
The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis


Abstract
:1. The Importance of ROS in Cellular Signaling and Gene Expression
2. The NADPH Oxidase Family
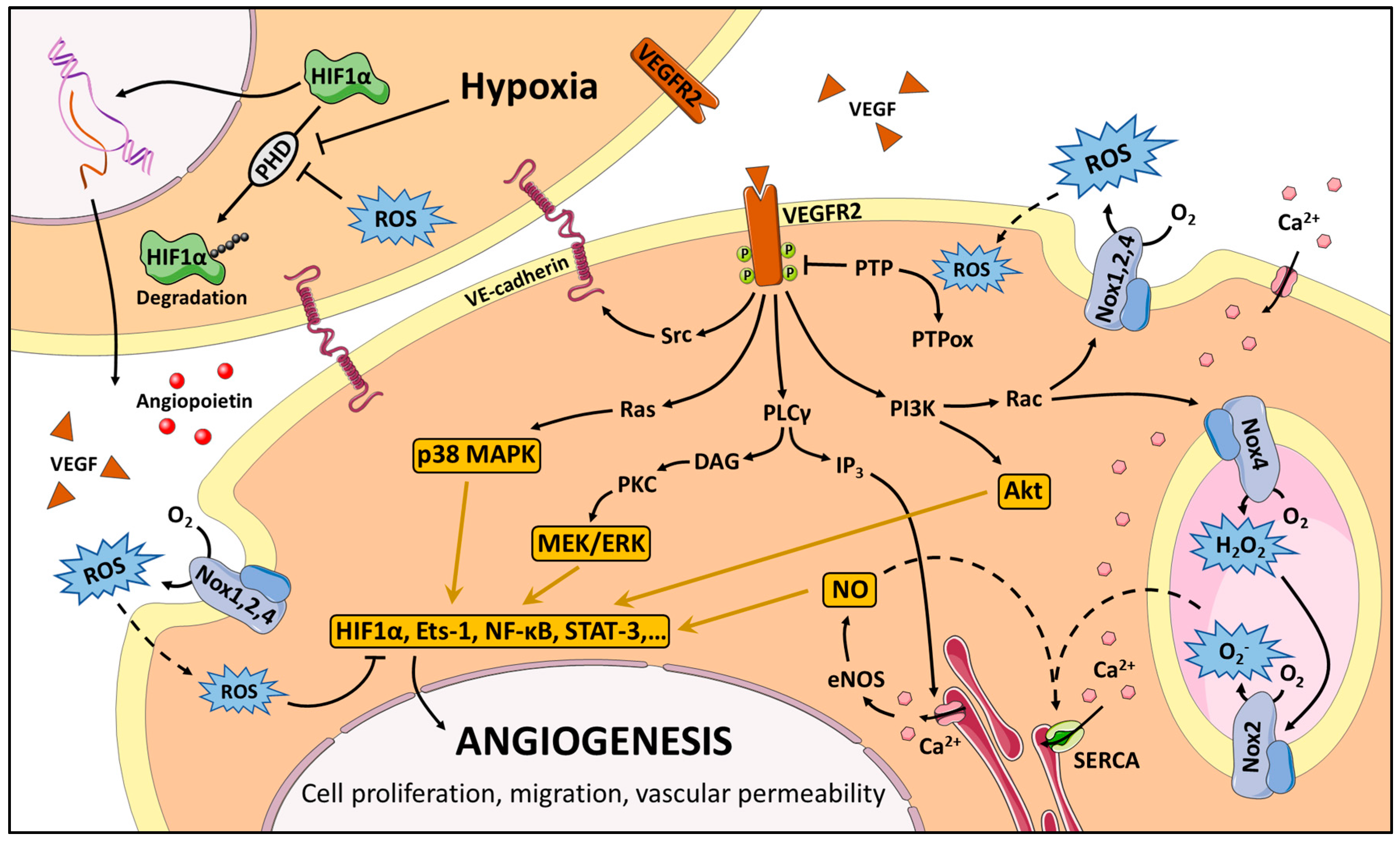
3. Redox Signaling in Angiogenesis
4. Therapeutic Opportunities
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Genestra, M. Oxyl Radicals, Redox-Sensitive Signalling Cascades and Antioxidants. Cell Signal. 2007, 19, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Jomova, K.; Rhodes, C.J.; Kuca, K.; Musilek, K. Redox- and Non-Redox-Metal-Induced Formation of Free Radicals and Their Role in Human Disease. Arch. Toxicol. 2016, 90, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.; Kauppila, J.H.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Sardina, J.L.; Lopez-Ruano, G.; Sanchez-Sanchez, B.; Llanillo, M.; Hernandez-Hernandez, A. Reactive Oxygen Species: Are They Important for Haematopoiesis? Crit. Rev. Oncol. Hematol. 2012, 81, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Galadari, S.; Rahman, A.; Pallichankandy, S.; Thayyullathil, F. Reactive Oxygen Species and Cancer Paradox: To Promote or to Suppress? Free Radic. Biol. Med. 2017, 104, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Tatla, S.; Woodhead, V.; Foreman, J.C.; Chain, B.M. The Role of Reactive Oxygen Species in Triggering Proliferation and IL-2 Secretion in T Cells. Free Radic. Biol. Med. 1999, 26, 14–24. [Google Scholar] [CrossRef]
- Goldstone, S.D.; Milligan, A.D.; Hunt, N.H. Oxidative Signalling and Gene Expression during Lymphocyte Activation. Biochim. Biophys. Acta 1996, 1314, 175–182. [Google Scholar] [CrossRef]
- Czech, M.P. Differential Effects of Sulfhydryl Reagents on Activation and Deactivation of the Fat Cell Hexose Transport System. J. Biol. Chem. 1976, 251, 1164–1170. [Google Scholar] [PubMed]
- Mukherjee, S.P.; Lane, R.H.; Lynn, W.S. Endogenous Hydrogen Peroxide and Peroxidative Metabolism in Adipocytes in Response to Insulin and Sulfhydryl Reagents. Biochem. Pharmacol. 1978, 27, 2589–2594. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen Peroxide as a Central Redox Signaling Molecule in Physiological Oxidative Stress: Oxidative Eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Bak, D.W.; Weerapana, E. Cysteine-Mediated Redox Signalling in the Mitochondria. Mol. Biosyst. 2015, 11, 678–697. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Sacco, F.; Perfetto, L.; Castagnoli, L.; Cesareni, G. The Human Phosphatase Interactome: An Intricate Family Portrait. FEBS Lett. 2012, 586, 2732–2739. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Serine/Threonine Phosphatases: Mechanism through Structure. Cell 2009, 139, 468–484. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y.; Wang, Y.; Dixon, J.E. Dissecting the Catalytic Mechanism of Protein-Tyrosine Phosphatases. Proc. Natl. Acad. Sci. USA 1994, 91, 1624–1627. [Google Scholar] [CrossRef] [PubMed]
- Barford, D.; Flint, A.J.; Tonks, N.K. Crystal Structure of Human Protein Tyrosine Phosphatase 1B. Science 1994, 263, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.C.; Fukada, T.; Tonks, N.K. Reversible Oxidation and Inactivation of Protein Tyrosine Phosphatases in Vivo. Mol. Cell 2002, 9, 387–399. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Dixon, J.E. Active Site Labeling of the Yersinia Protein Tyrosine Phosphatase: The Determination of the PKa of the Active Site Cysteine and the Function of the Conserved Histidine 402. Biochemistry 1993, 32, 9340–9345. [Google Scholar] [CrossRef] [PubMed]
- Barrett, W.C.; DeGnore, J.P.; Konig, S.; Fales, H.M.; Keng, Y.F.; Zhang, Z.Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B Via Glutathionylation of the Active Site Cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible Inactivation of Protein-Tyrosine Phosphatase 1B in A431 Cells Stimulated With Epidermal Growth Factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Qu, C.K.; Maeng, J.S.; Falahati, R.; Lee, C.; Williams, M.S. Receptor-Stimulated Oxidation of SHP-2 Promotes T-Cell Adhesion Through SLP-76-ADAP. EMBO J. 2005, 24, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Kumar, D.; Siddiqui, Z.; Basu, S.K.; Kumar, V.; Rao, K.V. The Strength of Receptor Signaling Is Centrally Controlled through a Cooperative Loop between Ca2+ and an Oxidant Signal. Cell 2005, 121, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Foley, T.D.; Petro, L.A.; Stredny, C.M.; Coppa, T.M. Oxidative Inhibition of Protein Phosphatase 2A Activity: Role of Catalytic Subunit Disulfides. Neurochem. Res. 2007, 32, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Carroll, K.S. Redox Regulation of Protein Kinases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 332–356. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; Cotter, T.G. Redox Regulation of Protein Kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef] [PubMed]
