
Curculigoside Attenuates Endoplasmic Reticulum Stress-Induced Epithelial Cell and Fibroblast Senescence by Regulating the SIRT1-P300 Signaling Pathway

Abstract
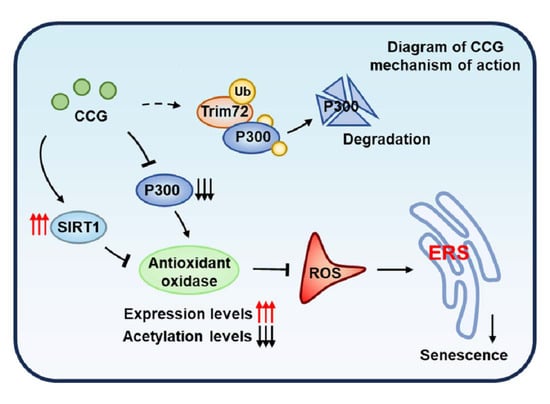
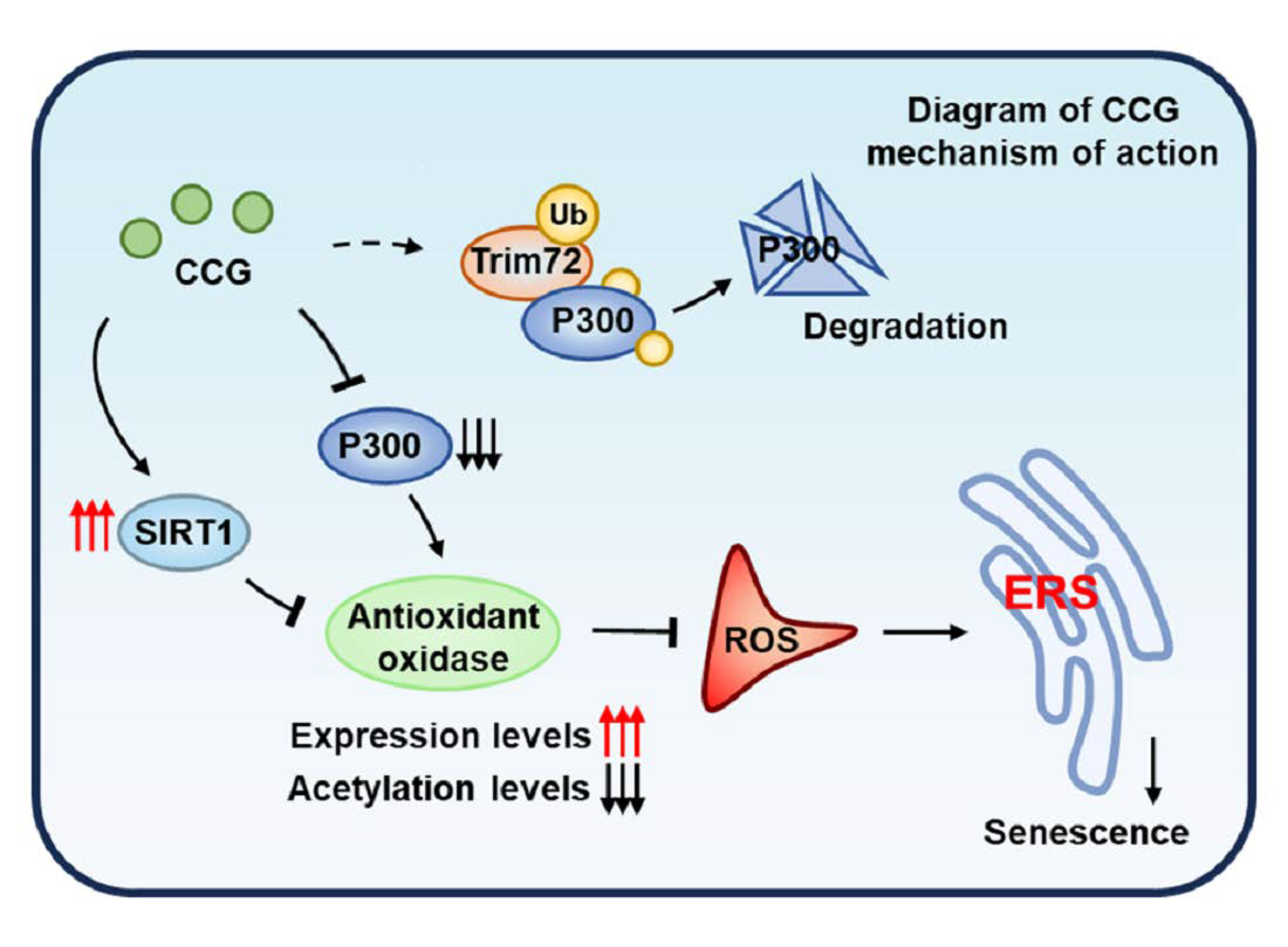
:
1. Introduction
2. Materials and Methods
2.1. Animals Experiments
2.2. Histological Analysis
2.3. Ashcroft Scores
2.4. Survival Rate
2.5. Measurement of Hydroxyproline Levels
2.6. Respiratory Function
2.7. Immunofluorescence
2.8. β-Galactosidase Staining
2.9. Western Blotting (WB)
2.10. Quantitative Real-Time PCR (qPCR)
2.11. siRNA Transfection
2.12. Immunoprecipitation (IP)
2.13. ROS Level
2.14. Detection of Calcium Influx
2.15. Network Pharmacology and Molecular Docking
2.16. Cellular Thermal Shift Assay (CESTA)
2.17. Cell Culture
2.18. Statistical Analysis
3. Results
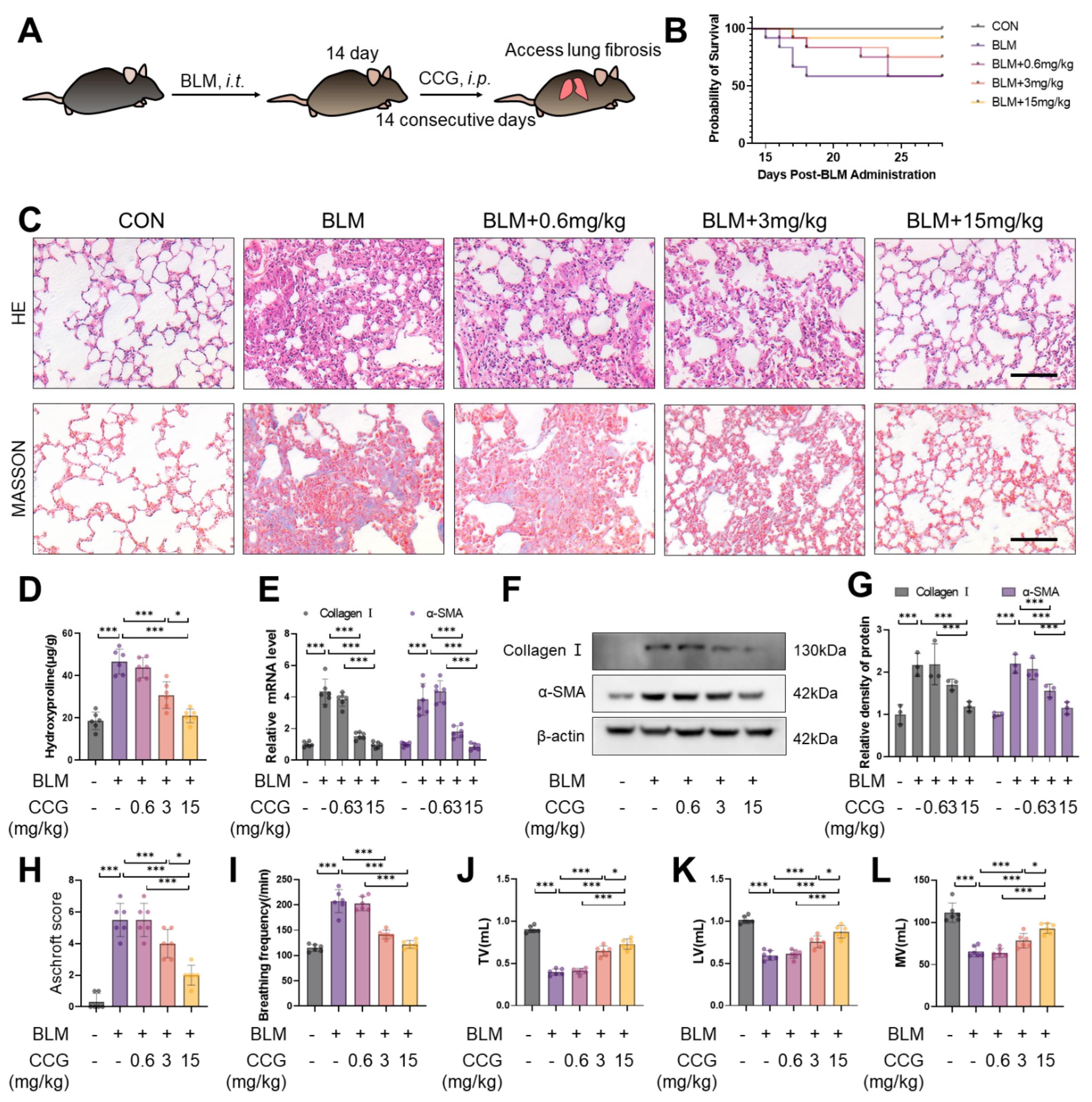
3.1. CCG Alleviated BLM-Induced Pulmonary Fibrosis in Mice
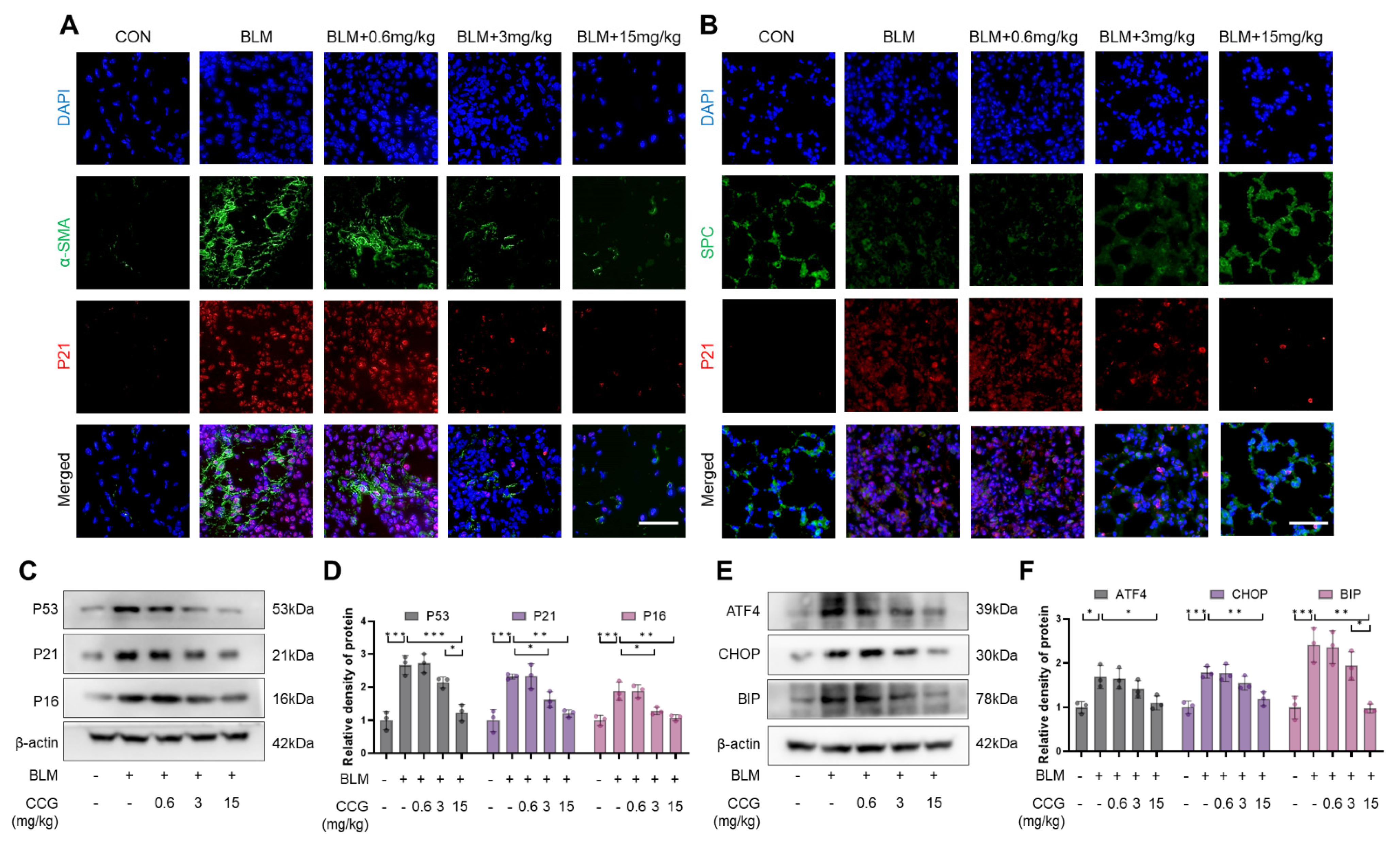
3.2. CCG Alleviated the Senescence of AECs and Fibroblasts Induced by BLM In Vivo
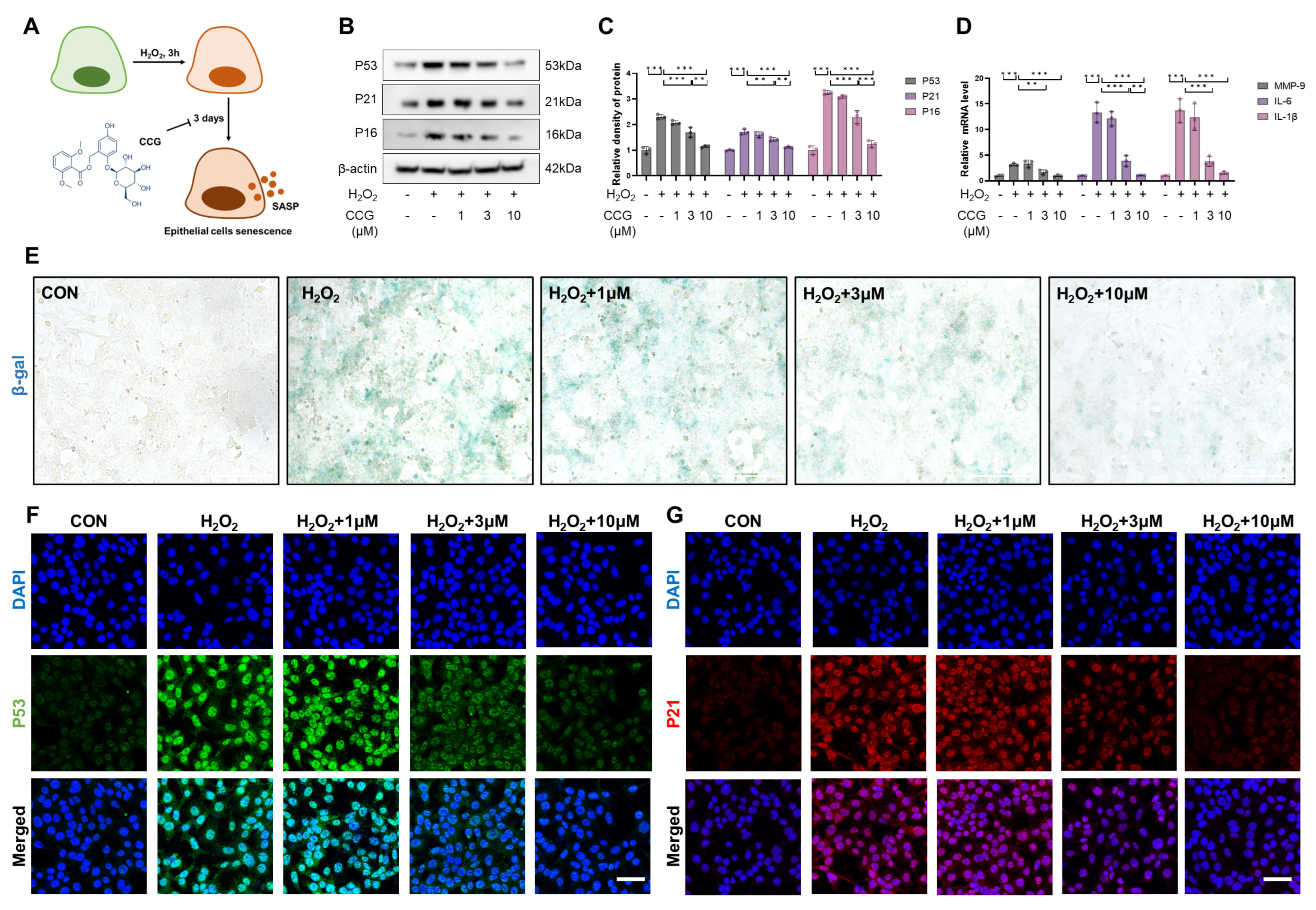
3.3. CCG Alleviated H2O2-Induced Senescence in AECs In Vitro
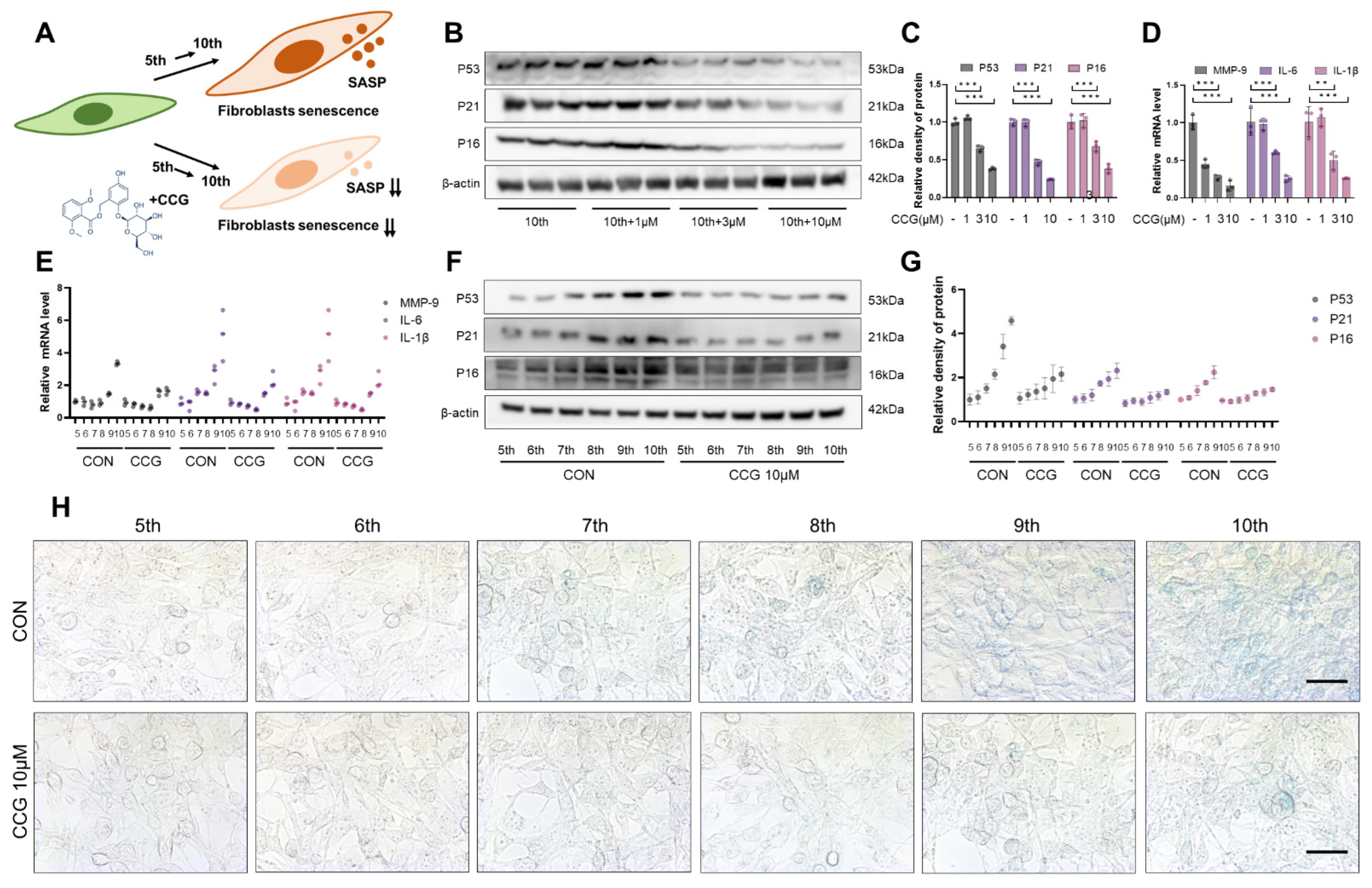
3.4. CCG Attenuated Natural Senescence in Primary Fibroblasts In Vitro
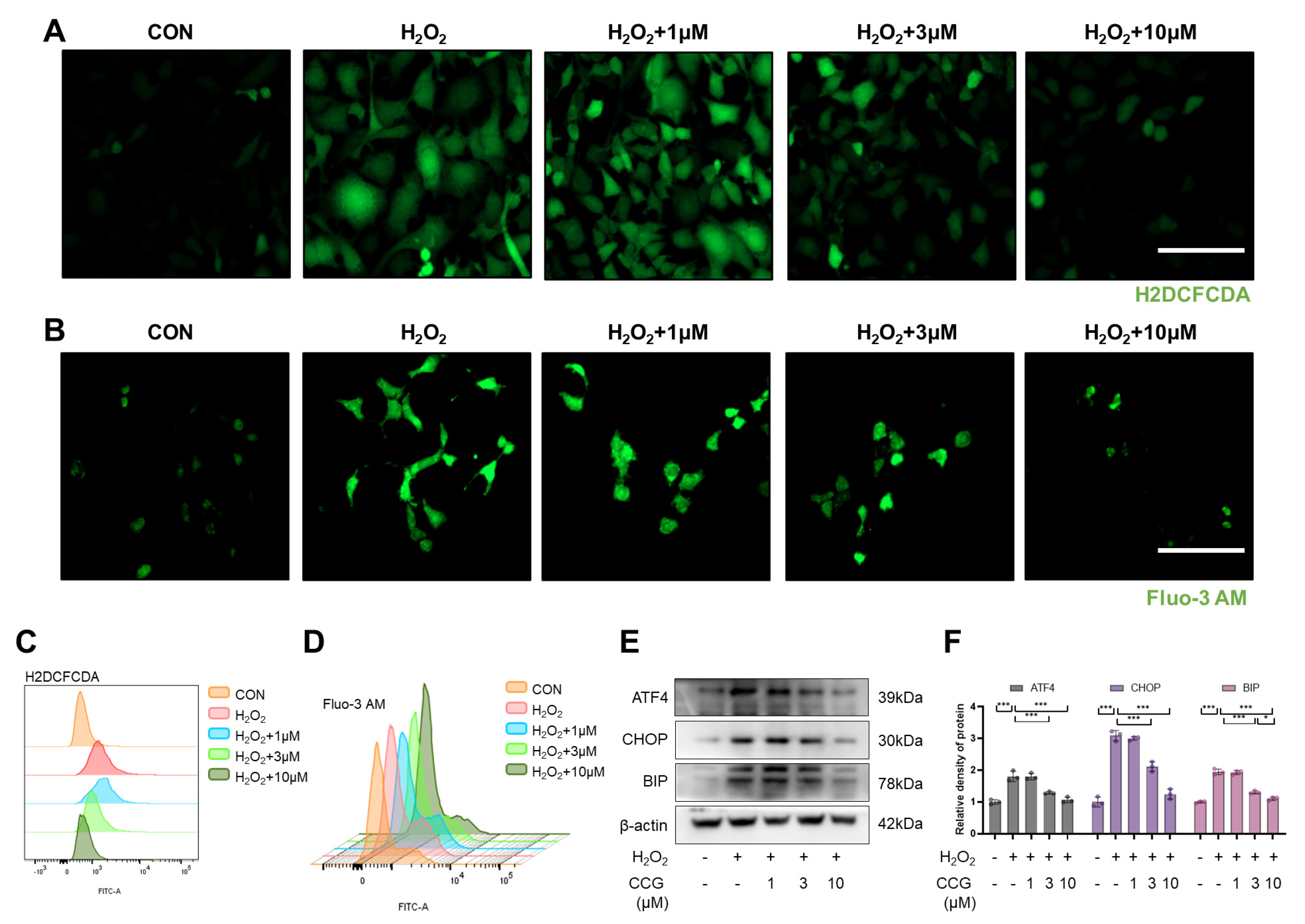
3.5. CCG Reduced ROS-Mediated ERS in AECs
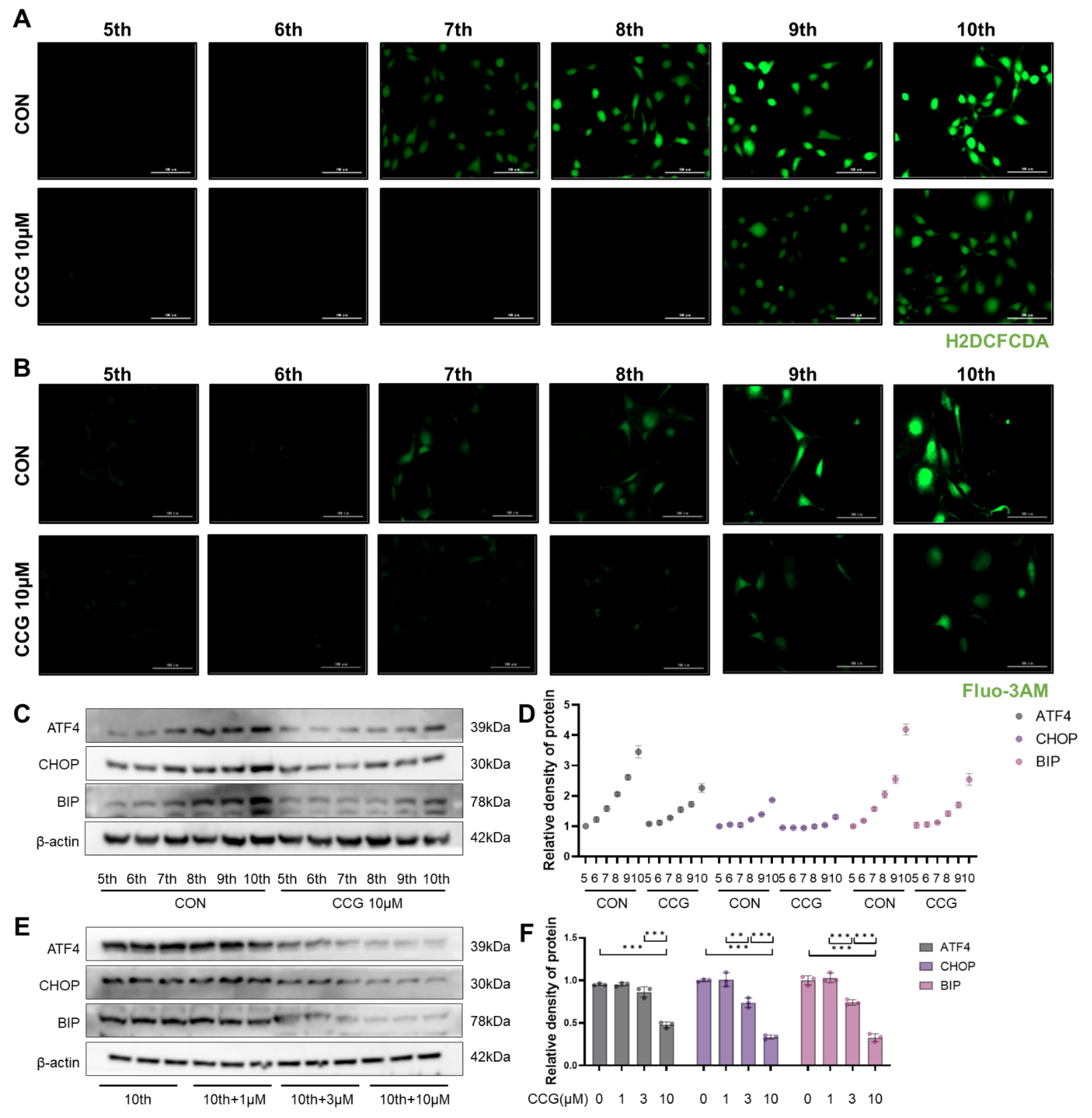
3.6. CCG Alleviated ROS-Mediated ERS in Naturally Senescent Fibroblasts
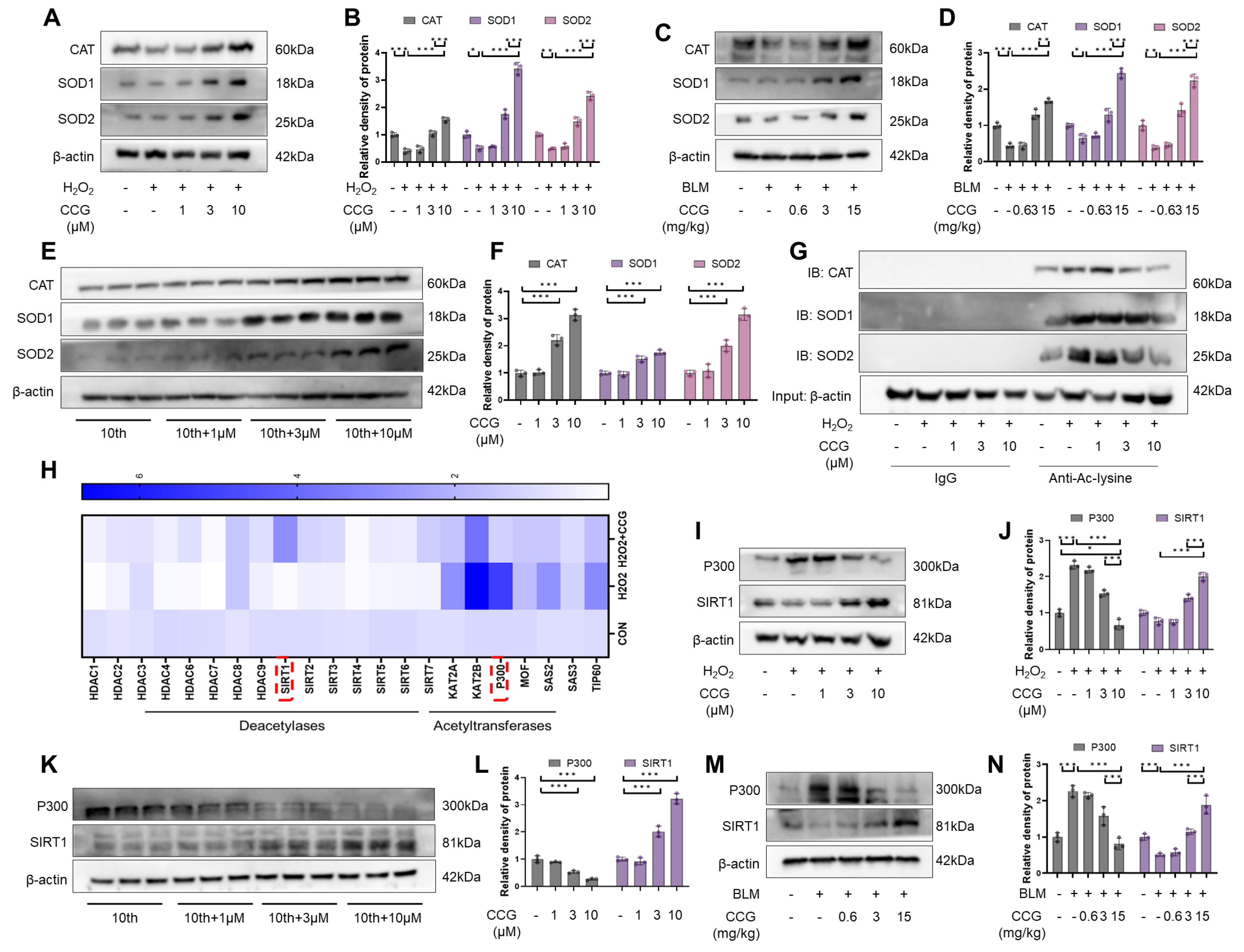
3.7. The SIRT1-P300 Signaling Pathway Mediated the Regulation of Antioxidant Enzymes through CCG
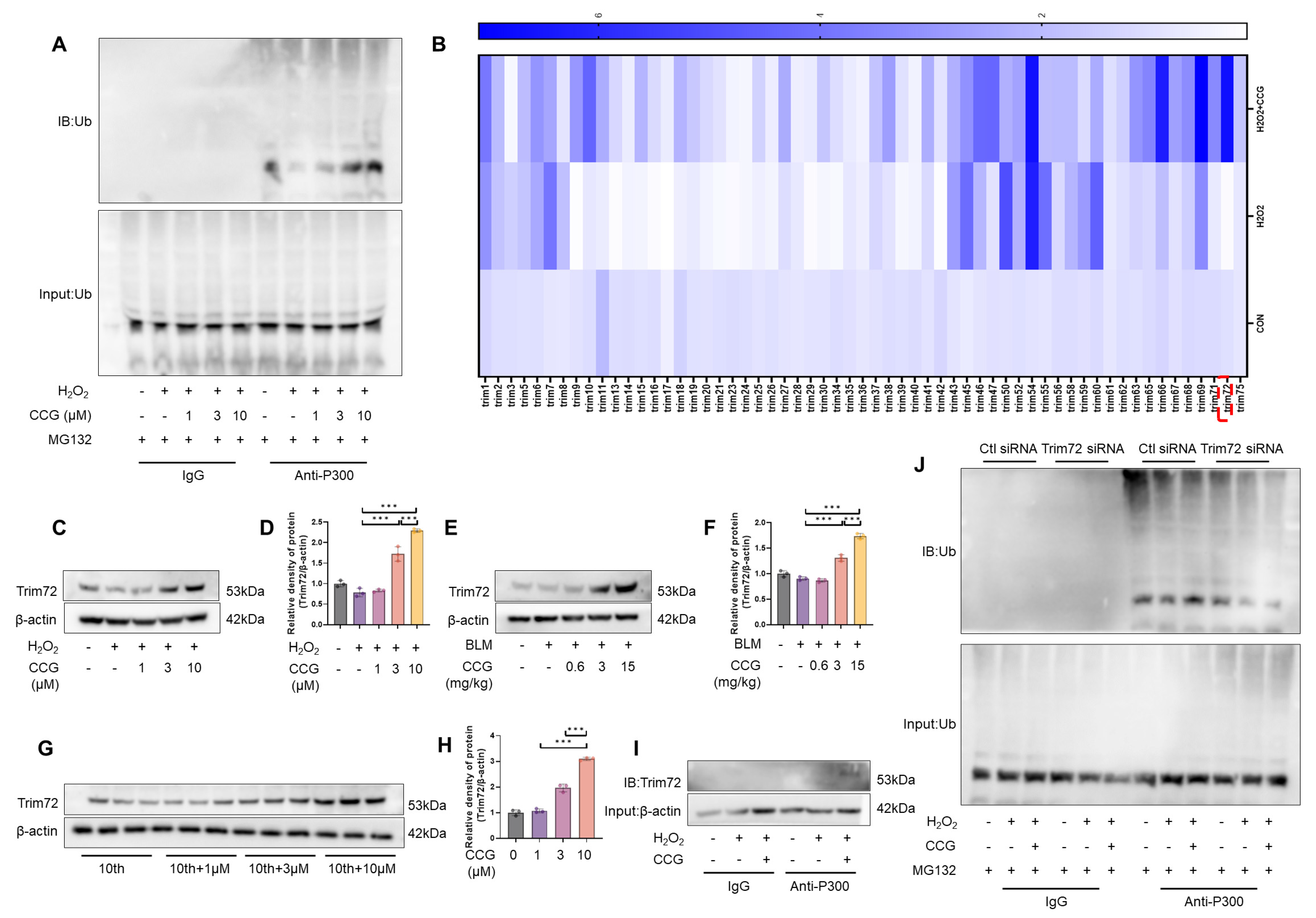
3.8. CCG Promoted the Ubiquitination and Degradation of P300 via Trim72
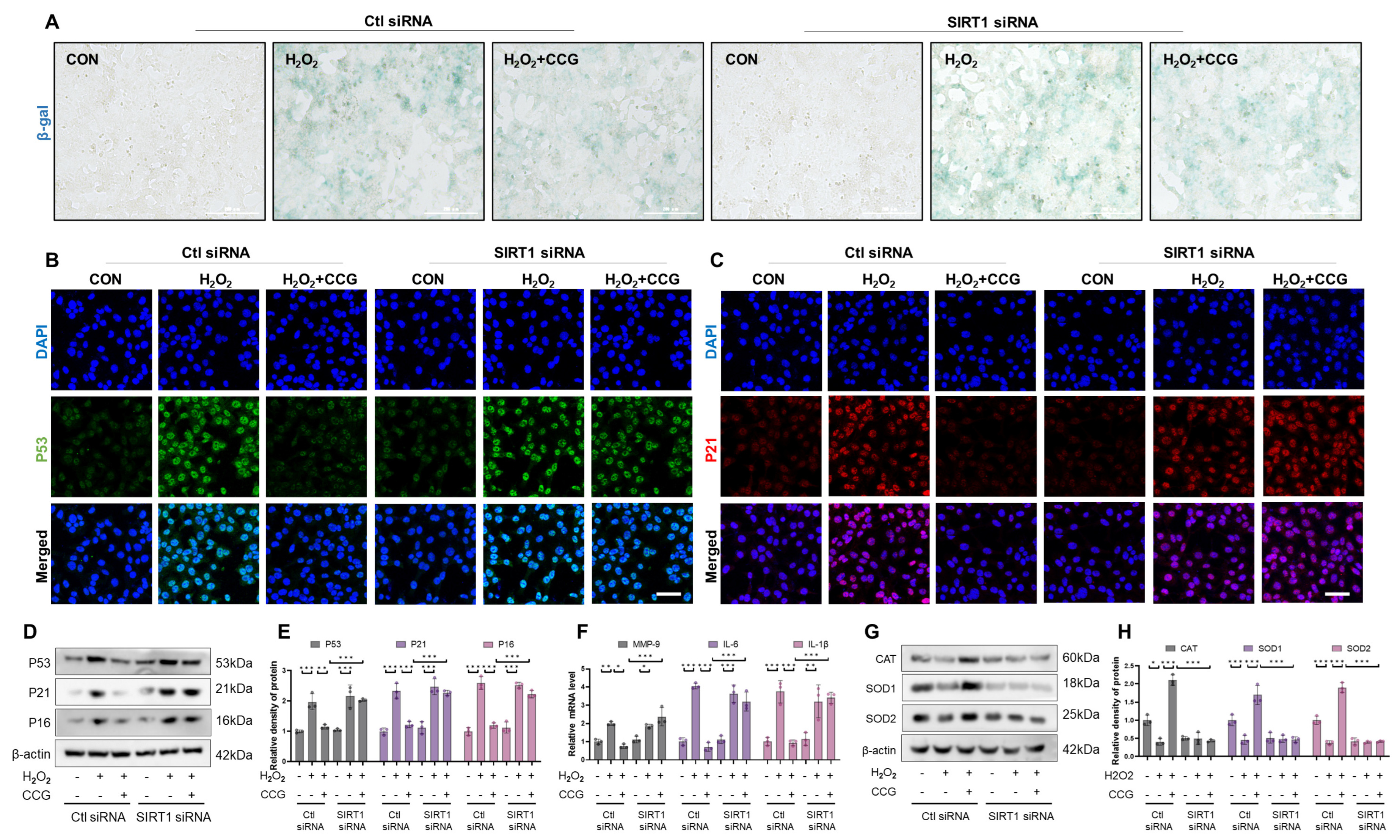
3.9. SIRT1 Mediated the Anti-Senescence Effect of CCG in AECs
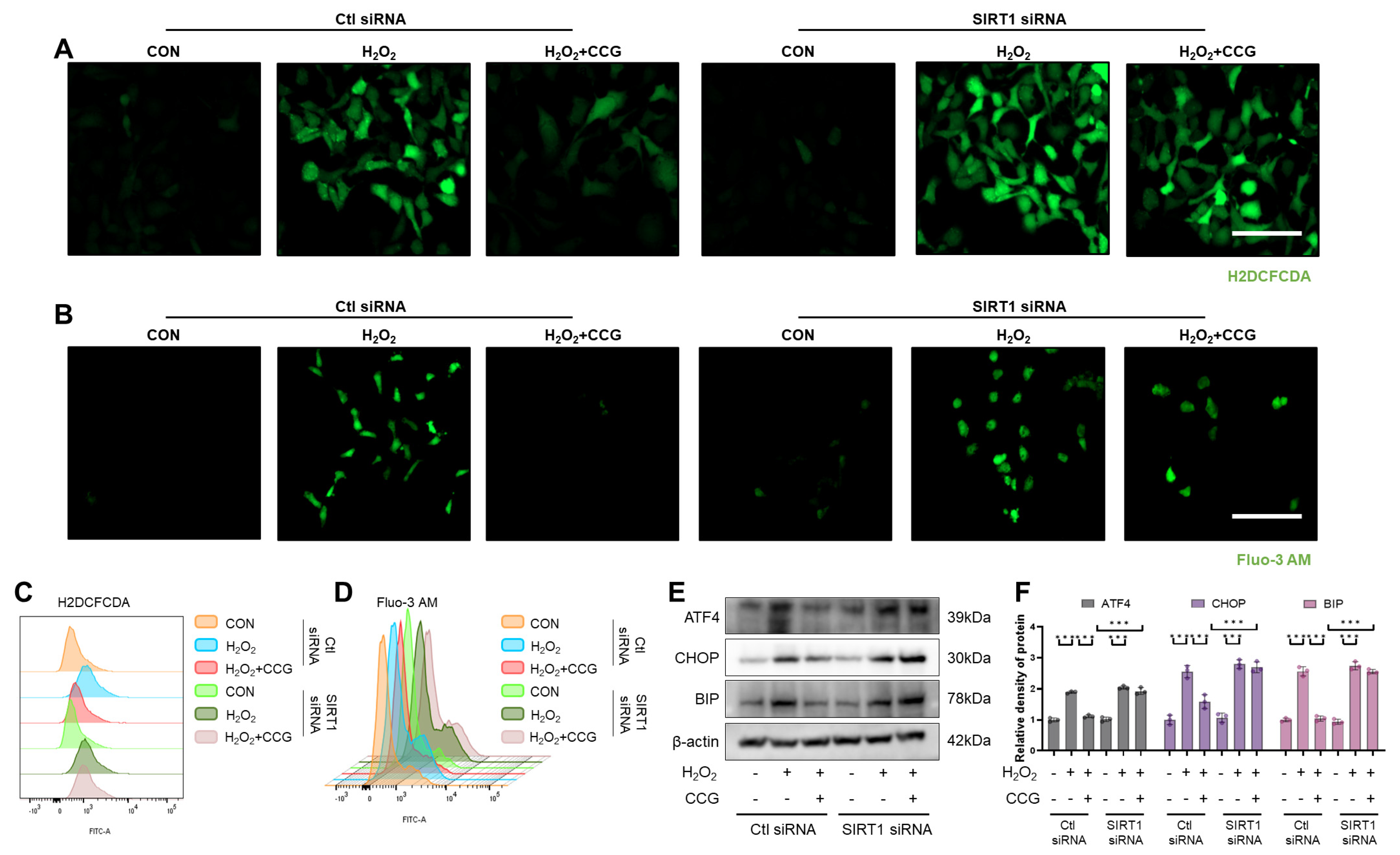
3.10. SIRT1 Mediated the Effect of CCG on ROS-Mediated ERS in AECs
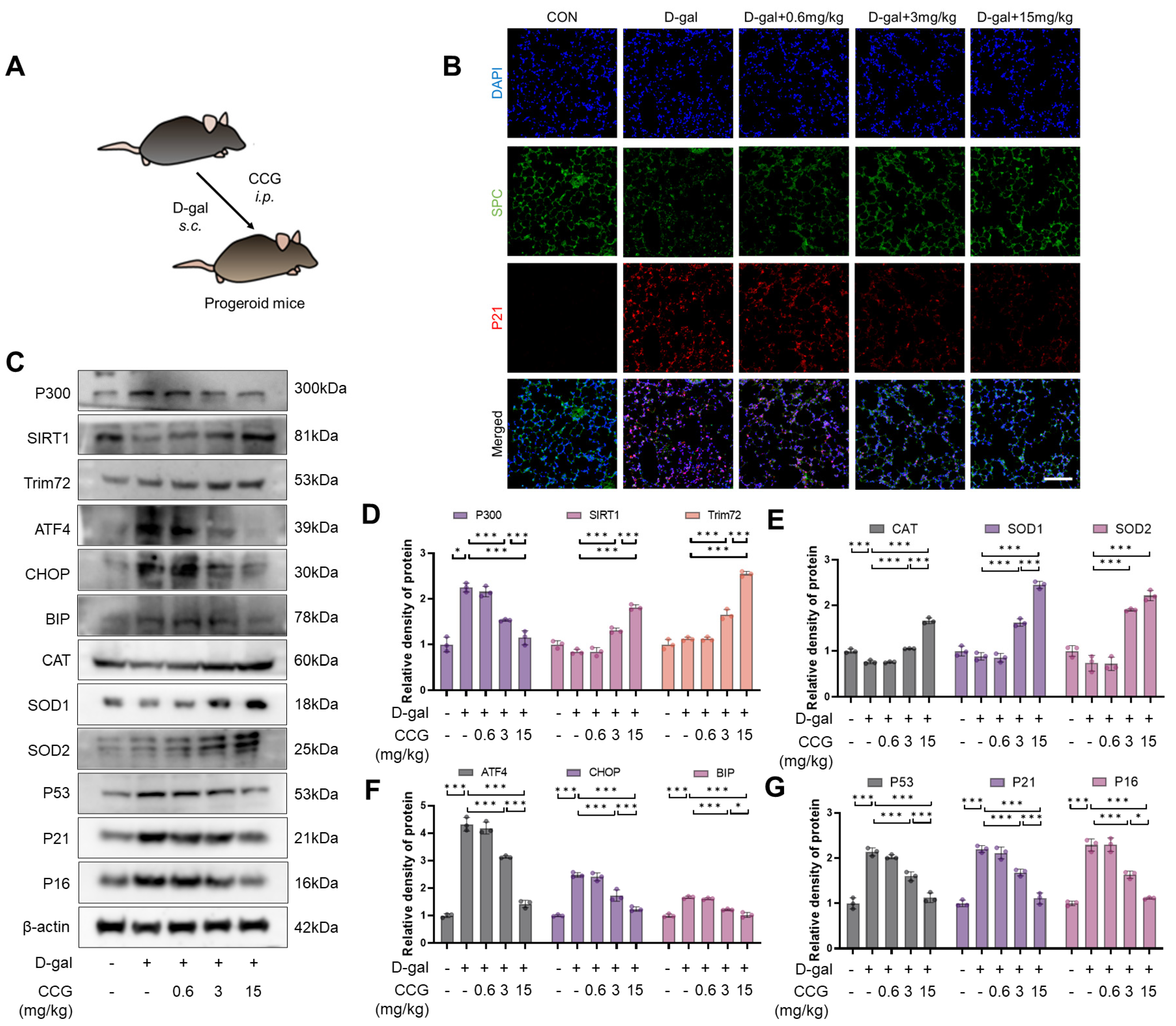
3.11. CCG Inhibited Lung Aging in Progeroid Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am. J. Respir. Crit. Care Med. 2019, 200, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.J.; Stout-Delgado, H.W. Aging and Lung Disease. Annu. Rev. Physiol. 2020, 82, 433–459. [Google Scholar] [CrossRef] [PubMed]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and consequences of endothelial cell senescence. Nat. Rev. Cardiol. 2023, 20, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Yousefzadeh, M.J.; Niedernhofer, L.J.; Robbins, P.D.; Zhu, Y. Cellular senescence: A key therapeutic target in aging and diseases. J. Clin. Investig. 2022, 132, e158450. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.; Ravichandran, R.; Sanborn, K.; Fleming, T.; Wheatcroft, S.B.; Kearney, M.T.; Tokman, S.; Walia, R.; Smith, M.A.; Flint, D.J.; et al. Loss of IGFBP2 mediates alveolar type 2 cell senescence and promotes lung fibrosis. Cell Rep. Med. 2023, 4, 100945. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Kamata, S.; Fujino, N.; Yamada, M.; Grime, K.; Suzuki, S.; Ota, C.; Tando, Y.; Okada, Y.; Sakurada, A.; Noda, M.; et al. Expression of cytochrome P450 mRNAs in Type II alveolar cells from subjects with chronic obstructive pulmonary disease. Pharmacol. Res. Perspect. 2018, 6, e00405. [Google Scholar] [CrossRef]
