
Flavonoid–Phenolic Acid Hybrids Are Potent Inhibitors of Ferroptosis via Attenuation of Mitochondrial Impairment

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Flavonoid–Phenolic Acid Hybrids
2.2. Cell Culture
2.3. Cell Viability
2.4. Cell Proliferation
2.5. Cell Death
2.6. (Mitochondrial) Lipid Peroxidation
2.7. Mitochondrial Superoxides (O2•−)
2.8. Mitochondrial Membrane Potential (Δψm)
2.9. Mitochondrial Ca2+
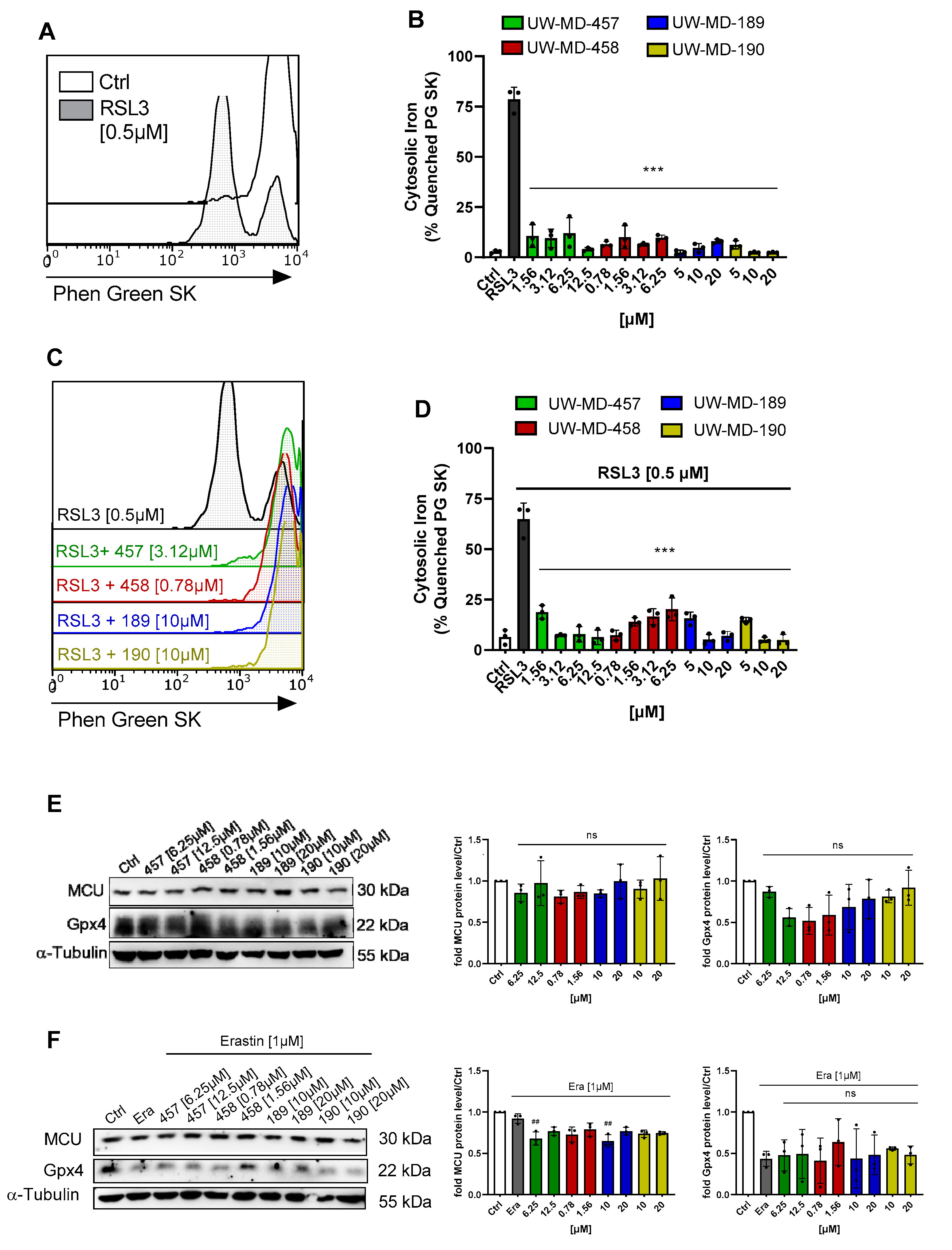
2.10. Cytosolic Iron
2.11. Protein Analysis
2.12. Seahorse Bioenergetic Flux Analysis
2.13. Radical Scavenging Activities
2.14. ATP Level
2.15. Mitochondrial Morphology
3. Results
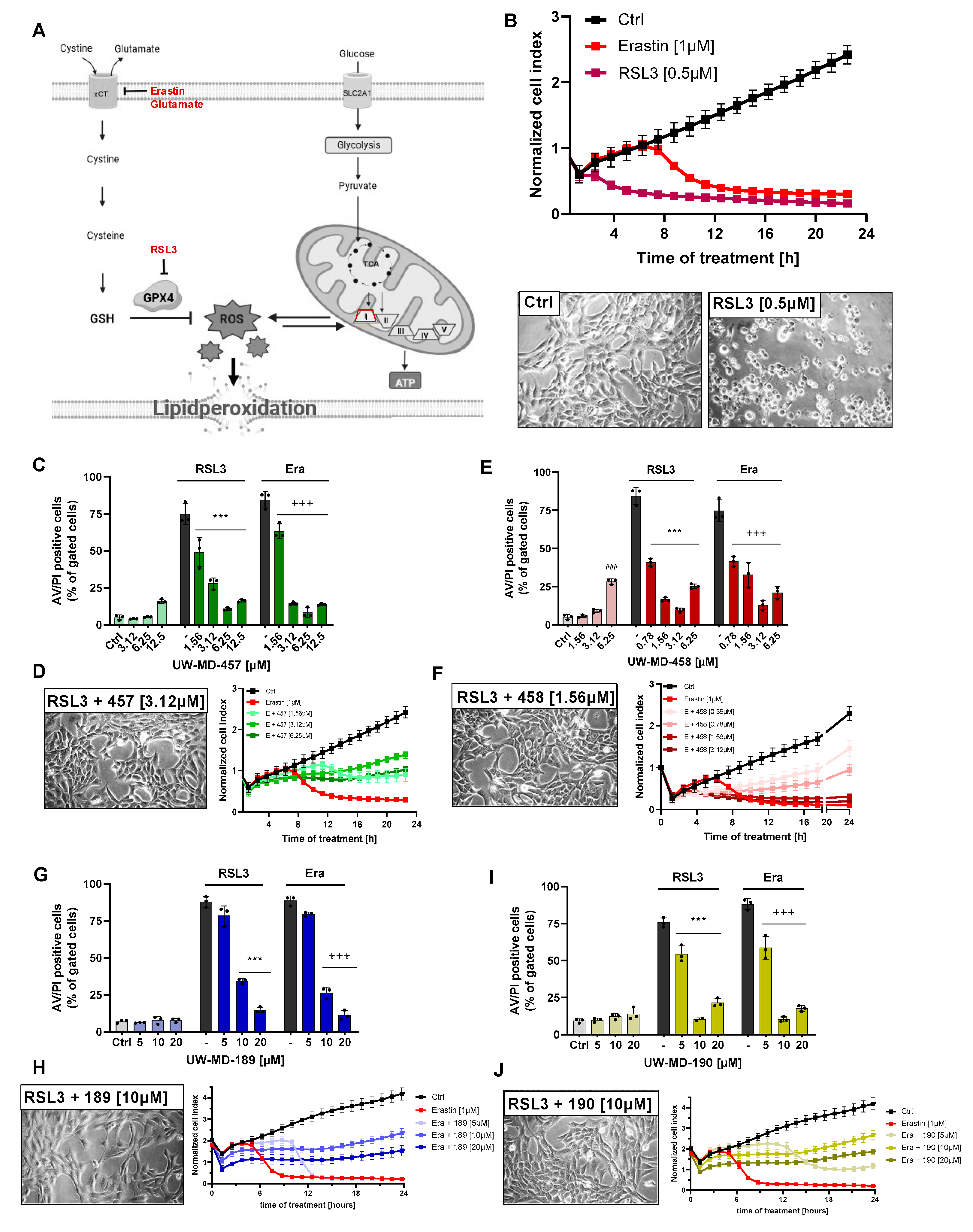
3.1. Flavonoid–Cinnamic and –Ferulic Acid Hybrids Protect HT22 Cells from Oxidative Stress-Induced Cell Death
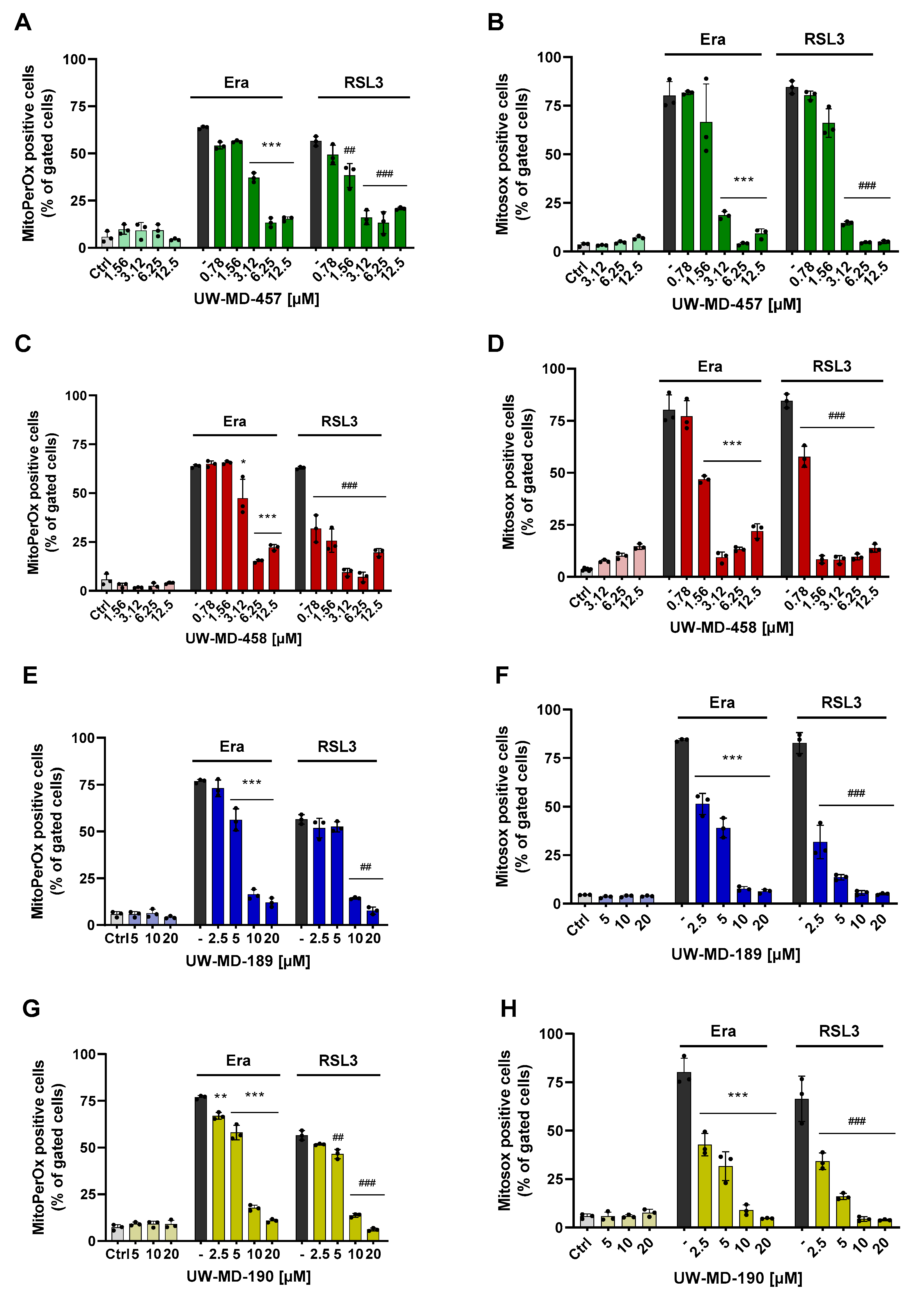
3.2. Flavonoid Amide and Ester Hybrids Suppress Intracellular ROS and Mitochondrial (Super)Oxide Production
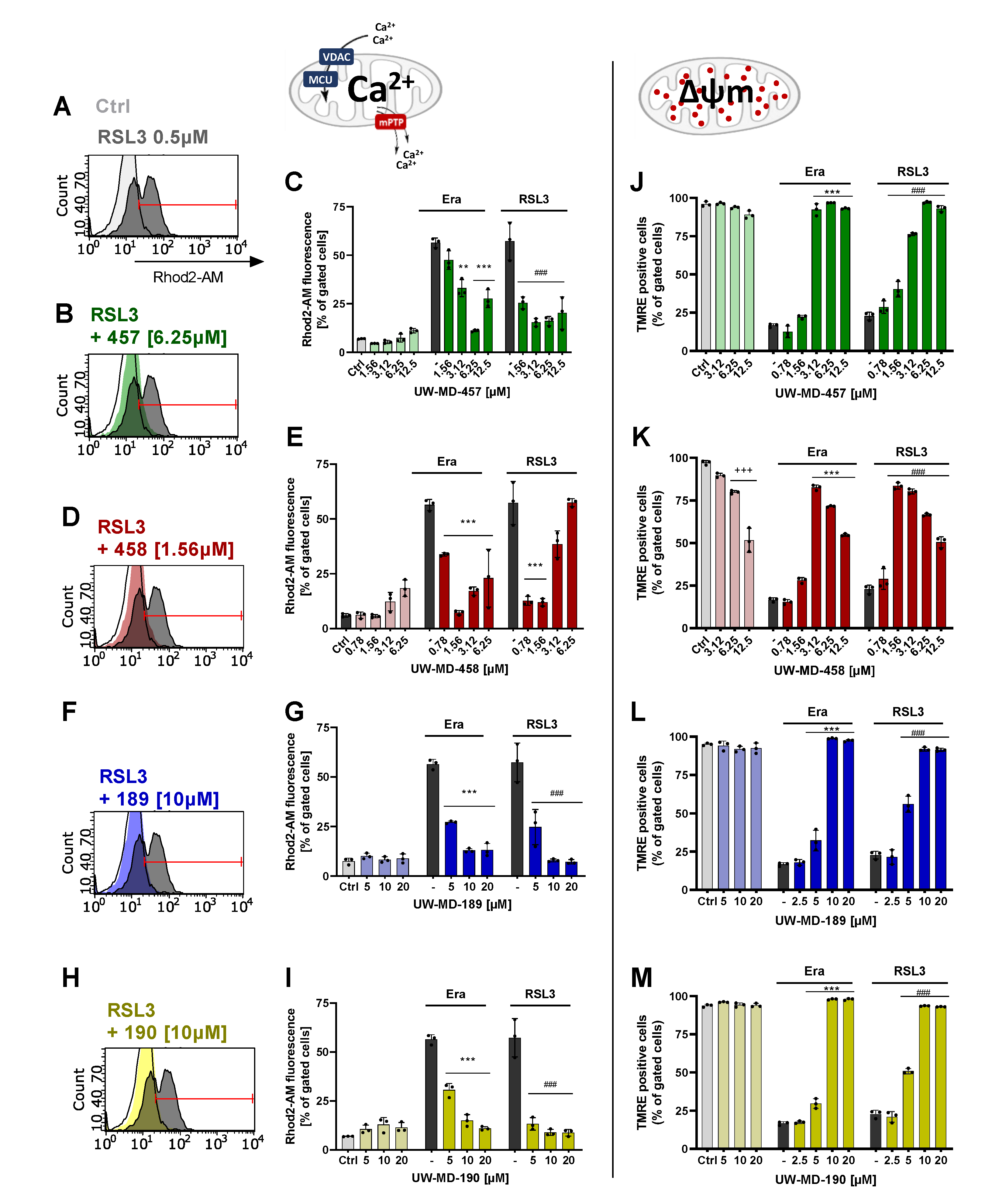
3.3. Flavonoid–Cinnamic and –Ferulic Acid Hybrids Both Preserve Mitochondrial Membrane Integrity and Maintain Mitochondrial Calcium Levels
3.4. Flavonoid–Phenolic Acid Hybrids Reduce Mitochondrial Oxidative Metabolism
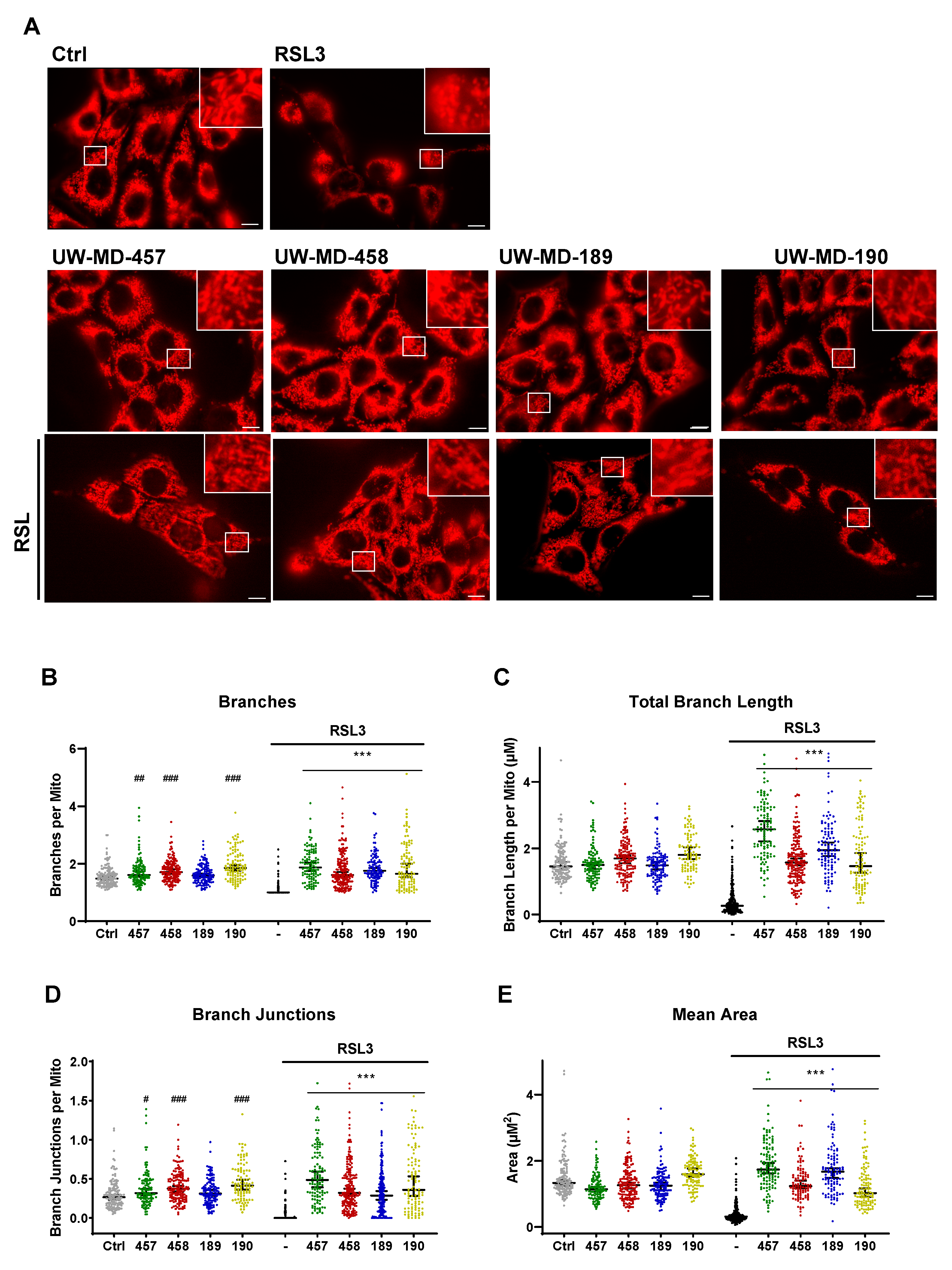
3.5. Flavonoid–Phenolic Acid Analogs Preserve Mitochondrial Morphology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Lima, T.; Li, T.Y.; Mottis, A.; Auwerx, J. Pleiotropic effects of mitochondria in aging. Nat. Aging 2022, 2, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Marchi, S.; Simoes, I.C.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [PubMed]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and links with diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jin, Y.; Lei, Y.; Liu, T.; Wan, Z.; Meng, H.; Wang, H. Ferroptosis Is Regulated by Mitochondria in Neurodegenerative Diseases. Neurodegener. Dis. 2020, 20, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Neitemeier, S.; Jelinek, A.; Laino, V.; Hoffmann, L.; Eisenbach, I.; Eying, R.; Ganjam, G.K.; Dolga, A.M.; Oppermann, S.; Culmsee, C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017, 12, 558–570. [Google Scholar] [CrossRef]