- Hempel, N.; Trebak, M. Crosstalk between Calcium and Reactive Oxygen Species Signaling in Cancer. Cell Calcium 2017. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schoneich, C.; Cohen, R.A. S-Glutathiolation by Peroxynitrite Activates SERCA During Arterial Relaxation by Nitric Oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Evangelista, A.M.; Thompson, M.D.; Bolotina, V.M.; Tong, X.; Cohen, R.A. Nox4- and Nox2-Dependent Oxidant Production Is Required for VEGF-Induced SERCA Cysteine-674 S-Glutathiolation and Endothelial Cell Migration. Free Radic. Biol. Med. 2012, 53, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schroder, K. Nox Family NADPH Oxidases: Molecular Mechanisms of Activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Maher, J.; Yamamoto, M. The Rise of Antioxidant Signaling—The Evolution and Hormetic Actions of Nrf2. Toxicol. Appl. Pharmacol. 2010, 244, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Milner, J. Redox Modulation of P53 Conformation and Sequence-Specific DNA Binding in Vitro. Cancer Res. 1993, 53, 4469–4473. [Google Scholar] [PubMed]
- Lando, D.; Pongratz, I.; Poellinger, L.; Whitelaw, M.L. A Redox Mechanism Controls Differential DNA Binding Activities of Hypoxia-Inducible Factor (HIF) 1alpha and the HIF-Like Factor. J. Biol. Chem. 2000, 275, 4618–4627. [Google Scholar] [CrossRef] [PubMed]
- Glineur, C.; Davioud-Charvet, E.; Vandenbunder, B. The Conserved Redox-Sensitive Cysteine Residue of the DNA-Binding Region in the C-Rel Protein Is Involved in the Regulation of the Phosphorylation of the Protein. Biochem. J. 2000, 352 Pt 2, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Janssen-Heininger, Y.M.; Poynter, M.E.; Baeuerle, P.A. Recent Advances Towards Understanding Redox Mechanisms in the Activation of Nuclear Factor KappaB. Free Radic. Biol. Med. 2000, 28, 1317–1327. [Google Scholar] [CrossRef]
- Tell, G.; Zecca, A.; Pellizzari, L.; Spessotto, P.; Colombatti, A.; Kelley, M.R.; Damante, G.; Pucillo, C. An ‘Environment to Nucleus’ Signaling System Operates in B Lymphocytes: Redox Status Modulates BSAP/Pax-5 Activation through Ref-1 Nuclear Translocation. Nucleic Acids Res. 2000, 28, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Kambe, F.; Ohmori, S.; Seo, H. Oxidoreductive Modification of Two Cysteine Residues in Paired Domain by Ref-1 Regulates DNA-Binding Activity of Pax-8. Biochem. Biophys. Res. Commun. 2002, 297, 288–293. [Google Scholar] [CrossRef]
- Huang, R.P.; Adamson, E.D. Characterization of the DNA-Binding Properties of the Early Growth Response-1 (Egr-1) Transcription Factor: Evidence for Modulation by a Redox Mechanism. DNA Cell Biol. 1993, 12, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.C.; Curran, T. Redox Activation of Fos-Jun DNA Binding Activity Is Mediated by a DNA Repair Enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [PubMed]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Doyle, K.; Fitzpatrick, F.A. Redox Signaling, Alkylation (Carbonylation) of Conserved Cysteines Inactivates Class I Histone Deacetylases 1, 2, and 3 and Antagonizes Their Transcriptional Repressor Function. J. Biol. Chem. 2010, 285, 17417–17424. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, S.A.; Turk, P.W.; Milkowski, D.H.; Kozlowski, K. Free Radical Adducts Induce Alterations in DNA Cytosine Methylation. Proc. Natl. Acad. Sci. USA 1994, 91, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Turk, P.W.; Laayoun, A.; Smith, S.S.; Weitzman, S.A. DNA Adduct 8-Hydroxyl-2’-Deoxyguanosine (8-Hydroxyguanine) Affects Function of Human DNA Methyltransferase. Carcinogenesis 1995, 16, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.D.; Chambers, P.A.; Lodge, J.P.; Pratt, J.R. Ischemia- Reperfusion Injury and Its Influence on the Epigenetic Modification of the Donor Kidney Genome. Transplantation 2008, 86, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative Damage to Methyl-CpG Sequences Inhibits the Binding of the Methyl-CpG Binding Domain (MBD) of Methyl-CpG Binding Protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef] [PubMed]
- Poyton, R.O.; Ball, K.A.; Castello, P.R. Mitochondrial Generation of Free Radicals and Hypoxic Signaling. Trends Endocrinol. Metab. 2009, 20, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Biological Roles for the NOX Family NADPH Oxidases. J. Biol. Chem. 2008, 283, 16961–16965. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Hably, C.; Meczner, A.; Timar, C.; Lakatos, G.; Enyedi, P.; Ligeti, E. Role of Nox2 in Elimination of Microorganisms. Semin. Immunopathol. 2008, 30, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Beaumel, S.; Grunwald, D.; Fieschi, F.; Stasia, M.J. Identification of NOX2 Regions for Normal Biosynthesis of Cytochrome B558 in Phagocytes Highlighting Essential Residues for P22phox Binding. Biochem. J. 2014, 464, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Heiple, J.M.; Simons, E.R.; Clark, R.A. Subcellular Localization of the B-Cytochrome Component of the Human Neutrophil Microbicidal Oxidase: Translocation during Activation. J. Cell. Biol. 1983, 97, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Lundqvist, H.; Follin, P.; Khalfan, L.; Dahlgren, C. Phorbol Myristate Acetate-Induced NADPH Oxidase Activity in Human Neutrophils: Only Half the Story Has Been Told. J. Leukoc. Biol. 1996, 59, 270–279. [Google Scholar] [PubMed]