- Guan, R.; Yuan, L.; Li, J.; Wang, J.; Li, Z.; Cai, Z.; Guo, H.; Fang, Y.; Lin, R.; Liu, W.; et al. Bone morphogenetic protein 4 inhibits pulmonary fibrosis by modulating cellular senescence and mitophagy in lung fibroblasts. Eur. Respir. J. 2022, 60, 2102307. [Google Scholar] [CrossRef]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Baek, A.R.; Hong, J.; Song, K.S.; Jang, A.S.; Kim, D.J.; Chin, S.S.; Park, S.W. Spermidine attenuates bleomycin-induced lung fibrosis by inducing autophagy and inhibiting endoplasmic reticulum stress (ERS)-induced cell death in mice. Exp. Mol. Med. 2020, 52, 2034–2045. [Google Scholar] [CrossRef] [PubMed]
- Grumelli, S.; Corry, D.B.; Song, L.Z.; Song, L.; Green, L.; Huh, J.; Hacken, J.; Espada, R.; Bag, R.; Lewis, D.E.; et al. An immune basis for lung parenchymal destruction in chronic obstructive pulmonary disease and emphysema. PLoS Med. 2004, 1, e8. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A., Jr.; Hogaboam, C.M. Quercetin Enhances Ligand-induced Apoptosis in Senescent Idiopathic Pulmonary Fibrosis Fibroblasts and Reduces Lung Fibrosis In Vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Xu, K.; Han, B.; Bai, Y.; Ma, X.Y.; Ji, Z.N.; Xiong, Y.; Miao, S.K.; Zhang, Y.Y.; Zhou, L.M. MiR-451a suppressing BAP31 can inhibit proliferation and increase apoptosis through inducing ER stress in colorectal cancer. Cell Death Dis. 2019, 10, 152. [Google Scholar] [CrossRef]
- Lin, X.; Dai, Y.; Tong, X.; Xu, W.; Huang, Q.; Jin, X.; Li, C.; Zhou, F.; Zhou, H.; Lin, X.; et al. Excessive oxidative stress in cumulus granulosa cells induced cell senescence contributes to endometriosis-associated infertility. Redox Biol. 2020, 30, 101431. [Google Scholar] [CrossRef]
- Wei, Y.; Sun, L.; Liu, C.; Li, L. Naringin regulates endoplasmic reticulum stress and mitophagy through the ATF3/PINK1 signaling axis to alleviate pulmonary fibrosis. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2023, 396, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Zhong, W.J.; Liu, Y.B.; Duan, J.X.; Jiang, N.; Yang, H.H.; Ma, S.C.; Jin, L.; Hong, J.R.; Zhou, Y.; et al. EETs alleviate alveolar epithelial cell senescence by inhibiting endoplasmic reticulum stress through the Trim25/Keap1/Nrf2 axis. Redox Biol. 2023, 63, 102765. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, J.; Sun, S.; Lin, W.; Li, Y.; Lu, Q.; Li, F.; Yang, Z.; Lu, Y.; Liu, W. Mecheliolide elicits ROS-mediated ERS driven immunogenic cell death in hepatocellular carcinoma. Redox Biol. 2022, 54, 102351. [Google Scholar] [CrossRef]
- Huang, Z.; Gan, S.; Zhuang, X.; Chen, Y.; Lu, L.; Wang, Y.; Qi, X.; Feng, Q.; Huang, Q.; Du, B.; et al. Artesunate Inhibits the Cell Growth in Colorectal Cancer by Promoting ROS-Dependent Cell Senescence and Autophagy. Cells 2022, 11, 2472. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Z.; Liu, Y.; Zhan, Z.; Yang, L.; Wang, C.; Jiang, Q.; Ran, H.; Li, P.; Wang, Z. ROS-responsive liposomes as an inhaled drug delivery nanoplatform for idiopathic pulmonary fibrosis treatment via Nrf2 signaling. J. Nanobiotechnology 2022, 20, 213. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, J.; Zheng, C.; Zhang, P.; Zhou, W.; Cui, W.; Mo, X.; Li, L.; Xu, L.; Gao, J. Nrf2 antioxidant pathway suppresses Numb-mediated epithelial-mesenchymal transition during pulmonary fibrosis. Cell Death Dis. 2018, 9, 83. [Google Scholar] [CrossRef]
