.; Greco, B.A.; Umanath, K.; Packham, D.; Fox, J.W.; Peterson, R.; Broome, B.R.; Greene, L.E.; Sika, M.; Lewis, J.B. Pyridoxamine dihydrochloride in diabetic nephropathy (PIONEER-CSG-17): Lessons learned from a pilot study. Nephron 2015, 129, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Haesen, S.; Verghote, E.; Heeren, E.; Wolfs, E.; Deluyker, D.; Bito, V. Pyridoxamine attenuates doxorubicin-induced cardiomyopathy without affecting its antitumor effect on rat mammary tumor cells. Cells 2024, 13, 120. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CTRL | DOX | DOX+PM | CTRL+PM | |
|---|---|---|---|---|
| Conventional echocardiography | ||||
| LVEF (%) | 86.0 ± 1.1 | 61.0 ± 3.3 **** | 73.5 ± 1.9 †,‡‡‡‡ | 84.6 ± 1.0 |
| Longitudinal LVFS (%) | 26.9 ± 0.8 | 14.7 ± 0.9 **** | 19.1± 0.8 ‡‡ | 25.2 ± 1 |
| Radial LVFS (%) | 56.0 ± 1.4 | 33.3 ± 2.2 **** | 43.4 ± 1.5 †††,‡‡‡ | 53.0 ± 1.5 |
| LV cardiac index (mL/min/m2) | 0.14 ± 0.01 | 0.14 ± 0.01 | 0.15 ± 0.01 | 0.15 ± 0.01 |
| LVESV/BSA (µL/cm2) | 0.059 ± 0.004 | 0.300 ± 0.035 **** | 0.166 ± 0.014 ‡‡ | 0.080 ± 0.007 |
| LVEDV/BSA (µL/cm2) | 0.504 ± 0.015 | 0.746 ± 0.041 *** | 0.620 ± 0.026 ‡ | 0.517 ± 0.020 |
| LVSV index (µL/cm2) | 0.43 ± 0.01 | 0.45 ± 0.02 | 0.45 ± 0.02 | 0.44 ± 0.01 |
| LV sphericity index | 0.18 ± 0.01 | 0.27 ± 0.03 ** | 0.19 ± 0.01 † | 0.17 ± 0.01 |
| LVAWd (mm) | 1.96 ± 0.05 | 1.85 ± 0.10 | 1.90 ± 0.06 | 1.77 ± 0.05 |
| LVPWd (mm) | 1.86 ± 0.05 | 1.74 ± 0.09 | 1.73 ± 0.05 | 1.84 ± 0.05 |
| E/A | 1.41 ± 0.08 | 1.43 ± 0.08 | 1.40 ± 0.05 | 1.38 ± 0.05 |
| E/E′ | −25.2 ± 1.6 | −28.1 ± 2.3 | −27.2 ± 1.3 | −26.4 ± 2.0 |
| HR (bpm) | 341.3 ± 11.3 | 307.2 ± 9.8 | 339.5 ± 5.2 † | 330.1 ± 7.1 |
| BSA (cm2) | 455.5 ± 3.3 | 403.7 ± 9.0 *** | 430.8 ± 5.2 | 447.9 ± 3.9 |
| Strain | ||||
| LVGLS (%) | −37.7 ± 1.8 | −24.0 ± 2.3 **** | −31.9 ± 1.5 †† | −35.9 ± 1.7 |
| LVGCS (%) | −39.6 ± 1.7 | −25.4 ± 2.2 **** | −31.2 ± 1.1 †,‡‡ | −39.1 ± 1.2 |
| LV peak radial strain (%) | 92.2 ± 7.2 | 47.1 ± 5.2 **** | 58.7 ± 4.7 ‡‡ | 86.9 ± 7.6 |
| Plasma | ||||
| BNP (ng/mL) | 0.16 ± 0.03 | 0.24 ± 0.04 | 0.18 ± 0.02 | 0.12 ± 0.01 |
| CTRL | DOX | DOX+PM | CTRL+PM | |
|---|---|---|---|---|
| Hemodynamic parameters | ||||
| LVESP (mmHg) | 99.8 ± 3.1 | 68.9 ± 5.4 **** | 95.1 ± 1.2 †††† | 95.3 ± 3.8 |
| LVEDP (mmHg) | 6.1 ± 1.6 | 8.6 ± 2.2 | 5.3 ± 0.7 | 7.3 ± 1.7 |
| LV dP/dtmax (mmHg/s) | 6750.0 ± 347.2 | 3322.0 ± 644.7 **** | 5718.0 ± 287.3 †† | 5861.0 ± 405.1 |
| LV dP/dtmin (mmHg/s) | −7895.0 ± 655.3 | −3199.0 ± 568.0 **** | −7182.0 ± 283.2 ††† | −6795.0 ± 119.4 |
| LV contractility index (1/s) | 114.8 ± 5.1 | 88.5 ± 7.0 * | 102.5 ± 2.4 | 114.1 ± 9.4 |
| Organ weight | ||||
| Lung wet-to-dry weight ratio | 5.03 ± 0.09 | 5.41 ± 0.16 | 5.27 ± 0.10 | 4.95 ± 0.09 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haesen, S.; Jager, M.M.; Brillouet, A.; de Laat, I.; Vastmans, L.; Verghote, E.; Delaet, A.; D’Haese, S.; Hamad, I.; Kleinewietfeld, M.; et al. Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity. Antioxidants 2024, 13, 112. https://doi.org/10.3390/antiox13010112
Haesen S, Jager MM, Brillouet A, de Laat I, Vastmans L, Verghote E, Delaet A, D’Haese S, Hamad I, Kleinewietfeld M, et al. Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity. Antioxidants. 2024; 13(1):112. https://doi.org/10.3390/antiox13010112
Chicago/Turabian StyleHaesen, Sibren, Manon Marie Jager, Aline Brillouet, Iris de Laat, Lotte Vastmans, Eline Verghote, Anouk Delaet, Sarah D’Haese, Ibrahim Hamad, Markus Kleinewietfeld, and et al. 2024. "Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity" Antioxidants 13, no. 1: 112. https://doi.org/10.3390/antiox13010112
APA StyleHaesen, S., Jager, M. M., Brillouet, A., de Laat, I., Vastmans, L., Verghote, E., Delaet, A., D’Haese, S., Hamad, I., Kleinewietfeld, M., Mebis, J., Mullens, W., Lambrichts, I., Wolfs, E., Deluyker, D., & Bito, V. (2024). Pyridoxamine Limits Cardiac Dysfunction in a Rat Model of Doxorubicin-Induced Cardiotoxicity. Antioxidants, 13(1), 112. https://doi.org/10.3390/antiox13010112