- Jones, S.A.; O’Donnell, V.B.; Wood, J.D.; Broughton, J.P.; Hughes, E.J.; Jones, O.T. Expression of Phagocyte NADPH Oxidase Components in Human Endothelial Cells. Am. J. Physiol. 1996, 271, H1626–H1634. [Google Scholar] [PubMed]
- Banfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two Novel Proteins Activate Superoxide Generation by the NADPH Oxidase NOX1. J. Biol. Chem. 2003, 278, 3510–3513. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.A.; Arnold, R.S.; Lassegue, B.; Shi, J.; Xu, X.; Sorescu, D.; Chung, A.B.; Griendling, K.K.; Lambeth, J.D. Cell Transformation by the Superoxide-Generating Oxidase Mox1. Nature 1999, 401, 79–82. [Google Scholar] [PubMed]
- Kobayashi, S.; Nojima, Y.; Shibuya, M.; Maru, Y. Nox1 Regulates Apoptosis and Potentially Stimulates Branching Morphogenesis in Sinusoidal Endothelial Cells. Exp. Cell Res. 2004, 300, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Lassegue, B.; Sorescu, D.; Szocs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel Gp91(Phox) Homologues in Vascular Smooth Muscle Cells : Nox1 Mediates Angiotensin II-Induced Superoxide Formation and Redox-Sensitive Signaling Pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, M.; Fan, C.; Yabe-Nishimura, C. NADPH Oxidase Is Involved in Prostaglandin F2alpha-Induced Hypertrophy of Vascular Smooth Muscle Cells: Induction of NOX1 by PGF2alpha. J. Biol. Chem. 2002, 277, 13438–13442. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Diebold, B.A.; Hughes, Y.; Lambeth, J.D. Nox1-Dependent Reactive Oxygen Generation Is Regulated by Rac1. J. Biol. Chem. 2006, 281, 17718–17726. [Google Scholar] [CrossRef] [PubMed]
- Banfi, B.; Malgrange, B.; Knisz, J.; Steger, K.; Dubois-Dauphin, M.; Krause, K.H. NOX3, a Superoxide-Generating NADPH Oxidase of the Inner Ear. J. Biol. Chem. 2004, 279, 46065–46072. [Google Scholar] [CrossRef] [PubMed]
- Paffenholz, R.; Bergstrom, R.A.; Pasutto, F.; Wabnitz, P.; Munroe, R.J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J.; et al. Vestibular Defects in Head-Tilt Mice Result From Mutations in Nox3, Encoding an NADPH Oxidase. Genes Dev. 2004, 18, 486–491. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.; Takeya, R.; Miyano, K.; Kikuchi, H.; Sumimoto, H. The NADPH Oxidase Nox3 Constitutively Produces Superoxide in a P22phox-Dependent Manner: Its Regulation by Oxidase Organizers and Activators. J. Biol. Chem. 2005, 280, 23328–23339. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Longo-Guess, C.M.; Bergstrom, D.E.; Nauseef, W.M.; Jones, S.M.; Banfi, B. Mutation of the Cyba Gene Encoding P22phox Causes Vestibular and Immune Defects in Mice. J. Clin. Investig. 2008, 118, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Kiss, P.J.; Knisz, J.; Zhang, Y.; Baltrusaitis, J.; Sigmund, C.D.; Thalmann, R.; Smith, R.J.; Verpy, E.; Banfi, B. Inactivation of NADPH Oxidase Organizer 1 Results in Severe Imbalance. Curr. Biol. 2006, 16, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, J.P.; Spruce, C.A.; Fairfield, H.E.; Bergstrom, D.E. Generation of a Conditional Null Allele of NADPH Oxidase Activator 1 (NOXA1). Genesis 2010, 48, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 As the Major Catalytic Component of an Endothelial NAD(P)H Oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Kopp, J.B.; Varnai, P.; Leto, T.L. Identification of Renox, an NAD(P)H Oxidase in Kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed]
- Ambasta, R.K.; Kumar, P.; Griendling, K.K.; Schmidt, H.H.; Busse, R.; Brandes, R.P. Direct Interaction of the Novel Nox Proteins With P22phox Is Required for the Formation of a Functionally Active NADPH Oxidase. J. Biol. Chem. 2004, 279, 45935–45941. [Google Scholar] [CrossRef] [PubMed]
- Martyn, K.D.; Frederick, L.M.; von Loehneysen, K.; Dinauer, M.C.; Knaus, U.G. Functional Analysis of Nox4 Reveals Unique Characteristics Compared to Other NADPH Oxidases. Cell Signal. 2006, 18, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The Superoxide-Producing NAD(P)H Oxidase Nox4 in the Nucleus of Human Vascular Endothelial Cells. Genes Cells 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Mandal, C.C.; Ganapathy, S.; Gorin, Y.; Mahadev, K.; Block, K.; Abboud, H.E.; Harris, S.E.; Ghosh-Choudhury, G.; Ghosh-Choudhury, N. Reactive Oxygen Species Derived From Nox4 Mediate BMP2 Gene Transcription and Osteoblast Differentiation. Biochem. J. 2011, 433, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Gorin, Y.; Ricono, J.M.; Kim, N.H.; Bhandari, B.; Choudhury, G.G.; Abboud, H.E. Nox4 Mediates Angiotensin II-Induced Activation of Akt/Protein Kinase B in Mesangial Cells. Am. J. Physiol. Renal Physiol. 2003, 285, F219–F229. [Google Scholar] [CrossRef] [PubMed]
- Serrander, L.; Jaquet, V.; Bedard, K.; Plastre, O.; Hartley, O.; Arnaudeau, S.; Demaurex, N.; Schlegel, W.; Krause, K.H. NOX5 Is Expressed at the Plasma Membrane and Generates Superoxide in Response to Protein Kinase C Activation. Biochimie 2007, 89, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Brar, S.S.; Corbin, Z.; Kennedy, T.P.; Hemendinger, R.; Thornton, L.; Bommarius, B.; Arnold, R.S.; Whorton, A.R.; Sturrock, A.B.; Huecksteadt, T.P.; et al. NOX5 NAD(P)H Oxidase Regulates Growth and Apoptosis in DU 145 Prostate Cancer Cells. Am. J. Physiol. Cell Physiol. 2003, 285, C353–C369. [Google Scholar] [CrossRef] [PubMed]