- Xiong, S.; Salazar, G.; San Martin, A.; Ahmad, M.; Patrushev, N.; Hilenski, L.; Nazarewicz, R.R.; Ma, M.; Ushio-Fukai, M.; Alexander, R.W. PGC-1 alpha serine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J. Biol. Chem. 2010, 285, 2474–2487. [Google Scholar] [CrossRef]
- Bai, L.L.; Zhang, L.Q.; Ma, J.; Li, J.; Tian, M.; Cao, R.J.; He, X.X.; He, Z.X.; Yu, H.L.; Zhu, X.J. DIP2A is involved in SOD-mediated antioxidative reactions in murine brain. Free Radic. Biol. Med. 2021, 168, 6–15. [Google Scholar] [CrossRef]
- Olmos, Y.; Sánchez-Gómez, F.J.; Wild, B.; García-Quintans, N.; Cabezudo, S.; Lamas, S.; Monsalve, M. SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxid. Redox Signal. 2013, 19, 1507–1521. [Google Scholar] [CrossRef]
- Huang, H.; Tang, S.; Ji, M.; Tang, Z.; Shimada, M.; Liu, X.; Qi, S.; Locasale, J.W.; Roeder, R.G.; Zhao, Y.; et al. p300-Mediated Lysine 2-Hydroxyisobutyrylation Regulates Glycolysis. Mol. Cell 2018, 70, 663–678.e666. [Google Scholar] [CrossRef]
- Zaini, M.A.; Müller, C.; de Jong, T.V.; Ackermann, T.; Hartleben, G.; Kortman, G.; Gührs, K.H.; Fusetti, F.; Krämer, O.H.; Guryev, V.; et al. A p300 and SIRT1 Regulated Acetylation Switch of C/EBPα Controls Mitochondrial Function. Cell Rep. 2018, 22, 497–511. [Google Scholar] [CrossRef]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. SIRT1 is regulated by a PPAR{γ}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef]
- Zeng, Z.; Cheng, S.; Chen, H.; Li, Q.; Hu, Y.; Wang, Q.; Zhu, X.; Wang, J. Activation and overexpression of Sirt1 attenuates lung fibrosis via P300. Biochem. Biophys. Res. Commun. 2017, 486, 1021–1026. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cui, K.; Guo, J.; Mu, W. Curculigoside attenuates osteoporosis through regulating DNMT1 mediated osteoblast activity. Vitr. Cell. Dev. Biol. Anim. 2023, 59, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Liu, S.; Zhang, Q.; Fang, Y.; Yu, Y.; Zhu, L.; Liu, Y.; Gong, W.; Zhao, L.; Qin, L.; et al. Curculigoside attenuates oxidative stress and osteoclastogenesis via modulating Nrf2/NF-κB signaling pathway in RAW264.7 cells. J. Ethnopharmacol. 2021, 275, 114129. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.K.; Hong, Y.J.; Wei, M.; Wu, Y.; Huang, Z.Q.; Chen, R.Z.; Chen, H.Z. Curculigoside attenuates human umbilical vein endothelial cell injury induced by H2O2. J. Ethnopharmacol. 2010, 132, 233–239. [Google Scholar] [CrossRef]
- Kolb, P.; Upagupta, C.; Vierhout, M.; Ayaub, E.; Bellaye, P.S.; Gauldie, J.; Shimbori, C.; Inman, M.; Ask, K.; Kolb, M.R.J. The importance of interventional timing in the bleomycin model of pulmonary fibrosis. Eur. Respir. J. 2020, 55, 1901105. [Google Scholar] [CrossRef] [PubMed]
