
Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection
 ,
,  and
and 
Abstract
:1. Introduction
2. Basic Mechanisms of Nitric Oxide (•NO) Regulation
2.1. •NO Synthesis in the Body
2.2. Mechanisms of •NO Cytotoxicity
2.3. Involvement of •NO in the Formation of Mitochondrial Dysfunction and Mitoptosis
3. Effects of •NO
3.1. Apoptosis and •NO
3.2. Anti-Apoptotic Effects of •NO
4. •NO in Health and Disease: Interactions, Clinical Relevance, and Therapeutic Implications
4.1. •NO and Superoxide Anion
4.2. •NO and Arterial Hypertension
4.3. •NO and the Thiol–Disulfide System of Neurons
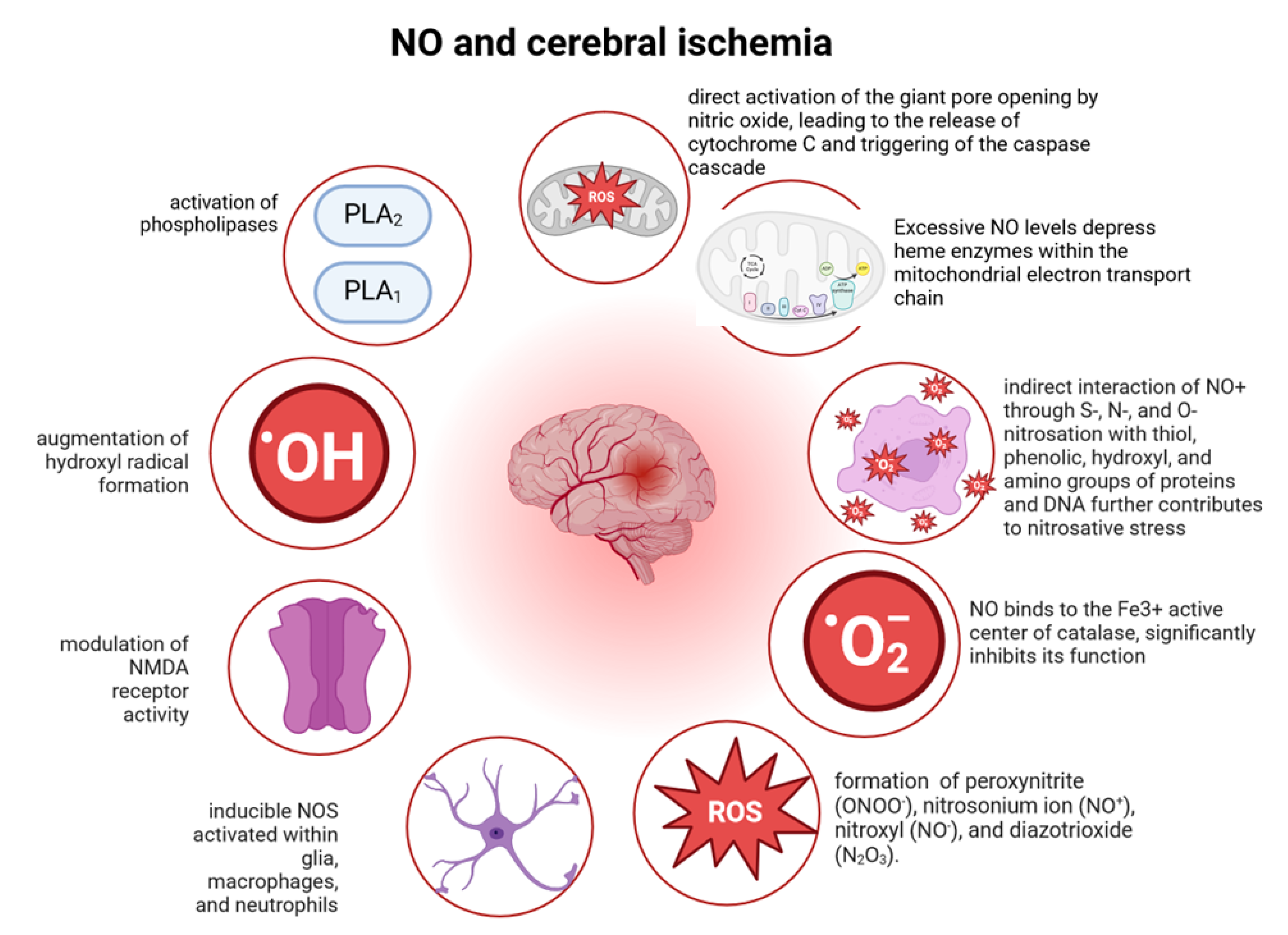
4.4. •NO and Cerebral Ischemia
4.5. •NO and Endothelial Dysfunction
4.6. Pharmacological Modulation of the Nitroxidergic System
4.7. Inhibitors of NOS Isoforms and Their Cytoprotective Effect of Neurons
4.8. Exogenous •NO
- -
- -
- Nitrites (amyl nitrite, NaNO2),
- -
- Nitrosothiols and substances that form various complexes with •NO: S-nitrosoglutathione (GSNO), S-nitroso-N-acetylpenicillamine (SNAP), diethylamine-NO (DEA-NO).
5. •NO Scavengers
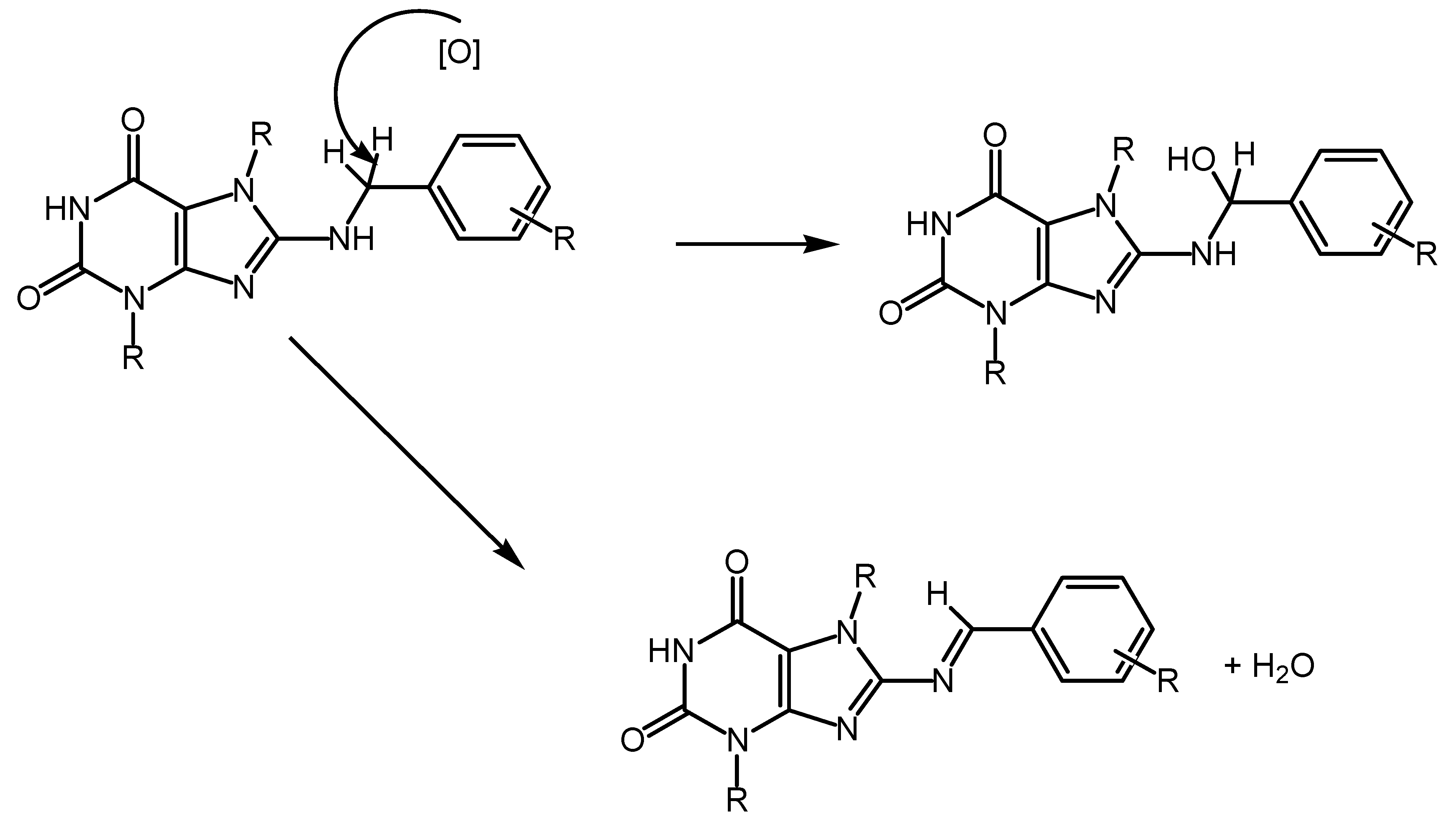
5.1. Xanthine Derivatives
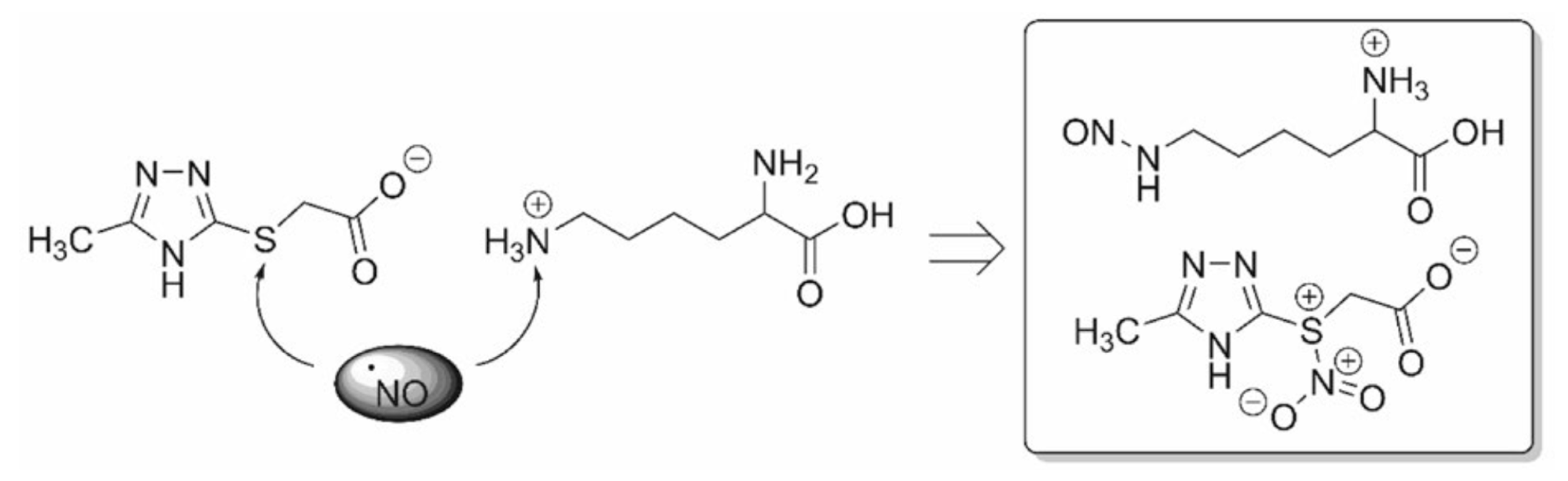
5.2. 1,2,4-Triazole Derivatives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BH4 | tetrahydrobiopterin |
| FAD | flavinadenine dinucleotide |
| HSP | heat shock proteins |
| IFN | interferon |
| IL-1 | interleukin-1 |
| L-NMMA | NG-monomethyl-L-arginine |
| NOS | nitric oxide synthase |
| PARP | poly(ADP-ribose) polymerase |
| TNF | tumour necrosis factor |
| TNFR | tumour necrosis factor receptor |
| ROS | reactive oxygen species |
| PCD | programmed cell death |
| sGC | soluble guanylate cyclase |
| cGMP | cyclic guanosine monophosphate |
References
- Andrabi, S.M.; Sharma, N.S.; Karan, A.; Shahriar, S.M.S.; Cordon, B.; Ma, B.; Xie, J. Nitric Oxide: Physiological Functions, Delivery, and Biomedical Applications. Adv. Sci. 2023, 10, e2303259. [Google Scholar] [CrossRef]
- Rosselli, M.; Keller, P.J.; Dubey, R.K. Role of nitric oxide in the biology, physiology and pathophysiology of reproduction. Hum. Reprod. Update 1998, 4, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Chehelgerdi, M.; Chehelgerdi, M.; Allela, O.Q.B.; Pecho, R.D.C.; Jayasankar, N.; Rao, D.P.; Thamaraikani, T.; Vasanthan, M.; Viktor, P.; Lakshmaiya, N.; et al. Progressing nanotechnology to improve targeted cancer treatment: Overcoming hurdles in its clinical implementation. Mol. Cancer 2023, 22, 169. [Google Scholar] [CrossRef]
- Esplugues, J.V. NO as a signalling molecule in the nervous system. Br. J. Pharmacol. 2002, 135, 1079–1095. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, M.L.; Ellis, C.G.; Goldman, D.; Stephenson, A.H.; Dietrich, H.H.; Sprague, R.S. Erythrocytes: Oxygen sensors and modulators of vascular tone. Physiology 2009, 24, 107–116. [Google Scholar] [CrossRef]
- Belenichev, I.; Gorbachova, S.; Pavlov, S.; Bukhtiyarova, N.; Puzyrenko, A.; Brek, O. Neurochemical Status of Nitric Oxide in the Settings of the Norm, Ischemic Event of Central Nervous System, and Pharmacological BN Intervention. Georgian Med. News 2021, 315, 169–176. [Google Scholar]
- Nicholls, P.; Hildebrandt, V. Binding of ligands and spectral shifts in cytochrome c oxidase. Biochem. J. 1978, 173, 65–72. [Google Scholar] [CrossRef]
- Ismail, A.G. Miniaturized Devices for Bioanalysis: Case of Nitric Oxide Stored as S-Nitrosothiols in Biological Fluids. Analytical Chemistry. Ph.D. Thesis, Université Pierre et Marie Curie. Paris VI, Paris, France, 2016. (In English). [Google Scholar]
- Khan, F.H.; Dervan, E.; Bhattacharyya, D.D.; McAuliffe, J.D.; Miranda, K.M.; Glynn, S.A. The Role of Nitric Oxide in Cancer: Master Regulator or NOt? Int. J. Mol. Sci. 2020, 21, 9393. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.G.; Sahni, A.; Pei, D. Understanding Cell Penetration of Cyclic Peptides. Chem. Rev. 2019, 119, 10241–10287. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A. Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application. Pharmaceutics 2022, 14, 543. [Google Scholar] [CrossRef]
- Campos, K.L.; Giovanelli, J.; Kaufman, S. Characteristics of the nitric oxide synthase-catalyzed conversion of arginine to N-hydroxyarginine, the first oxygenation step in the enzymic synthesis of nitric oxide. J. Biol. Chem. 1995, 270, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147 (Suppl. S1), S193–S201. [Google Scholar] [CrossRef] [PubMed]
- Alkaitis, M.S.; Crabtree, M.J. Recoupling the cardiac nitric oxide synthases: Tetrahydrobiopterin synthesis and recycling. Curr. Heart Fail. Rep. 2012, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Bredt, D.S.; Hwang, P.M.; Snyder, S.H. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature 1990, 347, 768–770. [Google Scholar] [CrossRef]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.A.; Billiar, T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998, 17, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic. Biol. Med. 2010, 49, 307–316. [Google Scholar] [CrossRef]
- Gilchrist, M.; McCauley, S.D.; Befus, A. Dean Expression, localization, and regulation of NOS in human mast cell lines: Effects on leukotriene production. Blood 2004, 104, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Mattila, J.T.; Thomas, A.C. Nitric oxide synthase: Non-canonical expression patterns. Front. Immunol. 2014, 5, 478. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Lotz, M.; König, T.; Ménard, S.; Gütle, D.; Bogdan, C.; Hornef, M.W. Cytokine-mediated control of lipopolysaccharide-induced activation of small intestinal epithelial cells. Immunology 2007, 122, 306–315. [Google Scholar] [CrossRef]
- Salim, T.; Sershen, C.L.; May, E.E. Investigating the Role of TNF-α and IFN-γ Activation on the Dynamics of iNOS Gene Expression in LPS Stimulated Macrophages. PLoS ONE 2016, 11, e0153289. [Google Scholar] [CrossRef]
- Ghosh, S.; Wolan, D.; Adak, S.; Crane, B.R.; Kwon, N.S.; Tainer, J.A.; Getzoff, E.D.; Stuehr, D.J. Mutational Analysis of the Tetrahydrobiopterin-binding Site in Inducible Nitric-oxide Synthase. J. Biol. Chem. 1999, 274, 24100–24112. [Google Scholar] [CrossRef]
- Iyanagi, T.; Xia, C.; Kim, J.J. NADPH-cytochrome P450 oxidoreductase: Prototypic member of the diflavin reductase family. Arch. Biochem. Biophys. 2012, 528, 72–89. [Google Scholar] [CrossRef]
- Tengan, C.H.; Rodrigues, G.S.; Godinho, R.O. Nitric oxide in skeletal muscle: Role on mitochondrial biogenesis and function. Int. J. Mol. Sci. 2012, 13, 17160–17184. [Google Scholar] [CrossRef]
- Gonçalves, D.A.; Jasiulionis, M.G.; Melo, F.H.M.d. The Role of the BH4 Cofactor in Nitric Oxide Synthase Activity and Cancer Progression: Two Sides of the Same Coin. Int. J. Mol. Sci. 2021, 22, 9546. [Google Scholar] [CrossRef]
- McMurry, J.L.; Chrestensen, C.A.; Scott, I.M.; Lee, E.W.; Rahn, A.M.; Johansen, A.M.; Forsberg, B.J.; Harris, K.D.; Salerno, J.C. Rate, affinity and calcium dependence of nitric oxide synthase isoform binding to the primary physiological regulator calmodulin. FEBS J. 2011, 278, 4943–4954. [Google Scholar] [CrossRef]
- Aoyagi, M.; Arvai, A.S.; Tainer, J.A.; Getzoff, E.D. Structural basis for endothelial nitric oxide synthase binding to calmodulin. EMBO J. 2003, 22, 766–775. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Rao, C.V. iNOS-selective inhibitors for cancer prevention: Promise and progress. Future Med. Chem. 2012, 4, 2193–2204. [Google Scholar] [CrossRef]
- Kolodziejski, P.J.; Koo, J.S.; Eissa, N.T. Regulation of inducible nitric oxide synthase by rapid cellular turnover and cotranslational down-regulation by dimerization inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18141–18146. [Google Scholar] [CrossRef]