- Kamiguti, A.S.; Serrander, L.; Lin, K.; Harris, R.J.; Cawley, J.C.; Allsup, D.J.; Slupsky, J.R.; Krause, K.H.; Zuzel, M. Expression and Activity of NOX5 in the Circulating Malignant B Cells of Hairy Cell Leukemia. J. Immunol. 2005, 175, 8424–8430. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Fu, X.; Behar, J.; Wands, J.; Beer, D.G.; Souza, R.F.; Spechler, S.J.; Lambeth, D.; Cao, W. NADPH Oxidase NOX5-S Mediates Acid-Induced Cyclooxygenase-2 Expression Via Activation of NF-KappaB in Barrett’s Esophageal Adenocarcinoma Cells. J. Biol. Chem. 2007, 282, 16244–16255. [Google Scholar] [CrossRef] [PubMed]
- Banfi, B.; Molnar, G.; Maturana, A.; Steger, K.; Hegedus, B.; Demaurex, N.; Krause, K.H. A Ca2+-Activated NADPH Oxidase in Testis, Spleen, and Lymph Nodes. J. Biol. Chem. 2001, 276, 37594–37601. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Cao, Z.; Xu, X.; van Meir, E.G.; Lambeth, J.D. Homologs of Gp91phox: Cloning and Tissue Expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar] [CrossRef]
- De, D.X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of Two Human Thyroid CDNAs Encoding New Members of the NADPH Oxidase Family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a Novel Flavoprotein Involved in the Thyroid NADPH Oxidase. Cloning of the Porcine and Human Cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed]
- Morand, S.; Dos Santos, O.F.; Ohayon, R.; Kaniewski, J.; Noel-Hudson, M.S.; Virion, A.; Dupuy, C. Identification of a Truncated Dual Oxidase 2 (DUOX2) Messenger Ribonucleic Acid (MRNA) in Two Rat Thyroid Cell Lines. Insulin and Forskolin Regulation of DUOX2 MRNA Levels in FRTL-5 Cells and Porcine Thyrocytes. Endocrinology 2003, 144, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, H.; Refetoff, S. Identification of the Maturation Factor for Dual Oxidase. Evolution of an Eukaryotic Operon Equivalent. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [PubMed]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct Subcellular Localizations of Nox1 and Nox4 in Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Filali, M.; Huss, G.J.; Stanic, B.; Chamseddine, A.; Barna, T.J.; Lamb, F.S. Cytokine Activation of Nuclear Factor Kappa B in Vascular Smooth Muscle Cells Requires Signaling Endosomes Containing Nox1 and ClC-3. Circ. Res. 2007, 101, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Petry, A.; Djordjevic, T.; Weitnauer, M.; Kietzmann, T.; Hess, J.; Gorlach, A. NOX2 and NOX4 Mediate Proliferative Response in Endothelial Cells. Antioxid. Redox Signal. 2006, 8, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Hahn, N.E.; Meischl, C.; Wijnker, P.J.; Musters, R.J.; Fornerod, M.; Janssen, H.W.; Paulus, W.J.; van Rossum, A.C.; Niessen, H.W.; Krijnen, P.A. NOX2, P22phox and P47phox Are Targeted to the Nuclear Pore Complex in Ischemic Cardiomyocytes Colocalizing With Local Reactive Oxygen Species. Cell. Physiol. Biochem. 2011, 27, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH Oxidases and the Endoplasmic Reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Xu, S.; Quan, X.; Nguyen, T.T.; Kong, I.D.; Chung, C.H.; Lee, E.Y.; Cha, S.K.; Park, K.S. Upregulation of Mitochondrial Nox4 Mediates TGF-Beta-Induced Apoptosis in Cultured Mouse Podocytes. Am. J. Physiol. Renal Physiol. 2014, 306, F155–F167. [Google Scholar] [CrossRef] [PubMed]
- Roumenina, L.T.; Rayes, J.; Frimat, M.; Fremeaux-Bacchi, V. Endothelial Cells: Source, Barrier, and Target of Defensive Mediators. Immunol. Rev. 2016, 274, 307–329. [Google Scholar] [CrossRef] [PubMed]
- Ando, J.; Yamamoto, K. Flow Detection and Calcium Signalling in Vascular Endothelial Cells. Cardiovasc. Res. 2013, 99, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W. Endothelium—Role in Regulation of Coagulation and Inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Lenting, P.J.; Christophe, O.D.; Denis, C.V. Von Willebrand Factor Biosynthesis, Secretion, and Clearance: Connecting the Far Ends. Blood 2015, 125, 2019–2028. [Google Scholar] [CrossRef] [PubMed]
- Osborn, L.; Hession, C.; Tizard, R.; Vassallo, C.; Luhowskyj, S.; Chi-Rosso, G.; Lobb, R. Direct Expression Cloning of Vascular Cell Adhesion Molecule 1, a Cytokine-Induced Endothelial Protein That Binds to Lymphocytes. Cell 1989, 59, 1203–1211. [Google Scholar] [CrossRef]