- Dorry, S.J.; Ansbro, B.O.; Ornitz, D.M.; Mutlu, G.M.; Guzy, R.D. FGFR2 Is Required for AEC2 Homeostasis and Survival after Bleomycin-induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 2020, 62, 608–621. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.T.; Kim, Y.O.; Yan, X.Z.; Abe, H.; Aslam, M.; Park, K.S.; Zhao, X.Y.; Jia, J.D.; Klein, T.; You, H.; et al. Fibroblast Activation Protein Activates Macrophages and Promotes Parenchymal Liver Inflammation and Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 841–867. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Van Westphal, C.; Carpenter-Thompson, R.; Dillip, K.M.; Vij, N. Nicotine exposure induces bronchial epithelial cell apoptosis and senescence via ROS mediated autophagy-impairment. Free Radic. Biol. Med. 2016, 97, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, J.; Lee, R.; Lee, E.; Choi, T.G.; Lee, A.S.; Yoon, Y.I.; Park, G.C.; Namgoong, J.M.; Lee, S.G.; et al. Therapeutic strategies for liver diseases based on redox control systems. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 156, 113764. [Google Scholar] [CrossRef]
- Adcock, I.M.; Lee, K.Y. Abnormal histone acetylase and deacetylase expression and function in lung inflammation. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2006, 55, 311–321. [Google Scholar] [CrossRef]
- Luo, G.; Liu, B.; Fu, T.; Liu, Y.; Li, B.; Li, N.; Geng, Q. The Role of Histone Deacetylases in Acute Lung Injury-Friend or Foe. Int. J. Mol. Sci. 2023, 24, 97876. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Wang, Y.; Chao, Y.; Zhang, J.; Jia, Y.; Tie, J.; Hu, D. Regulation of SIRT1 and Its Roles in Inflammation. Front. Immunol. 2022, 13, 831168. [Google Scholar] [CrossRef]
- Miao, J.; Liu, J.; Niu, J.; Zhang, Y.; Shen, W.; Luo, C.; Liu, Y.; Li, C.; Li, H.; Yang, P.; et al. Wnt/β-catenin/RAS signaling mediates age-related renal fibrosis and is associated with mitochondrial dysfunction. Aging Cell 2019, 18, e13004. [Google Scholar] [CrossRef]
- Sun, K.; Yang, P.; Zhao, R.; Bai, Y.; Guo, Z. Matrine Attenuates D-Galactose-Induced Aging-Related Behavior in Mice via Inhibition of Cellular Senescence and Oxidative Stress. Oxidative Med. Cell. Longev. 2018, 2018, 7108604. [Google Scholar] [CrossRef]
- Raghu, G.; Remy-Jardin, M.; Richeldi, L.; Thomson, C.C.; Inoue, Y.; Johkoh, T.; Kreuter, M.; Lynch, D.A.; Maher, T.M.; Martinez, F.J.; et al. Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2022, 205, e18–e47. [Google Scholar] [CrossRef]
- Raghu, G.; Chen, S.Y.; Yeh, W.S.; Maroni, B.; Li, Q.; Lee, Y.C.; Collard, H.R. Idiopathic pulmonary fibrosis in US Medicare beneficiaries aged 65 years and older: Incidence, prevalence, and survival, 2001-11. Lancet. Respir. Med. 2014, 2, 566–572. [Google Scholar] [CrossRef]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.I.; Dauti, S.; Kim, H.J.; Park, S.H.; Park, J.S.; Lee, C.W. Risk factors for interstitial lung disease: A 9-year Nationwide population-based study. BMC Pulm. Med. 2018, 18, 96. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dollé, M.E.; et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Parimon, T.; Hohmann, M.S.; Yao, C. Cellular Senescence: Pathogenic Mechanisms in Lung Fibrosis. Int. J. Mol. Sci. 2021, 22, 6214. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Álvarez, D.; Cárdenes, N.; Sellarés, J.; Bueno, M.; Corey, C.; Hanumanthu, V.S.; Peng, Y.; D’Cunha, H.; Sembrat, J.; Nouraie, M.; et al. IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L1164–L1173. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Chen, B.; Cai, H.R.; Xue, S.; You, W.J.; Liu, B.; Jiang, H.D. Bile acids induce activation of alveolar epithelial cells and lung fibroblasts through farnesoid X receptor-dependent and independent pathways. Respirology (Carlton Vic.) 2016, 21, 1075–1080. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, Q.; Li, M.; Duan, Q.; Zhao, Y.; Zhang, H. The effects of endothelium-specific CYP2J2 overexpression on the attenuation of retinal ganglion cell apoptosis in a glaucoma rat model. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2019, 33, 11194–11209. [Google Scholar] [CrossRef]
- Tian, Y.; Shi, H.; Zhang, D.; Wang, C.; Zhao, F.; Li, L.; Xu, Z.; Jiang, J.; Li, J. Nebulized inhalation of LPAE-HDAC10 inhibits acetylation-mediated ROS/NF-κB pathway for silicosis treatment. J. Control. Release Off. J. Control. Release Soc. 2023, 364, 618–631. [Google Scholar] [CrossRef]
- Roman, J.; Zhu, J.; Ritzenthaler, J.D.; Zelko, I.N. Epigenetic regulation of EC-SOD expression in aging lung fibroblasts: Role of histone acetylation. Free Radic. Biol. Med. 2017, 112, 212–223. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, L.; Shen, Y.; He, Y.; Cheng, G.; Yin, M.; Zhang, Q.; Qin, L. Curculigoside Protects against Excess-Iron-Induced Bone Loss by Attenuating Akt-FoxO1-Dependent Oxidative Damage to Mice and Osteoblastic MC3T3-E1 Cells. Oxidative Med. Cell. Longev. 2019, 2019, 9281481. [Google Scholar] [CrossRef]
- Du, P.; Zhang, X.; Luo, K.; Li, Y.; Fu, C.; Xiao, J.; Xiao, Q. Curculigoside mitigates hepatic ischemia/reperfusion-induced oxidative stress, inflammation, and apoptosis via activation of the Nrf-2/HO-1 pathway. Hum. Exp. Toxicol. 2022, 41, 9603271221087146. [Google Scholar] [CrossRef] [PubMed]
- Dang, R.; Wang, M.; Li, X.; Wang, H.; Liu, L.; Wu, Q.; Zhao, J.; Ji, P.; Zhong, L.; Licinio, J.; et al. Edaravone ameliorates depressive and anxiety-like behaviors via Sirt1/Nrf2/HO-1/Gpx4 pathway. J. Neuroinflamm. 2022, 19, 41. [Google Scholar] [CrossRef]
- An, Y.; Li, Y.; Hou, Y.; Huang, S.; Pei, G. Alzheimer’s Amyloid-β Accelerates Cell Senescence and Suppresses the SIRT1/NRF2 Pathway in Human Microglial Cells. Oxidative Med. Cell. Longev. 2022, 2022, 3086010. [Google Scholar] [CrossRef]
- Xu, J.J.; Cui, J.; Lin, Q.; Chen, X.Y.; Zhang, J.; Gao, E.H.; Wei, B.; Zhao, W. Protection of the enhanced Nrf2 deacetylation and its downstream transcriptional activity by SIRT1 in myocardial ischemia/reperfusion injury. Int. J. Cardiol. 2021, 342, 82–93. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef]
- Ota, H.; Eto, M.; Kano, M.R.; Kahyo, T.; Setou, M.; Ogawa, S.; Iijima, K.; Akishita, M.; Ouchi, Y. Induction of endothelial nitric oxide synthase, SIRT1, and catalase by statins inhibits endothelial senescence through the Akt pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2205–2211. [Google Scholar] [CrossRef]
- Cong, X.; Nagre, N.; Herrera, J.; Pearson, A.C.; Pepper, I.; Morehouse, R.; Ji, H.L.; Jiang, D.; Hubmayr, R.D.; Zhao, X. TRIM72 promotes alveolar epithelial cell membrane repair and ameliorates lung fibrosis. Respir. Res. 2020, 21, 132. [Google Scholar] [CrossRef]
- Jeffers, A.; Qin, W.; Owens, S.; Koenig, K.B.; Komatsu, S.; Giles, F.J.; Schmitt, D.M.; Idell, S.; Tucker, T.A. Glycogen Synthase Kinase-3β Inhibition with 9-ING-41 Attenuates the Progression of Pulmonary Fibrosis. Sci. Rep. 2019, 9, 18925. [Google Scholar] [CrossRef]