- Albina, J.E.; Reichner, J.S. Role of nitric oxide in mediation of macrophage cytotoxicity and apoptosis. Cancer Metastasis Rev. 1998, 17, 39–53. [Google Scholar] [CrossRef]
- Liew, F.Y.; Li, Y.; Millott, S. Tumour necrosis factor (TNF-alpha) in leishmaniasis. II. TNF-alpha-induced macrophage leishmanicidal activity is mediated by nitric oxide from L-arginine. Immunology 1990, 71, 556–559. [Google Scholar]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Bajgar, A.; Krejčová, G. On the origin of the functional versatility of macrophages. Front. Physiol. 2023, 14, 1128984. [Google Scholar] [CrossRef]
- Cavinato, L.; Genise, E.; Luly, F.R.; Di Domenico, E.G.; Del Porto, P.; Ascenzioni, F. Escaping the Phagocytic Oxidative Burst: The Role of SODB in the Survival of Pseudomonas aeruginosa Within Macrophages. Front. Microbiol. 2020, 11, 326. [Google Scholar] [CrossRef] [PubMed]
- Somasundaram, V.; Basudhar, D.; Bharadwaj, G.; No, J.H.; Ridnour, L.A.; Cheng, R.Y.; Fujita, M.; Thomas, D.D.; Anderson, S.K.; McVicar, D.W.; et al. Molecular Mechanisms of Nitric Oxide in Cancer Progression, Signal Transduction, and Metabolism. Antioxid. Redox Signal. 2019, 30, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Worrell, A.; McDevitt, E.; Henein, L.; Howell, G.E. 3rd. Alterations in macrophage phagocytosis and inflammatory tone following exposure to the organochlorine compounds oxychlordane and trans-nonachlor. Toxicol. In Vitro 2020, 65, 104791. [Google Scholar] [CrossRef]
- Wink, D.A.; Hanbauer, I.; Krishna, M.C.; DeGraff, W.; Gamson, J.; Mitchell, J.B. Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc. Natl. Acad. Sci. USA 1993, 90, 9813–9817. [Google Scholar] [CrossRef]
- Stojanović, S.; Stanić, D.; Nikolić, M.; Spasić, M.; Niketić, V. Iron catalyzed conversion of NO into nitrosonium (NO+) and nitroxyl (HNO/NO−) species. Nitric Oxide 2004, 11, 256–262. [Google Scholar] [CrossRef]
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813. [Google Scholar] [CrossRef]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef]
- Barrera, G. Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncol. 2012, 2012, 137289. [Google Scholar] [CrossRef]
- Reis, A.K.C.A.; Stern, A.; Monteiro, H.P. S-nitrosothiols and H2S donors: Potential chemo-therapeutic agents in cancer. Redox Biol. 2019, 27, 101190. [Google Scholar] [CrossRef]
- Ali, A.A.; Coulter, J.A.; Ogle, C.H.; Migaud, M.M.; Hirst, D.G.; Robson, T.; McCarthy, H.O. The contribution of N2O3 to the cytotoxicity of the nitric oxide donor DETA/NO: An emerging role for S-nitrosylation. Biosci. Rep. 2013, 33, e00031. [Google Scholar] [CrossRef]
- Vašková, J.; Kočan, L.; Vaško, L.; Perjési, P. Glutathione-Related Enzymes and Proteins: A Review. Molecules 2023, 28, 1447. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- Kobayashi, S.; Homma, T.; Fujii, J. Nitric oxide produced by NOS2 copes with the cytotoxic effects of superoxide in macrophages. Biochem. Biophys. Rep. 2021, 26, 100942. [Google Scholar] [CrossRef]
- Mason, M.G.; Nicholls, P.; Wilson, M.T.; Cooper, C.E. Nitric oxide inhibition of respiration involves both competitive (heme) and noncompetitive (copper) binding to cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2006, 103, 708–713. [Google Scholar] [CrossRef]
- Szabó, C.; Módis, K. Pathophysiological roles of peroxynitrite in circulatory shock. Shock 2010, 34 (Suppl. S1), 4–14. [Google Scholar] [CrossRef]
- Jourd’heuil, D.; Jourd’heuil, F.L.; Kutchukian, P.S.; Musah, R.A.; Wink, D.A.; Grisham, M.B. Reaction of superoxide and nitric oxide with peroxynitrite. Implications for peroxynitrite-mediated oxidation reactions in vivo. J. Biol. Chem. 2001, 276, 28799–28805. [Google Scholar] [CrossRef]
- Radi, R. Peroxynitrite, a stealthy biological oxidant. J. Biol. Chem. 2013, 288, 26464–26472. [Google Scholar] [CrossRef]
- Crack, J.C.; Balasiny, B.K.; Bennett, S.P.; Rolfe, M.D.; Froes, A.; MacMillan, F.; Green, J.; Cole, J.A.; Le Brun, N.E. The Di-Iron Protein YtfE Is a Nitric Oxide-Generating Nitrite Reductase Involved in the Management of Nitrosative Stress. J. Am. Chem. Soc. 2022, 144, 7129–7145. [Google Scholar] [CrossRef]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, M.; Robinson, G.L.; Cain, K. Glucose—A sweet way to die: Metabolic switching modulates tumor cell death. Cell Cycle 2012, 11, 3919–3925. [Google Scholar] [CrossRef] [PubMed]
- Kawano, T.; Zoga, V.; Kimura, M.; Liang, M.Y.; Wu, H.E.; Gemes, G.; McCallum, J.B.; Kwok, W.M.; Hogan, Q.H.; Sarantopoulos, C.D. Nitric oxide activates ATP-sensitive potassium channels in mammalian sensory neurons: Action by direct S-nitrosylation. Mol. Pain 2009, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Brüne, B. Nitric oxide: NO apoptosis or turning it ON? Cell Death Differ. 2003, 10, 864–869. [Google Scholar] [CrossRef]
- Islam, B.U.; Habib, S.; Ahmad, P.; Allarakha, S.; Moinuddin; Ali, A. Pathophysiological Role of Peroxynitrite Induced DNA Damage in Human Diseases: A Special Focus on Poly(ADP-ribose) Polymerase (PARP). Indian J. Clin. Biochem. 2015, 30, 368–385. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Song, B.J. Functional roles of protein nitration in acute and chronic liver diseases. Oxid. Med. Cell Longev. 2014, 2014, 149627. [Google Scholar] [CrossRef]
- Romero-Puertas, M.C.; Laxa, M.; Mattè, A.; Zaninotto, F.; Finkemeier, I.; Jones, A.M.; Perazzolli, M.; Vandelle, E.; Dietz, K.J.; Delledonne, M. S-nitrosylation of peroxiredoxin II E promotes peroxynitrite-mediated tyrosine nitration. Plant Cell 2007, 19, 4120–4130. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 2014, 159, 623–634. [Google Scholar] [CrossRef]
- Kay, J.; Thadhani, E.; Samson, L.; Engelward, B. Inflammation-induced DNA damage, mutations and cancer. DNA Repair 2019, 83, 102673. [Google Scholar] [CrossRef]
- Parrish, M.C.; Chaim, I.A.; Nagel, Z.D.; Tannenbaum, S.R.; Samson, L.D.; Engelward, B.P. Nitric oxide induced S-nitrosation causes base excision repair imbalance. DNA Repair 2018, 68, 25–33. [Google Scholar] [CrossRef]
- Brunyanszki, A.; Szczesny, B.; Virág, L.; Szabo, C. Mitochondrial poly(ADP-ribose) polymerase: The Wizard of Oz at work. Free Radic. Biol. Med. 2016, 100, 257–270. [Google Scholar] [CrossRef]
- Szabo, C.; Martins, V.; Liaudet, L. Poly(ADP- ribose) polymerase inhibition in acute lung injury: A re- emerging concept. Am. J. Respir. Cell. Mol. Biol. 2020, 63, 571–590. [Google Scholar] [CrossRef]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Pozzoli, G.; Morrione, A.; Giordano, A.; Cenciarelli, C. p53 signaling in cancer progression and therapy. Cancer Cell Int. 2021, 21, 703. [Google Scholar] [CrossRef]
- Abuetabh, Y.; Wu, H.H.; Chai, C.; Al Yousef, H.; Persad, S.; Sergi, C.M.; Leng, R. DNA damage response revisited: The p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 2022, 54, 1658–1669. [Google Scholar] [CrossRef]
- Dai, C.Q.; Luo, T.T.; Luo, S.C.; Wang, J.Q.; Wang, S.M.; Bai, Y.H.; Yang, Y.L.; Wang, Y.Y. p53 and mitochondrial dysfunction: Novel insight of neurodegenerative diseases. J. Bioenerg. Biomembr. 2016, 48, 337–347. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; St Clair, D.K. ROS and p53, a versatile partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Borsos, B.N.; Pantazi, V.; Páhi, Z.G.; Majoros, H.; Ujfaludi, Z.; Berzsenyi, I.; Pankotai, T. The role of p53 in the DNA damage-related ubiquitylation of S2P RNAPII. PLoS ONE 2022, 17, e0267615. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Reddy, T.P.; Glynn, S.A.; Billiar, T.R.; Wink, D.A.; Chang, J.C. Targeting Nitric Oxide: Say NO to Metastasis. Clin. Cancer Res. 2023, 29, 1855–1868. [Google Scholar] [CrossRef]