- Renkonen, R. Regulation of Intercellular Adhesion Molecule-1 Expression on Endothelial Cells with Correlation to Lymphocyte-Endothelial Binding. Scand. J. Immunol. 1989, 29, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Golebiewska, E.M.; Poole, A.W. Platelet Secretion: From Haemostasis to Wound Healing and Beyond. Blood Rev. 2015, 29, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Kolluru, G.K.; Fang, K.; Kevil, C.G. Redox Balance Dynamically Regulates Vascular Growth and Remodeling. Semin. Cell Dev. Biol. 2012, 23, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS Signaling and Redox Biology in Endothelial Cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Brandes, R.P. The Nox Family of NADPH Oxidases: Friend or Foe of the Vascular System? Curr. Hypertens. Rep. 2012, 14, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Nakamura, Y. Reactive Oxygen Species and Angiogenesis: NADPH Oxidase as Target for Cancer Therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.; Davel, L.E.; Sacerdote, D.L. Hydrogen Peroxide Is Involved in Lymphocyte Activation Mechanisms to Induce Angiogenesis. Eur. J. Cancer 1997, 33, 676–682. [Google Scholar] [CrossRef]
- Yasuda, M.; Ohzeki, Y.; Shimizu, S.; Naito, S.; Ohtsuru, A.; Yamamoto, T.; Kuroiwa, Y. Stimulation of in Vitro Angiogenesis by Hydrogen Peroxide and the Relation With ETS-1 in Endothelial Cells. Life Sci. 1999, 64, 249–258. [Google Scholar] [CrossRef]
- Chen, W.; Gabel, S.; Steenbergen, C.; Murphy, E. A Redox-Based Mechanism for Cardioprotection Induced by Ischemic Preconditioning in Perfused Rat Heart. Circ. Res. 1995, 77, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Skyschally, A.; Schulz, R.; Gres, P.; Korth, H.G.; Heusch, G. Attenuation of Ischemic Preconditioning in Pigs by Scavenging of Free Oxyradicals With Ascorbic Acid. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H698–H703. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Weihrauch, D.; Kehl, F.; Ludwig, L.M.; LaDisa, J.F., Jr.; Kersten, J.R.; Pagel, P.S.; Warltier, D.C. Mechanism of Preconditioning by Isoflurane in Rabbits: A Direct Role for Reactive Oxygen Species. Anesthesiology 2002, 97, 1485–1490. [Google Scholar] [CrossRef] [PubMed]
- Brakenhielm, E.; Cao, R.; Cao, Y. Suppression of Angiogenesis, Tumor Growth, and Wound Healing by Resveratrol, a Natural Compound in Red Wine and Grapes. FASEB J. 2001, 15, 1798–1800. [Google Scholar] [CrossRef] [PubMed]
- Oak, M.H.; El Bedoui, J.; Schini-Kerth, V.B. Antiangiogenic Properties of Natural Polyphenols from Red Wine and Green Tea. J. Nutr. Biochem. 2005, 16, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M.; Ushio-Fukai, M.; Hilenski, L.; Dikalov, S.I.; Chen, Y.E.; Tojo, T.; Fukai, T.; Fujimoto, M.; Patrushev, N.A.; Wang, N.; et al. IQGAP1, a Novel Vascular Endothelial Growth Factor Receptor Binding Protein, Is Involved in Reactive Oxygen Species—Dependent Endothelial Migration and Proliferation. Circ. Res. 2004, 95, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Harfouche, R.; Malak, N.A.; Brandes, R.P.; Karsan, A.; Irani, K.; Hussain, S.N. Roles of Reactive Oxygen Species in Angiopoietin-1/Tie-2 Receptor Signaling. FASEB J. 2005, 19, 1728–1730. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Kim, K.E.; Koh, G.Y.; Ho, Y.S.; Lee, K.J. Hydrogen Peroxide Produced by Angiopoietin-1 Mediates Angiogenesis. Cancer Res. 2006, 66, 6167–6174. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Kant, S.; Keaney, J.F., Jr. Reactive Oxygen Species in Endothelial Function—From Disease to Adaptation. Circ. J. 2015, 79, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. VEGF Controls Endothelial-Cell Permeability by Promoting the Beta-Arrestin-Dependent Endocytosis of VE-Cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, L.; Moldovan, N.I.; Sohn, R.H.; Parikh, S.A.; Goldschmidt-Clermont, P.J. Redox Changes of Cultured Endothelial Cells and Actin Dynamics. Circ. Res. 2000, 86, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Yamaoka-Tojo, M.; Hilenski, L.; Patrushev, N.A.; Anwar, G.M.; Quinn, M.T.; Ushio-Fukai, M. IQGAP1 Regulates Reactive Oxygen Species-Dependent Endothelial Cell Migration Through Interacting With Nox2. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.F.; Gu, Y.; Xu, Y.C.; Nwariaku, F.E.; Terada, L.S. Vascular Endothelial Growth Factor Causes Translocation of P47phox to Membrane Ruffles Through WAVE1. J. Biol. Chem. 2003, 278, 36830–36840. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Kirber, M.T.; Xiao, H.; Yang, Y.; Keaney, J.F., Jr. Regulation of ROS Signal Transduction by NADPH Oxidase 4 Localization. J. Cell. Biol. 2008, 181, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH Oxidase 4 Promotes Endothelial Angiogenesis Through Endothelial Nitric Oxide Synthase Activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 Is a Protective Reactive Oxygen Species Generating Vascular NADPH Oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Alexander, R.W. Reactive Oxygen Species As Mediators of Angiogenesis Signaling: Role of NAD(P)H Oxidase. Mol. Cell. Biochem. 2004, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K. Novel NAD(P)H Oxidases in the Cardiovascular System. Heart 2004, 90, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Urao, N. Novel Role of NADPH Oxidase in Angiogenesis and Stem/Progenitor Cell Function. Antioxid. Redox Signal. 2009, 11, 2517–2533. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Kapelak, B.; Jopek, A.; Rudzinski, P.; Pillai, R.; Korbut, R.; Channon, K.M. Systemic Regulation of Vascular NAD(P)H Oxidase Activity and Nox Isoform Expression in Human Arteries and Veins. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Van Buul, J.D.; Fernandez-Borja, M.; Anthony, E.C.; Hordijk, P.L. Expression and Localization of NOX2 and NOX4 in Primary Human Endothelial Cells. Antioxid. Redox Signal. 2005, 7, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Compartmentalization of Redox Signaling Through NADPH Oxidase-Derived ROS. Antioxid. Redox Signal. 2009, 11, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, G.; Banfi, B.; Deffert, C.; Fiette, L.; Schappi, M.; Herrmann, F.; Krause, K.H. Decreased Blood Pressure in NOX1-Deficient Mice. FEBS Lett. 2006, 580, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 Is Involved in Angiotensin II-Mediated Hypertension: A Study in Nox1-Deficient Mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, C.E.; Chaubey, S.; Zeng, L.; Yu, B.; Ivetic, A.; Walker, S.J.; Vanhoutte, D.; Heymans, S.; Grieve, D.J.; Cave, A.C.; et al. Endothelial NADPH Oxidase-2 Promotes Interstitial Cardiac Fibrosis and Diastolic Dysfunction Through Proinflammatory Effects and Endothelial-Mesenchymal Transition. J. Am. Coll. Cardiol. 2014, 63, 2734–2741. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Schnelle, M.; Zhang, J.; Murdoch, C.E.; Kossmann, S.; Protti, A.; Santos, C.X.; Sawyer, G.J.; Zhang, X.; Mongue-Din, H.; et al. Distinct Regulatory Effects of Myeloid Cell and Endothelial Cell Nox2 on Blood Pressure. Circulation 2017. [Google Scholar] [CrossRef]
- Montezano, A.C.; Tsiropoulou, S.; Dulak-Lis, M.; Harvey, A.; Camargo, L.L.; Touyz, R.M. Redox Signaling, Nox5 and Vascular Remodeling in Hypertension. Curr. Opin. Nephrol. Hypertens. 2015, 24, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson-Berka, J.L.; Rana, I.; Armani, R.; Agrotis, A. Reactive Oxygen Species, Nox and Angiotensin II in Angiogenesis: Implications for Retinopathy. Clin. Sci. (Lond.) 2013, 124, 597–615. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; et al. Calcium-Dependent NOX5 Nicotinamide Adenine Dinucleotide Phosphate Oxidase Contributes to Vascular Oxidative Stress in Human Coronary Artery Disease. J. Am. Coll. Cardiol. 2008, 52, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Manea, S.A.; Gan, A.M.; Constantin, A.; Fenyo, I.M.; Raicu, M.; Muresian, H.; Simionescu, M. Human Monocytes and Macrophages Express NADPH Oxidase 5; a Potential Source of Reactive Oxygen Species in Atherosclerosis. Biochem. Biophys. Res. Commun. 2015, 461, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Helmcke, I.; Palfi, K.; Krause, K.H.; Busse, R.; Brandes, R.P. Nox1 Mediates Basic Fibroblast Growth Factor-Induced Migration of Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, D.; Kato, M.; Nakayama, J.; Miyagawa, S.; Kamata, T. NADPH Oxidase 1 Plays a Critical Mediating Role in Oncogenic Ras-Induced Vascular Endothelial Growth Factor Expression. Oncogene 2008, 27, 4724–4732. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Urbani, S.; Jemelin, S.; Deffert, C.; Carnesecchi, S.; Basset, O.; Szyndralewiez, C.; Heitz, F.; Page, P.; Montet, X.; Michalik, L.; et al. Targeting Vascular NADPH Oxidase 1 Blocks Tumor Angiogenesis Through a PPARalpha Mediated Mechanism. PLoS ONE 2011, 6, e14665. [Google Scholar] [CrossRef]
- Peshavariya, H.; Dusting, G.J.; Jiang, F.; Halmos, L.R.; Sobey, C.G.; Drummond, G.R.; Selemidis, S. NADPH Oxidase Isoform Selective Regulation of Endothelial Cell Proliferation and Survival. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 380, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Djordjevic, T.; Petry, A.; Hatzelmann, A.; Tenor, H.; Hess, J.; Gorlach, A. Phosphodiesterase 2 Mediates Redox-Sensitive Endothelial Cell Proliferation and Angiogenesis by Thrombin Via Rac1 and NADPH Oxidase 2. Circ. Res. 2009, 104, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, R.W. Role of Gp91phox (Nox2)-Containing NAD(P)H Oxidase in Angiogenesis in Response to Hindlimb Ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of Nox2-Based NADPH Oxidase in Bone Marrow and Progenitor Cell Function Involved in Neovascularization Induced by Hindlimb Ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M.; Tang, Y.; Fukai, T.; Dikalov, S.I.; Ma, Y.; Fujimoto, M.; Quinn, M.T.; Pagano, P.J.; Johnson, C.; Alexander, R.W. Novel Role of Gp91(Phox)-Containing NAD(P)H Oxidase in Vascular Endothelial Growth Factor-Induced Signaling and Angiogenesis. Circ. Res. 2002, 91, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Sabrane, K.; Djordjevic, T.; Hess, J.; Gorlach, A. The HIF1 Target Gene NOX2 Promotes Angiogenesis Through Urotensin-II. J. Cell. Sci. 2012, 125, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, T.G.; Heymes, C.; You, D.; Blanc-Brude, O.; Mees, B.; Waeckel, L.; Duriez, M.; Vilar, J.; Brandes, R.P.; Levy, B.I.; et al. NADPH Oxidase-Derived Overproduction of Reactive Oxygen Species Impairs Postischemic Neovascularization in Mice With Type 1 Diabetes. Am. J. Pathol. 2006, 169, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Haddad, P.; Dussault, S.; Groleau, J.; Turgeon, J.; Maingrette, F.; Rivard, A. Nox2-Derived Reactive Oxygen Species Contribute to Hypercholesterolemia-Induced Inhibition of Neovascularization: Effects on Endothelial Progenitor Cells and Mature Endothelial Cells. Atherosclerosis 2011, 217, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Ellmark, S.H.; Dusting, G.J.; Fui, M.N.; Guzzo-Pernell, N.; Drummond, G.R. The Contribution of Nox4 to NADPH Oxidase Activity in Mouse Vascular Smooth Muscle. Cardiovasc. Res. 2005, 65, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; Peshavariya, H.; Dusting, G.J.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important Role of Nox4 Type NADPH Oxidase in Angiogenic Responses in Human Microvascular Endothelial Cells in Vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef] [PubMed]