- Jelinek, A.; Heyder, L.; Daude, M.; Plessner, M.; Krippner, S.; Grosse, R.; Diederich, W.E.; Culmsee, C. Mitochondrial rescue prevents glutathione peroxidase-dependent ferroptosis. Free Radic. Biol. Med. 2018, 117, 45–57. [Google Scholar] [CrossRef]
- Bhatti, G.K.; Gupta, A.; Pahwa, P.; Khullar, N.; Singh, S.; Navik, U.; Kumar, S.; Mastana, S.S.; Reddy, A.P.; Reddy, P.H.; et al. Targeting mitochondrial bioenergetics as a promising therapeutic strategy in metabolic and neurodegenerative diseases. Biomed. J. 2022, 45, 733–748. [Google Scholar] [CrossRef]
- Almikhlafi, M.A.; Karami, M.M.; Jana, A.; Alqurashi, T.M.; Majrashi, M.; Alghamdi, B.S.; Ashraf, G.M. Mitochondrial Medicine: A Promising Therapeutic Option Against Various Neurodegenerative Disorders. Curr. Neuropharmacol. 2023, 21, 1165–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, L.; Li, B.; Shi, J.; Xu, J.; Yuan, M. Targeting Mitochondrial Dysfunction in Neurodegenerative Diseases: Expanding the Therapeutic Approaches by Plant-Derived Natural Products. Pharmaceuticals 2023, 16, 277. [Google Scholar] [CrossRef] [PubMed]
- Du, M.-R.; Gao, Q.-Y.; Liu, C.-L.; Bai, L.-Y.; Li, T.; Wei, F.-L. Exploring the Pharmacological Potential of Metformin for Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 838173. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R. Alzheimer’s Disease: A Lesson from Mitochondrial Dysfunction. Antioxid. Redox Signal. 2007, 9, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tan, Y.; Ouyang, S.; He, J.; Liu, L. Resveratrol protects against myocardial ischemia-reperfusion injury via attenuating ferroptosis. Gene 2022, 808, 145968. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Quan, F.; Cao, Q.; Lin, Y.; Yue, C.; Bi, R.; Cui, X.; Yang, H.; Yang, Y.; Birnbaumer, L.; et al. Quercetin alleviates acute kidney injury by inhibiting ferroptosis. J. Adv. Res. 2021, 28, 231–243. [Google Scholar] [CrossRef]
- Jones, Q.R.; Warford, J.; Rupasinghe, H.V.; Robertson, G.S. Target-based selection of flavonoids for neurodegenerative disorders. Trends Pharmacol. Sci. 2012, 33, 602–610. [Google Scholar] [CrossRef]