- Urbanska, M.; Kazmierska-Grebowska, P.; Kowalczyk, T.; Caban, B.; Nader, K.; Pijet, B.; Kalita, K.; Gozdz, A.; Devijver, H.; Lechat, B.; et al. GSK3β activity alleviates epileptogenesis and limits GluA1 phosphorylation. eBioMedicine 2019, 39, 377–387. [Google Scholar] [CrossRef]
- Su, P.P.; Liu, D.W.; Zhou, S.J.; Chen, H.; Wu, X.M.; Liu, Z.S. Down-regulation of Risa improves podocyte injury by enhancing autophagy in diabetic nephropathy. Mil. Med. Res. 2022, 9, 23. [Google Scholar] [CrossRef]
- Li, Y.; Cui, W.; Song, B.; Ye, X.; Li, Z.; Lu, C. Autophagy-Sirtuin1(SIRT1) Alleviated the Coronary Atherosclerosis (AS)in Mice through Regulating the Proliferation and Migration of Endothelial Progenitor Cells (EPCs) via wnt/β-catenin/GSK3β Signaling Pathway. J. Nutr. Health Aging 2022, 26, 297–306. [Google Scholar] [CrossRef]
- Xu, T.; Wang, S.; Li, X.; Li, X.; Qu, K.; Tong, H.; Zhang, R.; Bai, S.; Fan, J. Lithium chloride represses abdominal aortic aneurysm via regulating GSK3β/SIRT1/NF-κB signaling pathway. Free Radic. Biol. Med. 2021, 166, 1–10. [Google Scholar] [CrossRef]
- Khan, H.; Bangar, A.; Grewal, A.K.; Singh, T.G. Mechanistic Implications of GSK and CREB Crosstalk in Ischemia Injury. Neurotox. Res. 2023, 42, 1. [Google Scholar] [CrossRef]
- Monteserin-Garcia, J.; Al-Massadi, O.; Seoane, L.M.; Alvarez, C.V.; Shan, B.; Stalla, J.; Paez-Pereda, M.; Casanueva, F.F.; Stalla, G.K.; Theodoropoulou, M. Sirt1 inhibits the transcription factor CREB to regulate pituitary growth hormone synthesis. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 1561–1571. [Google Scholar] [CrossRef]
- Zhang, J.S.; Koenig, A.; Harrison, A.; Ugolkov, A.V.; Fernandez-Zapico, M.E.; Couch, F.J.; Billadeau, D.D. Mutant K-Ras increases GSK-3β gene expression via an ETS-p300 transcriptional complex in pancreatic cancer. Oncogene 2011, 30, 3705–3715. [Google Scholar] [CrossRef]
- Jin, P.; Qi, D.; Cui, Y.; Lenahan, C.; Deng, S.; Tao, X. Activation of LRP6 with HLY78 Attenuates Oxidative Stress and Neuronal Apoptosis via GSK3β/Sirt1/PGC-1α Pathway after ICH. Oxidative Med. Cell. Longev. 2022, 2022, 7542468. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Source | Dilution Ratio |
|---|---|---|
| Anti-Collagen Ⅰ Polyclonal antibody | Abcam | 1:5000 |
| Anti-α-SMA Polyclonal antibody | Proteintech | 1:4000 |
| Anti-P53 Monoclonal antibody | Proteintech | 1:10,000 |
| Anti-P21 Polyclonal antibody | Abcam | 1:5000 |
| Anti-P16 Polyclonal antibody | Abcam | 1:5000 |
| Anti-ATF4 Polyclonal antibody | Proteintech | 1:1000 |
| Anti-CHOP Polyclonal antibody | Proteintech | 1:1000 |
| Anti-BIP Polyclonal antibody | Proteintech | 1:6000 |
| Anti-Catalase Polyclonal antibody | Abcam | 1:2000 |
| Anti-Superoxide Dismutase 1 Polyclonal antibody | Abcam | 1:20,000 |
| Anti-SOD2 Polyclonal antibody | Abcam | 1:2000 |
| Anti-P300 Polyclonal antibody | Zen-Bio | 1:1000 |
| Anti-SIRT1 Polyclonal antibody | Proteintech | 1:2000 |
| Anti-Ubiquitin Polyclonal antibody | Abcam | 1:2000 |
| Anti-Trim72 Polyclonal antibody | Proteintech | 1:5000 |
| Anti-GSK3β Polyclonal antibody | Proteintech | 1:4000 |
| Anti-Phospho-GSK3β Monoclonal antibody | Proteintech | 1:5000 |