- Hakami, N.Y.; Ranjan, A.K.; Hardikar, A.A.; Dusting, G.J.; Peshavariya, H.M. Role of NADPH Oxidase-4 in Human Endothelial Progenitor Cells. Front. Physiol. 2017, 8, 150. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiao, J.; Kuroda, J.; Ago, T.; Sadoshima, J.; Cohen, R.A.; Tong, X. Both Hydrogen Peroxide and Transforming Growth Factor Beta 1 Contribute to Endothelial Nox4 Mediated Angiogenesis in Endothelial Nox4 Transgenic Mouse Lines. Biochim. Biophys. Acta 2014, 1842, 2489–2499. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting Roles of NADPH Oxidase Isoforms in Pressure-Overload Versus Angiotensin II-Induced Cardiac Hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Brewer, A.C.; Schroder, K.; Santos, C.X.; Grieve, D.J.; Wang, M.; Anilkumar, N.; Yu, B.; Dong, X.; Walker, S.J.; et al. NADPH Oxidase-4 Mediates Protection Against Chronic Load-Induced Stress in Mouse Hearts by Enhancing Angiogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 18121–18126. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.; Kruse, C.; Zhang, M.; Schroder, K. Nox4 Supports Proper Capillary Growth in Exercise and Retina Neo-Vascularization. J. Physiol. 2015, 593, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Helfinger, V.; Henke, N.; Harenkamp, S.; Walter, M.; Epah, J.; Penski, C.; Mittelbronn, M.; Schroder, K. The NADPH Oxidase Nox4 Mediates Tumour Angiogenesis. Acta Physiol. (Oxf.) 2016, 216, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HO-1/CO System in Tumor Growth, Angiogenesis and Metabolism—Targeting HO-1 As an Anti-Tumor Therapy. Vascul. Pharmacol. 2015, 74, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.C.; Hamdy, R.C.; Chua, B.H. Upregulation of Vascular Endothelial Growth Factor by H2O2 in Rat Heart Endothelial Cells. Free Radic. Biol. Med. 1998, 25, 891–897. [Google Scholar] [CrossRef]
- Iwakami, S.; Misu, H.; Takeda, T.; Sugimori, M.; Matsugo, S.; Kaneko, S.; Takamura, T. Concentration-Dependent Dual Effects of Hydrogen Peroxide on Insulin Signal Transduction in H4IIEC Hepatocytes. PLoS ONE 2011, 6, e27401. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I.; Gliki, G. Signaling Transduction Mechanisms Mediating Biological Actions of the Vascular Endothelial Growth Factor Family. Cardiovasc. Res. 2001, 49, 568–581. [Google Scholar] [CrossRef]
- Yuan, S.; Kevil, C.G. Nitric Oxide and Hydrogen Sulfide Regulation of Ischemic Vascular Remodeling. Microcirculation 2016, 23, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Prata, C.; Caliceti, C.; Vieceli Dalla, S.F.; Zambonin, L.; Fiorentini, D.; Hakim, G. VEGF-Induced ROS Generation From NAD(P)H Oxidases Protects Human Leukemic Cells From Apoptosis. Int. J. Oncol. 2010, 36, 1581–1589. [Google Scholar] [PubMed]
- Soeda, S.; Shimada, T.; Koyanagi, S.; Yokomatsu, T.; Murano, T.; Shibuya, S.; Shimeno, H. An Attempt to Promote Neo-Vascularization by Employing a Newly Synthesized Inhibitor of Protein Tyrosine Phosphatase. FEBS Lett. 2002, 524, 54–58. [Google Scholar] [CrossRef]
- Corti, F.; Simons, M. Modulation of VEGF Receptor 2 Signaling by Protein Phosphatases. Pharmacol. Res. 2017, 115, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Jiang, W.G.; Ahmed, A.; Boulton, M. Vascular Endothelial Growth Factor-Induced Endothelial Cell Proliferation Is Regulated by Interaction Between VEGFR-2, SH-PTP1 and ENOS. Microvasc. Res. 2006, 71, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Abdelsaid, M.A.; El Remessy, A.B. S-Glutathionylation of LMW-PTP Regulates VEGF-Mediated FAK Activation and Endothelial Cell Migration. J. Cell Sci. 2012, 125, 4751–4760. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-Derived H2O2 Promotes VEGF Signaling in Caveolae/Lipid Rafts and Post-Ischemic Angiogenesis in Mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive Oxygen and NF-KappaB in VEGF-Induced Migration of Human Vascular Smooth Muscle Cells. Biochem. Biophys. Res. Commun. 2001, 285, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, Z.; Jiang, Y.; Hartnett, M.E. Endothelial NADPH Oxidase 4 Mediates Vascular Endothelial Growth Factor Receptor 2-Induced Intravitreal Neovascularization in a Rat Model of Retinopathy of Prematurity. Mol. Vis. 2014, 20, 231–241. [Google Scholar] [PubMed]