- Li, J.; Sun, M.; Cui, X.; Li, C. Protective Effects of Flavonoids against Alzheimer’s Disease: Pathological Hypothesis, Potential Targets, and Structure–Activity Relationship. Int. J. Mol. Sci. 2022, 23, 10020. [Google Scholar] [CrossRef]
- Hirohata, M.; Hasegawa, K.; Tsutsumi-Yasuhara, S.; Ohhashi, Y.; Ookoshi, T.; Ono, K.; Yamada, M.; Naiki, H. The Anti-Amyloidogenic Effect Is Exerted against Alzheimer’s β-Amyloid Fibrils in Vitro by Preferential and Reversible Binding of Flavonoids to the Amyloid Fibril Structure. Biochemistry 2007, 46, 1888–1899. [Google Scholar] [CrossRef]
- Ono, K.; Yoshiike, Y.; Takashima, A.; Hasegawa, K.; Naiki, H.; Yamada, M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 2003, 87, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Ushikubo, H.; Watanabe, S.; Tanimoto, Y.; Abe, K.; Hiza, A.; Ogawa, T.; Asakawa, T.; Kan, T.; Akaishi, T. 3,3′,4′,5,5′-Pentahydroxyflavone is a potent inhibitor of amyloid β fibril formation. Neurosci. Lett. 2012, 513, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, K.L.; Liu, R.H. Structure−Activity Relationships of Flavonoids in the Cellular Antioxidant Activity Assay. J. Agric. Food Chem. 2008, 56, 8404–8411. [Google Scholar] [CrossRef] [PubMed]
- Speisky, H.; Shahidi, F.; de Camargo, A.C.; Fuentes, J. Revisiting the Oxidation of Flavonoids: Loss, Conservation or Enhancement of Their Antioxidant Properties. Antioxidants 2022, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies2. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [PubMed]
- Thilakarathna, S.H.; Rupasinghe, H.P.V. Flavonoid Bioavailability and Attempts for Bioavailability Enhancement. Nutrients 2013, 5, 3367–3387. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhai, Y.; Chen, J.; Xu, X.; Wang, H. Kaempferol Ameliorates Oxygen-Glucose Deprivation/Reoxygenation-Induced Neuronal Ferroptosis by Activating Nrf2/SLC7A11/GPX4 Axis. Biomolecules 2021, 11, 923. [Google Scholar] [CrossRef]
- Cao, L.; Zhao, S.; Han, K.; Fan, L.; Zhao, C.; Yin, S.; Hu, H. Managing ferroptosis-related diseases with indirect dietary modulators of ferroptosis. J. Nutr. Biochem. 2023, 120, 109427. [Google Scholar] [CrossRef]