| Anti-β-Actin Recombinant antibody | Proteintech | 1:20,000 |
| Name | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| m-Colα1 | ATGTTCAGCTTTGTGGACCTC | CTGTACGCAGGTGATTGGTG |
| m-α-sma | TGGCTATTCAGGCTGTGCTGTC | CAATCTCACGCTCGGCAGTAGT |
| m-MMP-9 | AGACGCCCATTTCGACGATGAC | CAAACCGAGTTGGAACCACGAC |
| m-IL-6 | CCAGGAGCCCAGCTATGAAC | CCCAGGGAGAAGGCAACTG |
| m-IL-1β | CAGGCAGGCAGTATCACTCA | AGCTCATATGGGTCCGACAG |
| m-Trim1 | AGTTGTTTGAAGACCCCCTTCT | TGTAGGACACTGGAAAGCAGTAA |
| m-Trim2 | TGGACAGTTCAAAAGTCGTTTCG | AATGCTAACCCACTTGTTGTCAT |
| m-Trim3 | GCGTCTCAGCCCTACAAAACA | AAACTCCATTGTCTTGCCTTCA |
| m-Trim5 | AAGAAAGTTCCGAGCCCCTG | GTAGCGTTGAGCCTCTGTGA |
| m-Trim6 | ATGACTTCAACAGTCTTGGTGG | TTCCCAGGCTGATAGGAGGTC |
| m-Trim7 | ACAGAAACAGAATGAGAACCTGG | GCTCAGTGTGCTTTTGAACTCC |
| m-Trim8 | AGGGACACTCGGTGTGTGA | TGTCTGCCGCAAGTCTTCATC |
| m-Trim9 | CTTGGGCAATAACTGAAGGAGG | GCTGGAGTAGAAGTCGGGG |
| m-Trim10 | GGAACACGGGGAGAAAATCTAC | AGACACACGAGACACTTCTGT |
| m-Trim11 | GCCCTCATCTCCGAGCTTG | CGCAGCACTCAATGCAGAG |
| m-Trim13 | TGATGACCCCCGAGTGTTG | TTTCCTTACGGCAGGTAGGAC |
| m-Trim14 | GTGCGTGTGCAGAAGCTAATC | CTGCGTAAACCTTGAGCCTTT |
| m-Trim15 | CCTGAGCGAGACCTACTGTGA | AGAGCTTCTAACCGACTCCTG |
| m-Trim16 | TCTTGGGGCCAGCAGAGTAA | CTCACAGTAGTTCACCATGCAG |
| m-Trim17 | CTTGCCAGACGGTTACAAGAG | CTCAGCCACTTTTGTCAGGAG |
| m-Trim18 | CTGTGACGGCACCTGTCTC | AAACGGCTGACTGTTGGTCTT |
| m-Trim19 | CAGGCCCTAGAGCTGTCTAAG | ATACACTGGTACAGGGTGTGC |
| m-Trim20 | TCATCTGCTAAACACCCTGGA | GGGATCTTAGAGTGGCCCTTC |
| m-Trim21 | GGGAGGAGGTCACCTGTTCTA | CATTACCGTGTTCTTTTGCAGC |
| m-Trim23 | ACCAGAAGCTAATCAGATCCGA | TGGCTCACAGTCAAACTGCTG |
| m-Trim24 | TCAACAGGCCATAAAACAGTGG | GGCACTCGGGACATGAACTG |
| m-Trim25 | GATGAGACGTGGGTCGTCC | TCTGTGTGAGCCATTCCAATTC |
| m-Trim26 | TCGGCCAGTGGATACCTACAT | CTGCACTTGTGATTGTGGGG |
| m-Trim27 | GGAGCAAATCCAGAACCGACT | GCCCCGTTGATGCTGTTATAG |
| m-Trim28 | CGGCGCTATGGTGGATTGT | GGTTAGCATCCTGGGAATCAGAA |
| m-Trim29 | AGAATGGCACTAAAGCAGACAG | AAATAGGCCACTCTTCCCCTC |
| m-Trim30 | CTGTGAGTGCTGATTGTAACCA | ACTCGGCATACAGGGCAGT |
| m-Trim34 | GTAATAACGGTATCTTGGGCTCC | TGCGTTGTCTAACATCAAACCTT |
| m-Trim35 | TTCCGGGCCAAGTGTAAGAAC | CCAAGTCGTTTGCACCTCA |
| m-Trim36 | GGCTACATTATGGAATTGCTTGC | GGATCAGCGGGTGGGTAAAC |
| m-Trim37 | TCCAAGCTCTGTTGTTTCAGC | TTCCGCCCAACGACAGTTC |
| m-Trim38 | ATGGGCTCAGACTTTAGCACG | CTGTTTTTGGGCTGACATTGC |
| m-Trim39 | AACAGCTAATTGCGGATGTGA | ACAAACTTGACGCTTTTCCGAT |
| m-Trim40 | TCATCTGCTGGTCTTCTCCC | CAGGAGCTCCAAACCCCAAT |
| m-Trim41 | ATGAGCCGCATGTTTTGTCAG | GCCCCTAGTACACAGCAGT |
| m-Trim42 | ATGGAGACGGCTATGTGTGTC | GCACTTACAGTTGGGGTCATT |
| m-Trim43 | TGAAGGACTATAGGCGGTGGA | AGTGTTCACGTCCTATGCGG |
| m-Trim45 | TCAGGCAAGACTCATTGTCCT | ACGGATGTCCACTACTGAGAAT |
| m-Trim46 | GGTGAGGATATGCAGACCTTCA | TTGTGGGTACAAGGCAGCAC |
| m-Trim47 | GGTGAGCCAGATGTTTGCC | TCCCTCTTCGATGAACCCCAT |
| m-Trim50 | CCCATTTGCCTGGAGGTCTTC | CAGGACAGCATAGCTCGGAG |
| m-Trim52 | ATGCAGTCACTTCGGGAAGAA | CTATGGCTATGACCGACCCAC |
| m-Trim54 | GGAGAAGCAGCTCATTTGCC | CCTCCTGAAGACACCGTTGTG |
| m-Trim55 | AAAGCAACTGATCTGTCCCAT | TGTGGGTAAGTACGGGTTAGAG |
| m-Trim56 | CAGCGATTTCCTAGCCTGTAAA | GACCACCGATGTCCAGTTGT |
| m-Trim58 | AGTGGGACTGATGAGTGGGT | AATGAAGCCTCGGGCAGTAG |
| m-Trim59 | ATGCACAATTTTGAGGAGGAGT | GCAGTTAGGACACTTGAGTGGAA |