- Jiang, H.; Ji, P.; Shang, X.; Zhou, Y. Connection between Osteoarthritis and Nitric Oxide: From Pathophysiology to Therapeutic Target. Molecules 2023, 28, 1683. [Google Scholar] [CrossRef]
- Roufayel, R.; Younes, K.; Al-Sabi, A.; Murshid, N. BH3-Only Proteins Noxa and Puma Are Key Regulators of Induced Apoptosis. Life 2022, 12, 256. [Google Scholar] [CrossRef]
- Etti, I.C.; Rasedee, A.; Hashim, N.M.; Abdul, A.B.; Kadir, A.; Yeap, S.K.; Waziri, P.; Malami, I.; Lim, K.L.; Etti, C.J. Artonin E induces p53-independent G1 cell cycle arrest and apoptosis through ROS-mediated mitochondrial pathway and livin suppression in MCF-7 cells. Drug Des. Devel Ther. 2017, 11, 865–879. [Google Scholar] [CrossRef]
- Mintz, J.; Vedenko, A.; Rosete, O.; Shah, K.; Goldstein, G.; Hare, J.M.; Ramasamy, R.; Arora, H. Current Advances of Nitric Oxide in Cancer and Anticancer Therapeutics. Vaccines 2021, 9, 94. [Google Scholar] [CrossRef]
- Dat, N.T.; Binh, P.T.; Phuong Quynh, L.T.; Huong, H.T.; Minh, C.V. Sanggenon C and O inhibit NO production, iNOS expression and NF-κB activation in LPS-induced RAW264.7 cells. Immunopharmacol. Immunotoxicol. 2012, 34, 84–88. [Google Scholar] [CrossRef]
- Pourbagher-Shahri, A.M.; Farkhondeh, T.; Talebi, M.; Kopustinskiene, D.M.; Samarghandian, S.; Bernatoniene, J. An Overview of NO Signaling Pathways in Aging. Molecules 2021, 26, 4533. [Google Scholar] [CrossRef]
- Zhang, W.; Xiao, D.; Mao, Q.; Xia, H. Role of neuroinflammation in neurodegeneration development. Signal Transduct. Target. Ther. 2023, 8, 267. [Google Scholar] [CrossRef]
- Lin, H.-Y.; Shen, S.-C.; Lin, C.-W.; Yang, L.-Y.; Chen, Y.-C. Baicalein inhibition of hydrogen peroxide-induced apoptosis via ROS-dependent heme oxygenase 1 gene expression. Biochim. Biophys. Acta 2007, 1773, 1073–1086. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef]
- Gao, Q.; Zhou, Z.-Y.; He, Y.-N.; Dong, M.-H.; Wang, Z.-N.; Chen, H.-M. BDE-47 Induces Immunotoxicity in RAW264.7 Macrophages through the Reactive Oxygen Species-Mediated Mitochondrial Apoptotic Pathway. Molecules 2023, 28, 2036. [Google Scholar] [CrossRef]
- Zielinska, E.; Tukaj, C.; Radomski, M.W.; Inkielewicz-Stepniak, I. Molecular Mechanism of Silver Nanoparticles-Induced Human Osteoblast Cell Death: Protective Effect of Inducible Nitric Oxide Synthase Inhibitor. PLoS ONE 2016, 11, e0164137. [Google Scholar] [CrossRef]
- Naseri, M.H.; Mahdavi, M.; Davoodi, J.; Tackallou, S.H.; Goudarzvand, M.; Neishabouri, S.H. Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int. 2015, 15, 55. [Google Scholar] [CrossRef]
- Salvucci, O.; Carsana, M.; Bersani, I.; Tragni, G.; Anichini, A. Antiapoptotic role of endogenous nitric oxide in human melanoma cells. Cancer Res. 2001, 61, 318–326. [Google Scholar]
- Fulda, S.; Meyer, E.; Debatin, K.M. Inhibition of TRAIL-induced apoptosis by Bcl-2 overexpression. Oncogene 2002, 21, 2283–2294. [Google Scholar] [CrossRef]
- Qian, S.; Wei, Z.; Yang, W.; Huang, J.; Yang, Y.; Wang, J. The role of BCL-2 family proteins in regulating apoptosis and cancer therapy. Front. Oncol. 2022, 12, 985363. [Google Scholar] [CrossRef]
- Kurschat, C.; Metz, A.; Kirschnek, S.; Häcker, G. Importance of Bcl-2-family proteins in murine hematopoietic progenitor and early B cells. Cell Death Dis. 2021, 12, 784. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Monaco, G.; Terry, L.E.; Rosa, N.; Baker, M.R.; Parys, J.B.; Serysheva, I.I.; Yule, D.I.; Bultynck, G. Bcl-2-Protein Family as Modulators of IP3 Receptors and Other Organellar Ca2+ Channels. Cold Spring Harb. Perspect. Biol. 2020, 12, a035089. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef]
- Snyder, C.M.; Shroff, E.H.; Liu, J.; Chandel, N.S. Nitric oxide induces cell death by regulating anti-apoptotic BCL-2 family members. PLoS ONE 2009, 4, e7059. [Google Scholar] [CrossRef]
- Dorstyn, L.; Kumar, S. The p53-caspase-2 axis in the cell cycle and DNA damage response. Exp. Mol. Med. 2021, 53, 517–527. [Google Scholar]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 18, 1441–1449. [Google Scholar] [CrossRef]
- Tarr, J.M.; Eggleton, P.; Winyard, P.G. Nitric oxide and the regulation of apoptosis in tumour cells. Curr. Pharm. Des. 2006, 12, 4445–4468. [Google Scholar] [CrossRef]
- Barbosa, A.D.; Osório, H.; Sims, K.J.; Almeida, T.; Alves, M.; Bielawski, J.; Amorim, M.A.; Moradas-Ferreira, P.; Hannun, Y.A.; Costa, V. Role for Sit4p-dependent mitochondrial dysfunction in mediating the shortened chronological lifespan and oxidative stress sensitivity of Isc1p-deficient cells. Mol. Microbiol. 2011, 81, 515–527. [Google Scholar] [CrossRef]
- Lacza, Z.; Pankotai, E.; Busija, D.W. Mitochondrial nitric oxide synthase: Current concepts and controversies. Front. Biosci. (Landmark Ed.) 2009, 14, 4436–4443. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Bak, P.G.; Popazova, O.O.; Bukhtiyarova, N.V.; Yadlovsky, O.E. Nitric oxide-dependent mechanism of endothelial dysfunction formation is a promising target link for pharmacological management. Biopolym. Cell. 2022, 38, 145–157. [Google Scholar] [CrossRef]
- Yuryev, A.; Ono, M.; Goff, S.A.; Macaluso, F.; Wennogle, L.P. Isoform-specific localization of A-RAF in mitochondria. Mol. Cell Biol. 2000, 20, 4870–4878. [Google Scholar] [CrossRef]
- Lacza, Z.; Snipes, J.A.; Zhang, J.; Horváth, E.M.; Figueroa, J.P.; Szabó, C.; Busija, D.W. Mitochondrial nitric oxide synthase is not eNOS, nNOS or iNOS. Free Radic. Biol. Med. 2003, 35, 1217–1228. [Google Scholar] [CrossRef]
- Aitken, A. Post-translational modification of 14-3-3 isoforms and regulation of cellular function. Semin. Cell Dev. Biol. 2011, 22, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Gregorich, Z.R.; Cai, W.; Lin, Z.; Chen, A.J.; Peng, Y.; Kohmoto, T.; Ge, Y. Distinct sequences and post-translational modifications in cardiac atrial and ventricular myosin light chains revealed by top-down mass spectrometry. J. Mol. Cell Cardiol. 2017, 107, 13–21. [Google Scholar] [CrossRef]
- Huang, M.; Zhu, L.; Feng, L.; Zhan, L.; Zhao, Y.; Chen, X. Reforming Nitrate Metabolism for Enhancing L-Arginine Production in Corynebacterium crenatum Under Oxygen Limitation. Front. Microbiol. 2022, 13, 834311. [Google Scholar] [CrossRef]
- Meirelles, C.M.; Matsuura, C.; Silva, R.S., Jr.; Guimarães, F.F.; Gomes, P.S.C. Acute Effects of L-Arginine Supplementation on Oxygen Consumption Kinetics and Muscle Oxyhemoglobin and Deoxyhemoglobin during Treadmill Running in Male Adults. Int. J. Exerc. Sci. 2019, 12, 444–455. [Google Scholar]
- Hamdane, D.; Xia, C.; Im, S.C.; Zhang, H.; Kim, J.J.; Waskell, L. Structure and function of an NADPH-cytochrome P450 oxidoreductase in an open conformation capable of reducing cytochrome P450. J. Biol. Chem. 2009, 284, 11374–11384. [Google Scholar] [CrossRef]
- Pi, X.; Xie, L.; Portbury, A.L.; Kumar, S.; Lockyer, P.; Li, X.; Patterson, C. NADPH oxidase-generated reactive oxygen species are required for stromal cell-derived factor-1α-stimulated angiogenesis. Arter. Thromb. Vasc. Biol. 2014, 34, 2023–2032. [Google Scholar] [CrossRef]
- Nauseef, W.M. The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 2019, 60, 130–140. [Google Scholar] [CrossRef]
- O’Rourke, B. Mitochondrial ion channels. Annu. Rev. Physiol. 2007, 69, 19–49. [Google Scholar] [CrossRef]
- Patwardhan, G.A.; Beverly, L.J.; Siskind, L.J. Sphingolipids and mitochondrial apoptosis. J. Bioenerg. Biomembr. 2016, 48, 153–168. [Google Scholar] [CrossRef]
- Dayem, A.A.; Hossain, M.K.; Lee, S.B.; Kim, K.; Saha, S.K.; Yang, G.-M.; Choi, H.Y.; Cho, S.-G. The Role of Reactive Oxygen Species (ROS) in the Biological Activities of Metallic Nanoparticles. Int. J. Mol. Sci. 2017, 18, 120. [Google Scholar] [CrossRef]