- Gunesch, S.; Hoffmann, M.; Kiermeier, C.; Fischer, W.; Pinto, A.F.; Maurice, T.; Maher, P.; Decker, M. 7-O-Esters of taxifolin with pronounced and overadditive effects in neuroprotection, anti-neuroinflammation, and amelioration of short-term memory impairment in vivo. Redox Biol. 2020, 29, 101378. [Google Scholar] [CrossRef]
- Hofmann, J.; Spatz, P.; Walther, R.; Gutmann, M.; Maurice, T.; Decker, M. Synthesis and Biological Evaluation of Flavonoid-Cinnamic Acid Amide Hybrids with Distinct Activity against Neurodegeneration in Vitro and in Vivo. Chem. Eur. J. 2022, 28, e202200786. [Google Scholar] [CrossRef]
- Gunesch, S.; Soriano-Castell, D.; Lamer, S.; Schlosser, A.; Maher, P.; Decker, M. Development and Application of a Chemical Probe Based on a Neuroprotective Flavonoid Hybrid for Target Identification Using Activity-Based Protein Profiling. ACS Chem. Neurosci. 2020, 11, 3823–3837. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O. Calcium’s Role in Orchestrating Cancer Apoptosis: Mitochondrial-Centric Perspective. Int. J. Mol. Sci. 2023, 24, 8982. [Google Scholar] [CrossRef] [PubMed]
- Deniaud, A.; el Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2008, 27, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Galluzzi, L.; Kepp, O.; Kroemer, G. Adenine nucleotide translocase: A component of the phylogenetically conserved cell death machinery. Cell Death Differ. 2009, 16, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Deng, X.; Li, Y.; Hu, J.; Xie, L.; Shi, F.; Tang, M.; Bode, A.M.; Zhang, X.; Liao, W.; et al. Conformational change of adenine nucleotide translocase-1 mediates cisplatin resistance induced by EBV-LMP1. EMBO Mol. Med. 2021, 13, e14072. [Google Scholar] [CrossRef] [PubMed]
- Zille, M.; Kumar, A.; Kundu, N.; Bourassa, M.W.; Wong, V.S.C.; Willis, D.; Karuppagounder, S.S.; Ratan, R.R. Ferroptosis in Neurons and Cancer Cells Is Similar But Differentially Regulated by Histone Deacetylase Inhibitors. eNeuro 2019, 6, e0263-18. [Google Scholar] [CrossRef] [PubMed]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Manns, J.; Daubrawa, M.; Driessen, S.; Paasch, F.; Hoffmann, N.; Löffler, A.; Lauber, K.; Dieterle, A.; Alers, S.; Iftner, T.; et al. Triggering of a novel intrinsic apoptosis pathway by the kinase inhibitor staurosporine: Activation of caspase-9 in the absence of Apaf-1. FASEB J. 2011, 25, 3250–3261. [Google Scholar] [CrossRef]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The Hallmarks of Ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. [Google Scholar] [CrossRef]