| m-Trim60 | GCACAACTTCTGTTTTGCCTG | CAGTCATGTTACGGAACTGGTAG |
| m-Trim61 | CATCTTGCCCCCTGAAAGAAC | GGTCAGCATCAGCGGATCAC |
| m-Trim62 | CTTCGAGGAGTTGCAGAGAGA | GGCGTGAACATAATGCGGTC |
| m-Trim63 | GTGTGAGGTGCCTACTTGCTC | CTGCTCGCAGTAGATGCTCA |
| m-Trim65 | GAGGACGTGGTGACTTGCTC | GCTAGGCATGGGGTTTCGAT |
| m-Trim66 | CTTTGCCTTGTACTGCCCTCT | TTTTCCACGGGCCAAACAAAG |
| m-Trim67 | CCACTCTCTGCGAGCAATG | GCAGGCTCTTGGTAGAGGAC |
| m-Trim68 | TCCCAGAACTTGAGCTACACC | GCTCAGTCTTCTGTCCTTGGA |
| m-Trim69 | AACCACCACCCATTTACCCTC | ACGCCATGAATCCTGGATGC |
| m-Trim71 | CAAGCTGGAGAGCACCATCA | TGGATTTCTTATGTGCCACCTG |
| m-Trim72 | CCGCAGGCTCTAAGCACTAAC | GGTGGCTGAACTAGCCGAT |
| m-Trim75 | TTGGGTACCAACTGTCAGCC | AGACGGACCTTGTCTACAACA |
| m-β-actin | TTCCAGCCTTCCTTCTTG | GGGAGCCAGAGCAGTAATC |
| m-HDAC1 | AGTCTGTTACTACTACGACGGG | TGAGCAGCAAATTGTGAGTCAT |
| m-HDAC2 | GGAGGAGGCTACACAATCCG | TCTGGAGTGTTCTGGTTTGTCA |
| m-HDAC3 | GCCAAGACCGTGGCGTATT | GTCCAGCTCCATAGTGGAAGT |
| m-HDAC4 | CTGCAAGTGGCCCCTACAG | CTGCTCATGTTGACGCTGGA |
| m-HDAC5 | TGCAGCACGTTTTGCTCCT | GACAGCTCCCCAGTTTTGGT |
| m-HDAC6 | TCCACCGGCCAAGATTCTTC | CAGCACACTTCTTTCCACCAC |
| m-HDAC7 | GGCAGGCTTACACCAGCAA | TGGGCAGGCTGTAGGGAATA |
| m-HDAC8 | ACTATTGCCGGAGATCCAATGT | CCTCCTAAAATCAGAGTTGCCAG |
| m-SIRT1 | GCTGACGACTTCGACGACG | TCGGTCAACAGGAGGTTGTCT |
| m-SIRT2 | GCCTGGGTTCCCAAAAGGAG | GAGCGGAAGTCAGGGATACC |
| m-SIRT3 | ATCCCGGACTTCAGATCCCC | CAACATGAAAAAGGGCTTGGG |
| m-SIRT4 | GTGGAAGAATAAGAATGAGCGG A | GGCACAAATAACCCCGAGG |
| m-SIRT5 | CTCCGGGCCGATTCATTTCC | GCGTTCGCAAAACACTTCCG |
| m-SIRT6 | ATGTCGGTGAATTATGCAGCA | GCTGGAGGACTGCCACATTA |
| m-SIRT7 | CAGGTGTCACGCATCCTGAG | GCCCGTGTAGACAACCAAGT |
| m-KAT2A | CGAGTTGTGCCGTAGCTGTGA | ACCATTCCCAAGAGCCGGTTA |
| m-KAT2B | GAAGCCGCCATTTGAGAAGC | AGTTGATGCGGTTCAGAAACA |
| m-P300 | GCCCGTGTAGACAACCAAGT | GCCCGTGTAGACAACCAAGT |
| m-MOF | CTGGAAGGGCCAGCATGTTA | GGTTAGAGGCCAGGAAACCC |
| m-SAS2 | TTCGGCTCGCTGCTCATCC | GACTCTGCTCCCTCGCCAC |
| m-SAS3 | GCAGTCTCACCCAGACCACC | CAAAATGACAGCCGAAATTG |
| m-TIP60 | GGCTGGACTTAAAGA AGAT | GGACTTAAAGAAGATCCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, W.; Deng, L.; Qian, R.; Huang, X.; Liu, W.; Tang, S. Curculigoside Attenuates Endoplasmic Reticulum Stress-Induced Epithelial Cell and Fibroblast Senescence by Regulating the SIRT1-P300 Signaling Pathway. Antioxidants 2024, 13, 420. https://doi.org/10.3390/antiox13040420
Xie W, Deng L, Qian R, Huang X, Liu W, Tang S. Curculigoside Attenuates Endoplasmic Reticulum Stress-Induced Epithelial Cell and Fibroblast Senescence by Regulating the SIRT1-P300 Signaling Pathway. Antioxidants. 2024; 13(4):420. https://doi.org/10.3390/antiox13040420
Chicago/Turabian StyleXie, Weixi, Lang Deng, Rui Qian, Xiaoting Huang, Wei Liu, and Siyuan Tang. 2024. "Curculigoside Attenuates Endoplasmic Reticulum Stress-Induced Epithelial Cell and Fibroblast Senescence by Regulating the SIRT1-P300 Signaling Pathway" Antioxidants 13, no. 4: 420. https://doi.org/10.3390/antiox13040420
APA StyleXie, W., Deng, L., Qian, R., Huang, X., Liu, W., & Tang, S. (2024). Curculigoside Attenuates Endoplasmic Reticulum Stress-Induced Epithelial Cell and Fibroblast Senescence by Regulating the SIRT1-P300 Signaling Pathway. Antioxidants, 13(4), 420. https://doi.org/10.3390/antiox13040420