- Graceffa, V. Therapeutic Potential of Reactive Oxygen Species: State of the Art and Recent Advances. SLAS Technol. 2021, 26, 140–158. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef]
- Dubois, C.; Kondratskyi, A.; Bidaux, G.; Noyer, L.; Vancauwenberghe, E.; Farfariello, V.; Toillon, R.-A.; Roudbaraki, M.; Tierny, D.; Bonnal, J.-L.; et al. Co-targeting Mitochondrial Ca2+ Homeostasis and Autophagy Enhances Cancer Cells’ Chemosensitivity. iScience 2020, 23, 101263. [Google Scholar] [CrossRef]
- Giorgi, C.; Agnoletto, C.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Poletti, F.; Rimessi, A.; et al. Mitochondrial calcium homeostasis as potential target for mitochondrial medicine. Mitochondrion 2012, 12, 77–85. [Google Scholar] [CrossRef]
- Pesaresi, M.G.; Amori, I.; Giorgi, C.; Ferri, A.; Fiorenzo, P.; Gabanella, F.; Salvatore, A.M.; Giorgio, M.; Pelicci, P.G.; Pinton, P.; et al. Mitochondrial redox signalling by p66Shc mediates ALS-like disease through Rac1 inactivation. Hum. Mol. Genet. 2011, 20, 4196–4208. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Abramov, A.V.; Puzyrenko, A.; Bukhtiyarova, N.V.; Gorchakova, N.O.; Bak, P.G. Molecular mechanisms of myocardial damage in the hypertensive rats and hypertensive rats with metabolic disorders (diabetes mellitus, atherosclerosis). Res. Results Pharmacol. 2022, 8, 25–33. [Google Scholar] [CrossRef]
- Iova, O.M.; Marin, G.E.; Lazar, I.; Stanescu, I.; Dogaru, G.; Nicula, C.A.; Bulboacă, A.E. Nitric Oxide/Nitric Oxide Synthase System in the Pathogenesis of Neurodegenerative Disorders-An Overview. Antioxidants 2023, 12, 753. [Google Scholar] [CrossRef]
- Wang, Y.; Hong, F.; Yang, S. Roles of Nitric Oxide in Brain Ischemia and Reperfusion. Int. J. Mol. Sci. 2022, 23, 4243. [Google Scholar] [CrossRef]
- Liy, P.M.; Puzi, N.N.A.; Jose, S.; Vidyadaran, S. Nitric oxide modulation in neuroinflammation and the role of mesenchymal stem cells. Exp. Biol. Med. 2021, 246, 2399–2406. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenerg. Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef]
- Popazova, O.; Belenichev, I.; Bukhtiyarova, N.; Ryzhenko, V.; Oksenych, V.; Kamyshnyi, A. Cardioprotective Activity of Pharmacological Agents Affecting NO Production and Bioavailability in the Early Postnatal Period after Intrauterine Hypoxia in Rats. Biomedicines 2023, 11, 2854. [Google Scholar] [CrossRef]
- Uchi, J.; Ryu, S.-Y.; Jhun, B.S.; Hurst, S.; Sheu, S.-S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid. Redox Signal. 2014, 21, 987–1006. [Google Scholar] [CrossRef]
- Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef]
- Luiking, Y.C.; Engelen, M.P.; Deutz, N.E. Regulation of nitric oxide production in health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 97–104. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef]
- Paulo, M.; Costa, D.E.F.R.; Bonaventura, D.; Lunardi, C.N.; Bendhack, L.M. Nitric Oxide Donors as Potential Drugs for the Treatment of Vascular Diseases Due to Endothelium Dysfunction. Curr. Pharm. Des. 2020, 26, 3748–3759. [Google Scholar] [CrossRef]
- Zhang, Y.; Janssens, S.P.; Wingler, K.; Schmidt, H.H.; Moens, A.L. Modulating endothelial nitric oxide synthase: A new cardiovascular therapeutic strategy. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H634–H646. [Google Scholar] [CrossRef]
- Makinde, E.; Ma, L.; Mellick, G.D.; Feng, Y. Mitochondrial Modulators: The Defender. Biomolecules 2023, 13, 226. [Google Scholar] [CrossRef]
- Mimaki, M.; Wang, X.; McKenzie, M.; Thorburn, D.R.; Ryan, M.T. Understanding mitochondrial complex I assembly in health and disease. Biochim. Biophys. Acta 2012, 1817, 851–862. [Google Scholar] [CrossRef]
- Bolisetty, S.; Jaimes, E.A. Mitochondria and Reactive Oxygen Species: Physiology and Pathophysiology. Int. J. Mol. Sci. 2013, 14, 6306–6344. [Google Scholar] [CrossRef]
- Zhang, S.; Rao, S.; Yang, M.; Ma, C.; Hong, F.; Yang, S. Role of Mitochondrial Pathways in Cell Apoptosis during He-Patic Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2022, 23, 2357. [Google Scholar] [CrossRef]
- Long, R.T.; Peng, J.B.; Huang, L.L.; Jiang, G.P.; Liao, Y.J.; Sun, H.; Hu, Y.D.; Liao, X.H. Augmenter of Liver Regeneration Alleviates Renal Hypoxia-Reoxygenation Injury by Regulating Mitochondrial Dynamics in Renal Tubular Epithelial Cells. Mol. Cells 2019, 42, 893–905. [Google Scholar]
- Qajari, N.M.; Shafaroudi, M.M.; Gholami, M.; Khonakdar-Tarsi, A. Silibinin treatment results in reducing OPA1&MFN1 genes expression in a rat model hepatic ischemia-reperfusion. Mol. Biol. Rep. 2020, 47, 3271–3280. [Google Scholar]
- Qian, L.; Mehrabi Nasab, E.; Athari, S.M.; Athari, S.S. Mitochondria Signaling Pathways in Allergic Asthma. J. Investig. Med. 2022, 70, 863–882. [Google Scholar] [CrossRef]
- Martinvalet, D. Mitochondrial Entry of Cytotoxic Proteases: A New Insight into the Granzyme B Cell Death Pathway. Oxid. Med. Cell Longev. 2019, 2019, 9165214. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Rajesh, M.; Bátkai, S.; Kashiwaya, Y.; Haskó, G.; Liaudet, L.; Szabó, C.; Pacher, P. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1466–H1483. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Model. Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Cherniy, V.I.; Nagornaya, E.A.; Bukhtiyarova, N.V.; Kucherenko, V.I. Neuroprotection and Neuroplasticity; Polygraph Plus Ltd.: Kiev, Ukraine, 2015; Volume 510. [Google Scholar]
- Lubos, E.; Handy, D.E.; Loscalzo, J. Role of oxidative stress and nitric oxide in atherothrombosis. Front. Biosci. 2008, 13, 5323–5344. [Google Scholar] [CrossRef]
- Belenichev, I.; Aliyeva, O.; Popazova, O.; Bukhtiyarova, N. Molecular and biochemical mechanisms of diabetic encephalopathy. Acta Biochim. Pol. 2023, 70, 751–760. [Google Scholar] [CrossRef]
- Provenzano, F.; Torazza, C.; Bonifacino, T.; Bonanno, G.; Milanese, M. The Key Role of Astrocytes in Amyotrophic Lateral Sclerosis and Their Commitment to Glutamate Excitotoxicity. Int. J. Mol. Sci. 2023, 24, 15430. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; López-Blanch, R.; Jihad-Jebbar, A.; Vallés, S.L.; Estrela, J.M. Oxidative Stress, Neuroinflammation and Mitochondria in the Pathophysiology of Amyotrophic Lateral Sclerosis. Antioxidants 2020, 9, 901. [Google Scholar] [CrossRef]
- Perrelli, M.G.; Pagliaro, P.; Penna, C. Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species. World J. Cardiol. 2011, 3, 186–200. [Google Scholar] [CrossRef]
- Endlicher, R.; Drahota, Z.; Štefková, K.; Červinková, Z.; Kučera, O. The Mitochondrial Permeability Transition Pore-Current Knowledge of Its Structure, Function, and Regulation, and Optimized Methods for Evaluating Its Functional State. Cells 2023, 12, 1273. [Google Scholar] [CrossRef]
- De Zio, D.; Cianfanelli, V.; Cecconi, F. New insights into the link between DNA damage and apoptosis. Antioxid. Redox Signal. 2013, 19, 559–571. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Kahraman, S.; Siegel, A.; Polster, B.M.; Fiskum, G. Permeability transition pore-dependent and PARP-mediated depletion of neuronal pyridine nucleotides during anoxia and glucose deprivation. J. Bioenerg. Biomembr. 2015, 47, 53–61. [Google Scholar] [CrossRef]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Zhunina, O.A.; Yabbarov, N.G.; Grechko, A.V.; Starodubova, A.V.; Ivanova, E.; Nikiforov, N.G.; Orekhov, A.N. The Role of Mitochondrial Dysfunction in Vascular Disease, Tumorigenesis, and Diabetes. Front. Mol. Biosci. 2021, 8, 671908. [Google Scholar] [CrossRef]
- Armstrong, D. Diagnosis and nosology in primary care. Soc. Sci. Med. 2011, 73, 801–807. [Google Scholar] [CrossRef]