- Homma, T.; Kobayashi, S.; Sato, H.; Fujii, J. Superoxide produced by mitochondrial complex III plays a pivotal role in the execution of ferroptosis induced by cysteine starvation. Arch. Biochem. Biophys. 2021, 700, 108775. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, Y.; Shimizu, S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 2007, 12, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Cole-Ezea, P.; Swan, D.; Shanley, D.; Hesketh, J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelial cells. Free Radic. Biol. Med. 2012, 53, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Shi, R.; Luciani, D.S. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am. J. Physiol. Metab. 2020, 318, E87–E101. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Baidya, R.; Chakraborty, T.; Samanta, A.K.; Roy, S. Pharmacological basis and new insights of taxifolin: A comprehensive review. Biomed. Pharmacother. 2021, 142, 112004. [Google Scholar] [CrossRef]
- Cai, Y.-Z.; Sun, M.; Xing, J.; Luo, Q.; Corke, H. Structure–radical scavenging activity relationships of phenolic compounds from traditional Chinese medicinal plants. Life Sci. 2006, 78, 2872–2888. [Google Scholar] [CrossRef]
- Heim, K.E.; Tagliaferro, A.R.; Bobilya, D.J. Flavonoid antioxidants: Chemistry, metabolism and structure-activity relationships. J. Nutr. Biochem. 2002, 13, 572–584. [Google Scholar] [CrossRef]
- Leclerc, M.; Dudonné, S.; Calon, F. Can Natural Products Exert Neuroprotection without Crossing the Blood–Brain Barrier? Int. J. Mol. Sci. 2021, 22, 3356. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Liu, C.; Zhao, L.; Liu, H.; Li, W.; Guan, H.; Zhao, L.; Xiao, J. Effects of Taxifolin on Osteoclastogenesis in vitro and in vivo. Front. Pharmacol. 2018, 9, 1286. [Google Scholar] [CrossRef] [PubMed]