- Skulachev, V.P.; Vyssokikh, M.Y.; Chernyak, B.V.; Mulkidjanian, A.Y.; Skulachev, M.V.; Shilovsky, G.A.; Lyamzaev, K.G.; Borisov, V.B.; Severin, F.F.; Sadovnichii, V.A. Six Functions of Respiration: Isn’t It Time to Take Control over ROS Production in Mitochondria, and Aging Along with It? Int. J. Mol. Sci. 2023, 24, 12540. [Google Scholar] [CrossRef]
- Babizhayev, M.A.; Yegorov, Y.E. Reactive Oxygen Species and the Aging Eye: Specific Role of Metabolically Active Mitochondria in Maintaining Lens Function and in the Initiation of the Oxidation-Induced Maturity Onset Cataract—A Novel Platform of Mitochondria-Targeted Antioxidants With Broad Therapeutic Potential for Redox Regulation and Detoxification of Oxidants in Eye Diseases. Am. J. Ther. 2016, 23, e98–e117. [Google Scholar]
- Тitova, E.; Shagieva, G.; Ivanova, O.; Domnina, L.; Domninskaya, M.; Strelkova, O.; Khromova, N.; Kopnin, P.; Chernyak, B.; Skulachev, V.; et al. Mitochondria-targeted antioxidant SkQ1 suppresses fibrosarcoma and rhabdomyosarcoma tumour cell growth. Cell Cycle 2018, 17, 1797–1811. [Google Scholar] [CrossRef]
- Bielenichev, I.F.; Gorchakova, N.A.; Doroshenko, E.Y.; Samura, I.B.; Ryzhenko, V.P.; Bukhtiiarova, N.V. Use of metabolites, metabolithotropic agents and nutritional supplements in sports and sports medicine: A modern view on the problem. Mod. Med. Technol. 2023, 4, 76–88. (In Ukranian) [Google Scholar] [CrossRef]
- Shemarova, I.; Nesterov, V.; Emelyanova, L.; Korotkov, S. Mitochondrial mechanisms by which gasotransmitters (H2S, NO and CO) protect cardiovascular system against hypoxia. Front. Biosci. (Sch. Ed.). 2021, 13, 105–130. [Google Scholar]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef]
- Osei, D.; Baumgart-Vogt, E.; Ahlemeyer, B.; Herden, C. Tumor Necrosis Factor-α Receptor 1 Mediates Borna Disease Virus 1-Induced Changes in Peroxisomal and Mitochondrial Dynamics in Neurons. Int. J. Mol. Sci. 2024, 25, 1849. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noë, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Jensen, L.T.; Culotta, V.C. Activation of CuZn superoxide dismutases from Caenorhabditis elegans does not require the copper chaperone CCS. J. Biol. Chem. 2005, 280, 41373–41379. [Google Scholar] [CrossRef] [PubMed]
- Lob, H.E.; Vinh, A.; Li, L.; Blinder, Y.; Offermanns, S.; Harrison, D.G. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 2011, 58, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Aliyeva, O.G.; Popazova, O.O.; Bukhtiyarova, N.V. Involvement of heat shock proteins HSP70 in the mechanisms of endogenous neuroprotection: The prospect of using HSP70 modulators. Front. Cell Neurosci. 2023, 17, 1131683. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhai, R.; Xue, T.; Xu, X.; Ren, Y.; Ma, M.; Shi, F.; Wang, H.; Wang, N.; Zhou, F. HSP70 regulates cell proliferation and apoptosis in actinomycin-D-treated lung cancer cells. Transl. Cancer Res. 2020, 9, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Alvarez, B.; Radi, R. One- and two-electron oxidation of thiols: Mechanisms, kinetics and biological fates. Free Radic. Res. 2016, 50, 150–171. [Google Scholar] [CrossRef]
- Vanin, A.F. Physico-Chemistry of Dinitrosyl Iron Complexes as a Determinant of Their Biological Activity. Int. J. Mol. Sci. 2021, 22, 10356. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Chen, Y.-H.; Chiu, H.; Ko, Y.-H.; Wang, R.-T.; Wang, W.-P.; Chuang, Y.-J.; Huang, C.-C.; Lu, T.-T. Cell-Penetrating Delivery of Nitric Oxide by Biocompatible Dinitrosyl Iron Complex and Its Dermato-Physiological Implications. Int. J. Mol. Sci. 2021, 22, 10101. [Google Scholar] [CrossRef]
- Jandy, M.; Noor, A.; Nelson, P.; Dennys, C.N.; Karabinas, I.M.; Pestoni, J.C.; Singh, G.D.; Luc, L.; Devyldere, R.; Perdomo, N.; et al. Peroxynitrite nitration of Tyr 56 in Hsp90 induces PC12 cell death through P2X7R-dependent PTEN activation. Redox Biol. 2022, 50, 102247. [Google Scholar] [CrossRef]
- Kucharczyk, M.W.; Valiente, D.; Bannister, K. Developments in Understanding Diffuse Noxious Inhibitory Controls: Pharmacological Evidence from Pre-Clinical Research. J. Pain. Res. 2021, 14, 1083–1095. [Google Scholar] [CrossRef]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, Y.; Zhang, G.; Yang, Z.; Xu, W.; Chen, Q. The Applications and Mechanisms of Superoxide Dismutase in Medicine, Food, and Cosmetics. Antioxidants 2023, 12, 1675. [Google Scholar] [CrossRef]
- Lee, K.H.; Cha, M.; Lee, B.H. Neuroprotective Effect of Antioxidants in the Brain. Int. J. Mol. Sci. 2020, 21, 7152. [Google Scholar] [CrossRef]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef]
- Sakagami, H.; Satoh, K. Prooxidant action of two antioxidants: Ascorbic acid and gallic acid. Anticancer Res. 1997, 17, 221–224. [Google Scholar]
- Timoshnikov, V.A.; Selyutina, O.Y.; Polyakov, N.E.; Didichenko, V.; Kontoghiorghes, G.J. Mechanistic Insights of Chelator Complexes with Essential Transition Metals: Antioxidant/Pro-Oxidant Activity and Applications in Medicine. Int. J. Mol. Sci. 2022, 23, 1247. [Google Scholar] [CrossRef]
- Herb, M.; Schramm, M. Functions of ROS in Macrophages and Antimicrobial Immunity. Antioxidants 2021, 10, 313. [Google Scholar] [CrossRef]
- Kashfi, K.; Kannikal, J.; Nath, N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells 2021, 10, 3194. [Google Scholar] [CrossRef]
- Pigott, B.; Bartus, K.; Garthwaite, J. On the selectivity of neuronal NOS inhibitors. Br. J. Pharmacol. 2013, 168, 1255–1265. [Google Scholar] [CrossRef]
- Poh, W.H.; Rice, S.A. Recent Developments in Nitric Oxide Donors and Delivery for Antimicrobial and Anti-Biofilm Applications. Molecules 2022, 27, 674. [Google Scholar] [CrossRef]
- Melvin, A.C.; Jones, W.M.; Lutzke, A.; Allison, C.L.; Reynolds, M.M. S-Nitrosoglutathione exhibits greater stability than S-nitroso-N-acetylpenicillamine under common laboratory conditions: A comparative stability study. Nitric Oxide 2019, 92, 18–25. [Google Scholar] [CrossRef]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Shah, F.; Chekman, I.S.; Nagornaya, E.A.; Gorbacheva, S.V.; Gorchakova, N.A. Thiol-Disulfide System: Role in Endogenous Cyto-and Organoprotection, Pathways of Pharmacological Modulation; LLC “Vydavnytstvo” Yuston: Kyiv, Ukraine, 2020. [Google Scholar]
- Wang, Y.; Tang, B.; Long, L.; Luo, P.; Xiang, W.; Li, X.; Wang, H.; Jiang, Q.; Tan, X.; Luo, S.; et al. Improvement of obesity-associated disorders by a small-molecule drug targeting mitochondria of adipose tissue macrophages. Nat. Commun. 2021, 12, 102. [Google Scholar] [CrossRef] [PubMed]
- Poderoso, J.J.; Helfenberger, K.; Poderoso, C. The effect of nitric oxide on mitochondrial respiration. Nitric Oxide 2019, 88, 61–72. [Google Scholar] [CrossRef]
- Presley, T.; Vedam, K.; Liu, X.; Zweier, J.L.; Ilangovan, G. Activation of Hsp90/NOS and increased NO generation does not impair mitochondrial respiratory chain by competitive binding at cytochrome c oxidase in low oxygen concentrations. Cell Stress Chaperones 2009, 14, 611–627. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef]
- Moldoveanu, T.; Czabotar, P.E. BAX, BAK, and BOK: A Coming of Age for the BCL-2 Family Effector Proteins. Cold Spring Harb. Perspect. Biol. 2020, 12, a036319. [Google Scholar] [CrossRef]
- Brockhaus, F.; Brüne, B. Overexpression of CuZn superoxide dismutase protects RAW 264.7 macrophages against nitric oxide cytotoxicity. Biochem. J. 1999, 338 Pt 2, 295–303. [Google Scholar] [CrossRef]
- Lanzarin, G.A.B.; Félix, L.M.; Monteiro, S.M.; Ferreira, J.M.; Oliveira, P.A.; Venâncio, C. Anti-Inflammatory, Anti-Oxidative and Anti-Apoptotic Effects of Thymol and 24-Epibrassinolide in Zebrafish Larvae. Antioxidants 2023, 12, 1297. [Google Scholar] [CrossRef]
- Mazzei, L.; Docherty, N.G.; Manucha, W. Mediators and mechanisms of heat shock protein 70 based cytoprotection in obstructive nephropathy. Cell Stress Chaperones 2015, 20, 893–906. [Google Scholar] [CrossRef]
- Hussar, P. Apoptosis Regulators Bcl-2 and Caspase-3. Encyclopedia 2022, 2, 1624–1636. [Google Scholar] [CrossRef]
- Wali, G.; Kumar, K.R.; Liyanage, E.; Davis, R.L.; Mackay-Sim, A.; Sue, C.M. Mitochondrial Function in Hereditary Spastic Paraplegia: Deficits in SPG7 but Not SPAST Patient-Derived Stem Cells. Front. Neurosci. 2020, 14, 820. [Google Scholar] [CrossRef] [PubMed]
- Dudeja, V.; Mujumdar, N.; Phillips, P.; Chugh, R.; Borja-Cacho, D.; Dawra, R.K.; Vickers, S.M.; Saluja, A.K. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms. Gastroenterology 2009, 136, 1772–1782. [Google Scholar] [CrossRef]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Wei, Y.; Zhuang, Y.; Zhang, Y.; Luo, L.; Yu, B.; Zeng, J. Role of heat shock protein 70 in silibinin-induced apoptosis in bladder cancer. J. Cancer 2024, 15, 79–89. [Google Scholar] [CrossRef]
- Zhai, C.; Lv, J.; Wang, K.; Li, Q.; Qu, Y. HSP70 silencing aggravates apoptosis induced by hypoxia/reoxygenation in vitro. Exp. Ther. Med. 2019, 18, 1013–1020. [Google Scholar] [CrossRef]
- Eskandari, E.; Eaves, C.J. Paradoxical roles of caspase-3 in regulating cell survival, proliferation, and tumorigenesis. J. Cell Biol. 2022, 221, e202201159. [Google Scholar] [CrossRef]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Methela, N.J.; Islam, M.S.; Lee, D.-S.; Yun, B.-W.; Mun, B.-G. S-Nitrosoglutathione (GSNO)-Mediated Lead Detoxification in Soybean through the Regulation of ROS and Metal-Related Transcripts. Int. J. Mol. Sci. 2023, 24, 9901. [Google Scholar] [CrossRef]
- Yang, R.; Gao, Y.; Li, H.; Huang, W.; Tu, D.; Yang, M.; Liu, X.; Hong, J.-S.; Gao, H.-M. Posttranslational S-nitrosylation modification regulates HMGB1 secretion and promotes its proinflammatory and neurodegenerative effects. Cell Rep. 2022, 40, 111330. [Google Scholar] [CrossRef]
- He, M.T.; Park, H.S.; Kim, Y.S.; Lee, A.Y.; Cho, E.J. Protective Effect of Membrane-Free Stem Cells against Lipopolysaccharide and Interferon-Gamma-Stimulated Inflammatory Responses in RAW 264.7 Macrophages. Int. J. Mol. Sci. 2021, 22, 6894. [Google Scholar] [CrossRef]
- Wu, C.H.; Chen, T.L.; Chen, T.G.; Ho, W.P.; Chiu, W.T.; Chen, R.M. Nitric oxide modulates pro- and anti-inflammatory cytokines in lipopolysaccharide-activated macrophages. J. Trauma 2003, 55, 540–545. [Google Scholar] [CrossRef]
- Sangaran, P.G.; Ibrahim, Z.A.; Chik, Z.; Mohamed, Z.; Ahmadiani, A. Lipopolysaccharide Pre-conditioning Attenuates Pro-inflammatory Responses and Promotes Cytoprotective Effect in Differentiated PC12 Cell Lines via Pre-activation of Toll-Like Receptor-4 Signaling Pathway Leading to the Inhibition of Caspase-3/Nuclear Factor-κappa B Pathway. Front. Cell Neurosci. 2021, 14, 598453. [Google Scholar] [PubMed]
- Kim, J.Y.; Barua, S.; Huang, M.Y.; Park, J.; Yenari, M.A.; Lee, J.E. Heat Shock Protein 70 (HSP70) Induction: Chaperonotherapy for Neuroprotection after Brain Injury. Cells 2020, 9, 2020. [Google Scholar] [CrossRef] [PubMed]
- Szyller, J.; Bil-Lula, I. Heat Shock Proteins in Oxidative Stress and Ischemia/Reperfusion Injury and Benefits from Physical Exercises: A Review to the Current Knowledge. Oxid. Med. Cell Longev. 2021, 2021, 6678457. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Ohshima, S.; Pediaditakis, P.; Lemasters, J.J. Nitric oxide protects rat hepatocytes against reperfusion injury mediated by the mitochondrial permeability transition. Hepatology 2004, 39, 1533–1543. [Google Scholar] [CrossRef]
- Guedes, T.A.; Moreira-de-Sousa, C.; Lima, H.M.S.; Grella, T.C.; Socolowski, P.C.; Fontanetti, C.S. Cytoprotective and anti-apoptotic action of HSP70 stress protein in Oreochromis niloticus exposed to residual dilutions of insecticides with fipronil and ethiprole. J. Environ. Sci. Health B 2020, 55, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yu, Y.; Gorshkov, B.; Haigh, S.; Bordan, Z.; Weintraub, D.; Rudic, R.D.; Chakraborty, T.; Barman, S.A.; Verin, A.D.; et al. Hsp70 Suppresses Mitochondrial Reactive Oxygen Species and Preserves Pulmonary Microvascular Barrier Integrity Following Exposure to Bacterial Toxins. Front. Immunol. 2018, 9, 1309. [Google Scholar] [CrossRef]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, C.M.; Wu, J.; Huang, S.; Wang, G.L. Heat shock protein 32/heme oxygenase-1 protects mouse Sertoli cells from hyperthermia-induced apoptosis by CO activation of sGC signalling pathways. Cell Biol. Int. 2014, 38, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Mouawad, N.; Capasso, G.; Ruggeri, E.; Martinello, L.; Severin, F.; Visentin, A.; Facco, M.; Trentin, L.; Frezzato, F. Is It Still Possible to Think about HSP70 as a Therapeutic Target in Onco-Hematological Diseases? Biomolecules 2023, 13, 604. [Google Scholar] [CrossRef]
- Broniowska, K.A.; Hogg, N. The chemical biology of S-nitrosothiols. Antioxid. Redox Signal. 2012, 17, 969–980. [Google Scholar] [CrossRef]
- Desideri, E.; Ciccarone, F.; Ciriolo, M.R. Targeting Glutathione Metabolism: Partner in Crime in Anticancer Therapy. Nutrients. 2019, 11, 1926. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef]
- Baldelli, S.; Ciccarone, F.; Limongi, D.; Checconi, P.; Palamara, A.T.; Ciriolo, M.R. Glutathione and Nitric Oxide: Key Team Players in Use and Disuse of Skeletal Muscle. Nutrients 2019, 11, 2318. [Google Scholar] [CrossRef] [PubMed]
- Girard, P.M.; Peynot, N.; Lelièvre, J.M. Differential correlations between changes to glutathione redox state, protein ubiquitination, and stress-inducible HSPA chaperone expression after different types of oxidative stress. Cell Stress Chaperones 2018, 23, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gong, W.; Wu, S.; Perrett, S. Hsp70 in Redox Homeostasis. Cells 2022, 11, 829. [Google Scholar] [CrossRef]
- Collins, C.B.; Nguyen, T.T.; Leddy, R.S.; Alula, K.M.; Yeckes, A.R.; Strassheim, D.; Aherne, C.M.; Luck, M.E.; Karoor, V.; Jedlicka, P.; et al. Heat shock factor 1 drives regulatory T-cell induction to limit murine intestinal inflammation. Mucosal Immunol. 2024, 17, 94–110. [Google Scholar] [CrossRef]
- Belenichev, I.; Bila, Y. The effect of the heat shock protein HSP70 modulators on the energy metabolism of the rats brain in acute cerebral ischemia. Biol. Mark. Guid. 2019, 6, 51–62. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar]
- Grossini, E.; Bellofatto, K.; Farruggio, S.; Sigaudo, L.; Marotta, P.; Raina, G.; De Giuli, V.; Mary, D.; Pollesello, P.; Minisini, R.; et al. Levosimendan inhibits peroxidation in hepatocytes by modulating apoptosis/autophagy interplay. PLoS ONE 2015, 10, e0124742. [Google Scholar] [CrossRef]
- Mohammadinejad, R.; Moosavi, M.A.; Tavakol, S.; Vardar, D.Ö.; Hosseini, A.; Rahmati, M.; Dini, L.; Hussain, S.; Mandegary, A.; Klionsky, D.J. Necrotic, apoptotic and autophagic cell fates triggered by nanoparticles. Autophagy 2019, 15, 4–33. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Boiti, A.; Vallone, D.; Foulkes, N.S. Reactive Oxygen Species Signaling and Oxidative Stress: Transcriptional Regulation and Evolution. Antioxidants 2024, 13, 312. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Mayorov, V.I.; Panov, A.V. Physiological Levels of Nitric Oxide Diminish Mitochondrial Superoxide. Potential Role of Mitochondrial Dinitrosyl Iron Complexes and Nitrosothiols. Front. Physiol. 2017, 8, 907. [Google Scholar] [CrossRef] [PubMed]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta 2014, 1840, 768–780. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, A.J.P.O.; de Oliveira, J.C.P.L.; da Silva Pontes, L.V.; de Souza Júnior, J.F.; Gonçalves, T.A.F.; Dantas, S.H.; de Almeida Feitosa, M.S.; Silva, A.O.; de Medeiros, I.A. ROS: Basic Concepts, Sources, Cellular Signaling, and its Implications in Aging Pathways. Oxid. Med. Cell Longev. 2022, 2022, 1225578. [Google Scholar] [CrossRef] [PubMed]