- Suraweera, T.L.; Rupasinghe, H.P.V.; Dellaire, G.; Xu, Z. Regulation of Nrf2/ARE Pathway by Dietary Flavonoids: A Friend or Foe for Cancer Management? Antioxidants 2020, 9, 973. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, C.; Fornai, M.; Antonioli, L.; Blandizzi, C.; Calderone, V. Phytochemicals as Novel Therapeutic Strategies for NLRP3 Inflammasome-Related Neurological, Metabolic, and Inflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 2876. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.E. The interactions of flavonoids within neuronal signalling pathways. Genes Nutr. 2007, 2, 257–273. [Google Scholar] [CrossRef]
- Hsu, C.-L.; Yen, G.-C. Effects of Flavonoids and Phenolic Acids on the Inhibition of Adipogenesis in 3T3-L1 Adipocytes. J. Agric. Food Chem. 2007, 55, 8404–8410. [Google Scholar] [CrossRef]
- Gan, B. Mitochondrial regulation of ferroptosis. J. Cell Biol. 2021, 220, e202105043. [Google Scholar] [CrossRef]
- Chen, Q.; Ruan, D.; Shi, J.; Du, D.; Bian, C. The multifaceted roles of natural products in mitochondrial dysfunction. Front. Pharmacol. 2023, 14, 1093038. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K. Quercetin and Ferroptosis. Life 2023, 13, 1730. [Google Scholar] [CrossRef]
- Bayele, H.K.; Balesaria, S.; Srai, S.K. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free. Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef]
- Neitemeier, S.; Dolga, A.M.; Honrath, B.; Karuppagounder, S.S.; Alim, I.; Ratan, R.R.; Culmsee, C. Inhibition of HIF-prolyl-4-hydroxylases prevents mitochondrial impairment and cell death in a model of neuronal oxytosis. Cell Death Dis. 2016, 7, e2214. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Song, H.; Cho, J.H. Flavonoids mitigate neurodegeneration in aged Caenorhabditis elegans by mitochondrial uncoupling. Food Sci. Nutr. 2020, 8, 6633–6642. [Google Scholar] [CrossRef] [PubMed]
- Valdameri, G.; Herrerias, T.; Carnieri, E.G.S.; Cadena, S.M.S.C.; Martinez, G.R.; Rocha, M.E.M. Importance of the core structure of flavones in promoting inhibition of the mitochondrial respiratory chain. Chem. Interact. 2010, 188, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Fato, R.; Bergamini, C.; Bortolus, M.; Maniero, A.L.; Leoni, S.; Ohnishi, T.; Lenaz, G. Differential effects of mitochondrial Complex I inhibitors on production of reactive oxygen species. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Vranas, M.; Wohlwend, D.; Qiu, D.; Gerhardt, S.; Trncik, C.; Pervaiz, M.; Ritter, K.; Steimle, S.; Randazzo, A.; Einsle, O.; et al. Structural Basis for Inhibition of ROS-Producing Respiratory Complex I by NADH-OH. Angew. Chem. 2021, 60, 27277–27281. [Google Scholar] [CrossRef]
- Kotlyar, A.B.; Karliner, J.S.; Cecchini, G. A novel strong competitive inhibitor of complex I. FEBS Lett. 2005, 579, 4861–4866. [Google Scholar] [CrossRef]
- Chavda, V.; Lu, B. Reverse Electron Transport at Mitochondrial Complex I in Ischemic Stroke, Aging, and Age-Related Diseases. Antioxidants 2023, 12, 895. [Google Scholar] [CrossRef]
- Sadek, H.A.; Szweda, P.A.; Szweda, L.I. Modulation of Mitochondrial Complex I Activity by Reversible Ca2+ and NADH Mediated Superoxide Anion Dependent Inhibition. Biochemistry 2004, 43, 8494–8502. [Google Scholar] [CrossRef]
- Matsuzaki, S.; Szweda, L.I. Inhibition of Complex I by Ca2+ Reduces Electron Transport Activity and the Rate of Superoxide Anion Production in Cardiac Submitochondrial Particles. Biochemistry 2007, 46, 1350–1357. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 390473. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, L.; Liu, J.; Ma, L.; Zhang, W. Adenine nucleotide translocase: Current knowledge in post-translational modifications, regulations and pathological implications for human diseases. FASEB J. 2023, 37, e22953. [Google Scholar] [CrossRef] [PubMed]
- Brustovetsky, N.; Klingenberg, M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J. Biol. Chem. 1994, 269, 27329–27336. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H+ transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Baker, P.R.; Freeman, B.A.; Brookes, P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res. 2009, 82, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Nadtochiy, S.M.; Zhu, Q.; Urciuoli, W.; Rafikov, R.; Black, S.M.; Brookes, P.S. Nitroalkenes Confer Acute Cardioprotection via Adenine Nucleotide Translocase 1. J. Biol. Chem. 2011, 287, 3573–3580. [Google Scholar] [CrossRef]