- Fragoso-Morales, L.G.; Correa-Basurto, J.; Rosales-Hernández, M.C. Implication of Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase and Its Inhibitors in Alzheimer’s Disease Murine Models. Antioxidants 2021, 10, 218. [Google Scholar] [CrossRef] [PubMed]
- Shahani, N.; Sawa, A. Protein S-nitrosylation: Role for nitric oxide signaling in neuronal death. Biochim. Biophys. Acta 2012, 1820, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Qiu, H. Post-Translational S-Nitrosylation of Proteins in Regulating Cardiac Oxidative Stress. Antioxidants 2020, 9, 1051. [Google Scholar] [CrossRef]
- Mazur, I.; Belenichev, I.; Kucherenko, L.; Bukhtiyarova, N.; Puzyrenko, A.; Khromylova, O.; Bidnenko, O.; Gorchakova, N. Antihypertensive and cardioprotective effects of new compound 1-(β-phenylethyl)-4-amino-1, 2, 4-triazolium bromide (Hypertril). Eur. J. Pharmacol. 2019, 853, 336–344. [Google Scholar] [CrossRef]
- Vona, R.; Pallotta, L.; Cappelletti, M.; Severi, C.; Matarrese, P. The Impact of Oxidative Stress in Human Pathology: Focus on Gastrointestinal Disorders. Antioxidants 2021, 10, 201. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Gorbacheva, S.V.; Demchenko, A.V.; Bukhtiyarova, N.V. The thiol-disulfide balance and the nitric oxide system in the brain tissue of rats subjected to experimental acute impairment of cerebral blood flow: The therapeutic effects of nootropic drugs. Neurochem. J. 2014, 8, 24–27. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Burlaka, B.S.; Bukhtiyarova, N.V.; Aliyeva, E.G.; Suprun, E.V.; Ishchenko, A.M.; Simbirtsev, A.S. Pharmacological Correction of Thiol-Disulphide Imbalance in the Rat Brain by Intranasal Form of Il-1b Antagonist in a Model of Chronic Cerebral Ischemia. Neurochem. J. 2021, 15, 30–36. [Google Scholar] [CrossRef]
- Di Giacomo, G.; Rizza, S.; Montagna, C.; Filomeni, G. Established Principles and Emerging Concepts on the Interplay between Mitochondrial Physiology and S-(De)nitrosylation: Implications in Cancer and Neurodegeneration. Int. J. Cell Biol. 2012, 2012, 361872. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Loscalzo, J. Redox regulation of mitochondrial function. Antioxid. Redox Signal. 2012, 16, 1323–1367. [Google Scholar] [CrossRef]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Litvinenko, E.S.; Kamishny, A.M. Character of mRNA HIF-1α and HIF-3α expression, level of nitrotyrosine, cGMP and interleukins in the brain homogenate of Mongolian sand rats with acute cerebral blood flow disturbance and against the background of therapy with modulators of the glutathione system. Visnyk Probl. Biol. Medytsyny 2018, 1, 142. [Google Scholar]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef]
- Griswold-Prenner, I.; Kashyap, A.K.; Mazhar, S.; Hall, Z.W.; Fazelinia, H.; Ischiropoulos, H. Unveiling the human nitroproteome: Protein tyrosine nitration in cell signaling and cancer. J. Biol. Chem. 2023, 299, 105038. [Google Scholar] [CrossRef] [PubMed]
- Belenichev, I.F.; Bila, Y.V. The relationship between the concentration of HSP 70 activity of the thiol-disulfide system and the degree of neurological disorders in the modeling of acute cerebral ischemia. Bull. Probl. Biol. Med. 2017, 1, 86–91. [Google Scholar]
- Ray, A.; Maharana, K.C.; Meenakshi, S.; Singh, S. Endothelial dysfunction and its relation in different disorders: Recent update. Health Sci. Rev. 2023, 7, 100084. [Google Scholar] [CrossRef]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.S.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Matjuda, E.N.; Engwa, G.A.; Sewani-Rusike, C.R.; Nkeh-Chungag, B.N. An Overview of Vascular Dysfunction and Determinants: The Case of Children of African Ancestry. Front. Pediatr. 2021, 9, 769589. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, H.; Streese, L.; Vilser, W. Retinal vessel diameters and function in cardiovascular risk and disease. Prog. Retin. Eye Res. 2022, 91, 101095. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Moschetti, L.; Piantoni, S.; Vizzardi, E.; Sciatti, E.; Riccardi, M.; Franceschini, F.; Cavazzana, I. Endothelial Dysfunction in Systemic Lupus Erythematosus and Systemic Sclerosis: A Common Trigger for Different Microvascular Diseases. Front. Med. 2022, 9, 849086. [Google Scholar] [CrossRef]
- Steyers, C.M., 3rd; Miller, F.J., Jr. Endothelial Dysfunction in Chronic Inflammatory Diseases. Int. J. Mol. Sci. 2014, 15, 11324–11349. [Google Scholar] [CrossRef] [PubMed]
- Janaszak-Jasiecka, A.; Płoska, A.; Wierońska, J.M.; Dobrucki, L.W.; Kalinowski, L. Endothelial dysfunction due to eNOS uncoupling: Molecular mechanisms as potential therapeutic targets. Cell Mol. Biol. Lett. 2023, 28, 21. [Google Scholar] [CrossRef] [PubMed]
- Lorin, J.; Zeller, M.; Guilland, J.-C.; Cottin, Y.; Vergely, C.; Rochette, L. Arginine and nitric oxide synthase: Regulatory mechanisms and cardiovascular aspects. Mol. Nutr. Food Res. 2014, 58, 101–116. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Mazur, I.A.; Abramov, A.V.; Kucherenko, L.I.; Bukhtiyarova, N.V.; Egorov, A.A.; Belenicheva, O.I.; Polyakova, E.N. The endothelium-protective effect of 3-methyl-1, 2, 4-triazolyl-5-thioacetate (S)-2, 6-diaminohexanic acid (lysinium): Effects on the expression of vascular endothelial growth factor (VEGF) and the characteristics of the endothelio cytes of the cerebral vessels of animals with cerebral ischemia. Neurochem. J. 2013, 7, 296–302. [Google Scholar]
- Rajendran, S.; Shen, X.; Glawe, J.; Kolluru, G.K.; Kevil, C.G. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Growth and Remodeling. Compr. Physiol. 2019, 9, 1213–1247. [Google Scholar] [PubMed]
- Correia, M.J.; Pimpão, A.B.; Fernandes, D.G.F.; Morello, J.; Sequeira, C.O.; Calado, J.; Antunes, A.M.M.; Almeida, M.S.; Branco, P.; Monteiro, E.C.; et al. Cysteine as a Multifaceted Player in Kidney, the Cysteine-Related Thiolome and Its Implications for Precision Medicine. Molecules 2022, 27, 1416. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical interventions. J. Amino Acids. 2012, 2012, 736837. [Google Scholar] [CrossRef] [PubMed]
- Tiurenkov, I.N.; Voronkov, A.V.; Slietsans, A.A.; Volotova, E.V. Endothelial protection drugs--a new class of pharmacological agents. Vestn. Ross. Akad. Med. Nauk 2012, 7, 50–57. (In Russian) [Google Scholar] [CrossRef]
- De Leonardis, F.; Colalillo, G.; Finazzi Agrò, E.; Miano, R.; Fuschi, A.; Asimakopoulos, A.D. Endothelial Dysfunction, Erectile Deficit and Cardiovascular Disease: An Overview of the Pathogenetic Links. Biomedicines 2022, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. Molecular mechanisms underlying the activation of eNOS. Pflug. Arch. 2010, 459, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Everett, A.D.; Stoops, T.D.; Nairn, A.C.; Brautigan, D. Angiotensin II regulates phosphorylation of translation elongation factor-2 in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H161–H167. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M. The Endothelium: Part 1, Multiple Functions of the Endothelial Cells—Focus on Endothelium-Derived Vasoactive Mediators; Morgan Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Gorbacheva, S.V.; Belenichev, I.F. Indicators of thiol-disulfide system and nitrosative stress in neurons under conditions of modeling glutamate excitotoxicity in vitro and against the background of application of nos inhibitors of different selectivity. World Med. Biol. 2015, 11, 112–116. (In Russian) [Google Scholar]
- Gorbacheva, S.V.; Belenichev, I.F. Possible ways of interrupting NO-dependent pathways of neurodegeneration with the use of no-synthase inhibitors of different selectivity in conditions of experimental cerebral circulation disorder. Achiev. Biol. Med. 2015, 2, 21–25. (In Ukranian) [Google Scholar]
- Floryszak-Wieczorek, J.; Milczarek, G.; Arasimowicz, M.; Ciszewski, A. Do nitric oxide donors mimic endogenous NO-related response in plants? Planta 2006, 224, 1363–1372. [Google Scholar] [CrossRef]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay. Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef]
- Sun, L.; Liu, H.; Ye, Y.; Lei, Y.; Islam, R.; Tan, S.; Tong, R.; Miao, Y.B.; Cai, L. Smart nanoparticles for cancer therapy. Signal Transduct. Target. Ther. 2023, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Xu, J.; Singh, B.; Yu, X.; Wu, T.; Huang, Y. Nitrates for the prevention of cardiac morbidity and mortality in patients undergoing non-cardiac surgery. Cochrane Database Syst. Rev. 2016, 2016, CD010726. [Google Scholar] [CrossRef]
- Hottinger, D.G.; Beebe, D.S.; Kozhimannil, T.; Prielipp, R.C.; Belani, K.G. Sodium nitroprusside in 2014, A clinical concepts review. J. Anaesthesiol. Clin. Pharmacol. 2014, 30, 462–471. [Google Scholar] [PubMed]
- Broniowska, K.A.; Diers, A.R.; Hogg, N. S-nitrosoglutathione. Biochim. Biophys. Acta 2013, 1830, 3173–3181. [Google Scholar] [CrossRef]
- Goudie, M.J.; Brisbois, E.J.; Pant, J.; Thompson, A.; Potkay, J.A.; Handa, H. Characterization of an S-nitroso-N-acetylpenicillamine-based nitric oxide releasing polymer from a translational perspective. Int. J. Polym. Mater. 2016, 65, 769–778. [Google Scholar] [CrossRef]
- Sysel, A.M.; Dunphy, M.J.; Bauer, J.A. Antimicrobial properties of diethylamine NONOate, a nitric oxide donor, against Escherichia coli: A pilot study. J. Antibiot. 2021, 74, 260–265. [Google Scholar] [CrossRef]
- da Silva, G.M.; da Silva, M.C.; Nascimento, D.V.G.; Lima Silva, E.M.; Gouvêa, F.F.F.; de França Lopes, L.G.; Araújo, A.V.; Ferraz Pereira, K.N.; de Queiroz, T.M. Nitric Oxide as a Central Molecule in Hypertension: Focus on the Vasorelaxant Activity of New Nitric Oxide Donors. Biology 2021, 10, 1041. [Google Scholar] [CrossRef]
- Mastrolia, I.; Foppiani, E.M.; Murgia, A.; Candini, O.; Samarelli, A.V.; Grisendi, G.; Veronesi, E.; Horwitz, E.M.; Dominici, M. Challenges in Clinical Development of Mesenchymal Stromal/Stem Cells: Concise Review. Stem Cells Transl. Med. 2019, 8, 1135–1148. [Google Scholar] [CrossRef]
- Belenichev, I.; Bak, P.; Popazova, O.; Ryzhenko, V.; Bukhtiyarova, N.; Puzyrenko, A. Integrative and Biochemical Parameters in Rats in the Simulation of Doxorubicin Chronic Heart Failure and During the Use of β-Adrenergic Blockers. J. Fac. Pharm. Ank. Univ. 2023, 47, 228–238. [Google Scholar] [CrossRef]
- Goncharov, O.; Belenichev, I.; Abramov, A.; Popazova, O.; Kucherenko, L.; Bukhtiyarova, N.; Pavliuk, I. Influence of experimental heart failure therapy with different generations of β-adrenergic blockers on Cardiac Electrical Activity (ECG) and Autonomic Regulation of Heart Rhythm (ARHR). Pharmacia 2023, 70, 1157–1165. [Google Scholar] [CrossRef]
- Ryzhenko, V.P.; Belenichev, I.F.; Samura, I.B.; Ryzhov, O.A. Development of software for prediction and virtual screening of antioxidant activity of new synthesized azaheterocyclic compounds. Int. J. Basic. Clin. Pharmacol. 2019, 8, 1292–1296. [Google Scholar] [CrossRef]
- Ryzhenko, V.; Ryzhov, O.; Belenichev, I.F.; Levich, S.V. Study of Dependence of Xanthine Derivatives NO-Scavenger Properties from Energy Descriptors. Biol. Mark. Guid. Ther. 2018, 5, 37–46. [Google Scholar] [CrossRef]
- Ryzhov, O.A.; Ryzhenko, V.P.; Levich, S.V.; Belenichev, I.F. Analysis of influence of quantum chemical descriptors on no-scavenger properties among xanthine derivatives. Biol. Mark. Guid. Ther. 2017, 4, 39–48. [Google Scholar] [CrossRef]
- Belenichev, I.F.; Nosach, S.G.; Samura, I.B.; Levich, S.V. Some Aspects of Neuroprotective Action of a New Derivative of 3-Methylxanthine (Compound C-3) Under Conditions of Acute Disorder of Cerebral Circulation (ADCC) Modeling by Ischemic Stroke Type. Biol. Mark. Guid. Ther. 2018, 5, 63–73. [Google Scholar] [CrossRef]
- Belenichev, I.; Aleksandrova, K.; Shkoda, A.; Levich, S.; Nosach, S. Influence of 3-methylxanthine derivative on the morphological and functional characteristics of neurons of sensorimotor cortex of rats with experimental intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2016, 36, 787. [Google Scholar]
- Belenichev, I.F.; Aleksandrova, K.V.; Buhtiyarova, N.V.; Levich, S.V.; Nosach, S.G.; Sinchenko, D.N. Antioxidant effect of xanthinyl-7-acetic acid derivative on SOD activity under condition of nitrosative stress in vitro. Biol. Mark. Guid. Ther. 2016, 3, 139–145. [Google Scholar] [CrossRef]
- Ryzhenko, V.P. Optimization of purposeful search of NO scavengers in a number of xanthine derivatives. In The Dissertation on Competition of a Scientific Degree of the Candidate of Biological Sciences on a Specialty 14.03.05 “Pharmacology”; State Institution “Institute of Pharmacology and Toxicology of the National Academy of Medical Sciences of Ukraine”: Kyiv, Ukraine, 2020. [Google Scholar]
- Chekman, I.S.; Belenichev, I.F.; Syrova, A.O.; Gorchakova, N.A.; Bukhtiyarova, N.V.; Ryzhenko, V.P.; Chalenko, N.N. Aspects of creation of neuroprotective, anti-inflammatory drugs. Dopov. Nac. Akad. Nauk. Ukr. 2019, 9, 88–98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacological Agent | Primary Target | Pharmacological Effect |
|---|---|---|
| S-methylisothiourea (SMT) | Selective highly reactive iNOS inhibitor | Injection w/w to rats after occlusion of carotid arteries (1 mg/kg) over 4 days led to a reliable protective effect only from the 1st day of the experiment, reaching the maximum on the 4th day. SMT had a significant neuroprotective effect [182] |
| N-nitro-L-arginine methyl ester hydrochloride | Selective iNOS inhibitor | Incorporation of 40 μmol into the neuronal suspension prior to glutamate (100 μM) had a protective effect when incubated for 30 and 60 min (decreased nitrotyrosine, increased GSH, Cu-Zn-SOD) [182,262] |
| N-propyl-L-arginine hydrochloride | Selective nNOS inhibitor | Incorporation of 50 μmol into the neuronal suspension prior to glutamate (100 μM) had a protective effect when incubated for 30 (decreased nitrotyrosine, increased GSH, Cu-Zn-SOD), then the effect diminished. Injection w/w to rats after occlusion of carotid arteries (2.5 mg/kg) during 4 days for the first 12 h, a reliable effect [262] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belenichev, I.; Popazova, O.; Bukhtiyarova, N.; Savchenko, D.; Oksenych, V.; Kamyshnyi, O. Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection. Antioxidants 2024, 13, 504. https://doi.org/10.3390/antiox13050504
Belenichev I, Popazova O, Bukhtiyarova N, Savchenko D, Oksenych V, Kamyshnyi O. Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection. Antioxidants. 2024; 13(5):504. https://doi.org/10.3390/antiox13050504
Chicago/Turabian StyleBelenichev, Igor, Olena Popazova, Nina Bukhtiyarova, Dmytro Savchenko, Valentyn Oksenych, and Oleksandr Kamyshnyi. 2024. "Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection" Antioxidants 13, no. 5: 504. https://doi.org/10.3390/antiox13050504
APA StyleBelenichev, I., Popazova, O., Bukhtiyarova, N., Savchenko, D., Oksenych, V., & Kamyshnyi, O. (2024). Modulating Nitric Oxide: Implications for Cytotoxicity and Cytoprotection. Antioxidants, 13(5), 504. https://doi.org/10.3390/antiox13050504