- Carrer, A.; Tommasin, L.; Šileikytė, J.; Ciscato, F.; Filadi, R.; Urbani, A.; Forte, M.; Rasola, A.; Szabò, I.; Carraro, M.; et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat. Commun. 2021, 12, 4835. [Google Scholar] [CrossRef]
- Rabenau, M.; Unger, M.; Drewe, J.; Culmsee, C. Metabolic switch induced by Cimicifuga racemosa extract prevents mitochondrial damage and oxidative cell death. Phytomedicine 2019, 52, 107–116. [Google Scholar] [CrossRef]
- Rabenau, M.; Dillberger, B.; Günther, M.; Krippner, S.; Butterweck, V.; Boonen, G.; Drewe, J.; Eckert, G.P.; Culmsee, C. Cimicifuga racemosa Extract Ze 450 Re-Balances Energy Metabolism and Promotes Longevity. Antioxidants 2021, 10, 1432. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EC50 RSL3 | EC50 Erastin | EC50 Glutamate |
|---|---|---|---|
| UW-MD-189 | 3.23 µM | 1.53 µM | 1.50 µM |
| UW-MD-190 | 0.40 µM | 0.42 µM | 0.43 µM |
| UW-MD-457 | 5.85 µM | 6.34 µM | 5.94 µM |
| UW-MD-458 | 5.19 µM | 4.93 µM | 5.08 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Günther, M.; Dabare, S.; Fuchs, J.; Gunesch, S.; Hofmann, J.; Decker, M.; Culmsee, C. Flavonoid–Phenolic Acid Hybrids Are Potent Inhibitors of Ferroptosis via Attenuation of Mitochondrial Impairment. Antioxidants 2024, 13, 44. https://doi.org/10.3390/antiox13010044
Günther M, Dabare S, Fuchs J, Gunesch S, Hofmann J, Decker M, Culmsee C. Flavonoid–Phenolic Acid Hybrids Are Potent Inhibitors of Ferroptosis via Attenuation of Mitochondrial Impairment. Antioxidants. 2024; 13(1):44. https://doi.org/10.3390/antiox13010044
Chicago/Turabian StyleGünther, Madeline, Samentha Dabare, Jennifer Fuchs, Sandra Gunesch, Julian Hofmann, Michael Decker, and Carsten Culmsee. 2024. "Flavonoid–Phenolic Acid Hybrids Are Potent Inhibitors of Ferroptosis via Attenuation of Mitochondrial Impairment" Antioxidants 13, no. 1: 44. https://doi.org/10.3390/antiox13010044
APA StyleGünther, M., Dabare, S., Fuchs, J., Gunesch, S., Hofmann, J., Decker, M., & Culmsee, C. (2024). Flavonoid–Phenolic Acid Hybrids Are Potent Inhibitors of Ferroptosis via Attenuation of Mitochondrial Impairment. Antioxidants, 13(1), 44. https://doi.org/10.3390/antiox13010044