- Chen, X.L.; Tummala, P.E.; Olbrych, M.T.; Alexander, R.W.; Medford, R.M. Angiotensin II Induces Monocyte Chemoattractant Protein-1 Gene Expression in Rat Vascular Smooth Muscle Cells. Circ. Res. 1998, 83, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Tummala, P.E.; Chen, X.L.; Sundell, C.L.; Laursen, J.B.; Hammes, C.P.; Alexander, R.W.; Harrison, D.G.; Medford, R.M. Angiotensin II Induces Vascular Cell Adhesion Molecule-1 Expression in Rat Vasculature: A Potential Link Between the Renin-Angiotensin System and Atherosclerosis. Circulation 1999, 100, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Galis, Z.S.; Khatri, J.J. Matrix Metalloproteinases in Vascular Remodeling and Atherogenesis: The Good, the Bad, and the Ugly. Circ. Res. 2002, 90, 251–262. [Google Scholar] [PubMed]
- Alon, T.; Hemo, I.; Itin, A.; Pe’er, J.; Stone, J.; Keshet, E. Vascular Endothelial Growth Factor Acts As a Survival Factor for Newly Formed Retinal Vessels and Has Implications for Retinopathy of Prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Oliner, J.D.; Bready, J.; Nguyen, L.; Estrada, J.; Hurh, E.; Ma, H.; Pretorius, J.; Fanslow, W.; Nork, T.M.; Leedle, R.A.; et al. AMG 386, a Selective Angiopoietin 1/2-Neutralizing Peptibody, Inhibits Angiogenesis in Models of Ocular Neovascular Diseases. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2170–2180. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Willett, K.L.; Aspegren, O.P.; Smith, L.E. Suppression of Retinal Neovascularization by Erythropoietin SiRNA in a Mouse Model of Proliferative Retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.Q.; Xia, X.B.; Xu, H.Z.; Jiang, J. Suppression of Retinal Neovascularization by Small-Interference RNA Targeting Erythropoietin. Ophthalmologica 2009, 223, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Lima e Silva, R.; Shen, J.; Hackett, S.F.; Kachi, S.; Akiyama, H.; Kiuchi, K.; Yokoi, K.; Hatara, M.C.; Lauer, T.; Aslam, S.; et al. The SDF-1/CXCR4 Ligand/Receptor Pair Is an Important Contributor to Several Types of Ocular Neovascularization. FASEB J. 2007, 21, 3219–3230. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive Oxygen Species in the Control of Hypoxia-Inducible Factor-Mediated Gene Expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Calvani, M.; Comito, G.; Giannoni, E.; Chiarugi, P. Time-Dependent Stabilization of Hypoxia Inducible Factor-1alpha by Different Intracellular Sources of Reactive Oxygen Species. PLoS ONE 2012, 7, e38388. [Google Scholar] [CrossRef] [PubMed]
- Goyal, P.; Weissmann, N.; Grimminger, F.; Hegel, C.; Bader, L.; Rose, F.; Fink, L.; Ghofrani, H.A.; Schermuly, R.T.; Schmidt, H.H.; et al. Upregulation of NAD(P)H Oxidase 1 in Hypoxia Activates Hypoxia-Inducible Factor 1 Via Increase in Reactive Oxygen Species. Free Radic. Biol. Med. 2004, 36, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Diebold, I.; Petry, A.; Hess, J.; Gorlach, A. The NADPH Oxidase Subunit NOX4 Is a New Target Gene of the Hypoxia-Inducible Factor-1. Mol. Biol. Cell 2010, 21, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Buckle, A.M.; Cooke, J.P.; Ng, M.K. The Emerging Role of the Thioredoxin System in Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Lee, D.J.; Lee, K.W.; Park, Y.S.; Lee, J.Y.; Lee, S.H.; Koh, Y.J.; Koh, G.Y.; Choi, C.; Yu, D.Y.; et al. Peroxiredoxin II Is an Essential Antioxidant Enzyme That Prevents the Oxidative Inactivation of VEGF Receptor-2 in Vascular Endothelial Cells. Mol. Cell 2011, 44, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.W.; Byzova, T.V. Oxidative Stress in Angiogenesis and Vascular Disease. Blood 2014, 123, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Forstermann, U. Vascular Oxidative Stress, Nitric Oxide and Atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Qin, D.N.; Wang, F.X.; Ren, J.; Li, H.B.; Zhang, M.; Yang, Q.; Miao, Y.W.; Yu, X.J.; Qi, J.; et al. Inhibition of Reactive Oxygen Species in Hypothalamic Paraventricular Nucleus Attenuates the Renin-Angiotensin System and Proinflammatory Cytokines in Hypertension. Toxicol. Appl. Pharmacol. 2014, 276, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Rose, B.A.; Roberts, W.J.; He, F.; Banes-Berceli, A.K. Molecular Characterization of Reactive Oxygen Species in Systemic and Pulmonary Hypertension. Am. J. Hypertens. 2014, 27, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of Reactive Oxygen Species in the Progression of Type 2 Diabetes and Atherosclerosis. Mediat. Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Kurz, S.; Munzel, T.; Tarpey, M.; Freeman, B.A.; Griendling, K.K.; Harrison, D.G. Angiotensin II-Mediated Hypertension in the Rat Increases Vascular Superoxide Production Via Membrane NADH/NADPH Oxidase Activation. Contribution to Alterations of Vasomotor Tone. J. Clin. Investig. 1996, 97, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Landmesser, U.; Cai, H.; Dikalov, S.; McCann, L.; Hwang, J.; Jo, H.; Holland, S.M.; Harrison, D.G. Role of P47(Phox) in Vascular Oxidative Stress and Hypertension Caused by Angiotensin II. Hypertension 2002, 40, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M. Reactive Oxygen Species as Mediators of Calcium Signaling by Angiotensin II: Implications in Vascular Physiology and Pathophysiology. Antioxid. Redox Signal. 2005, 7, 1302–1314. [Google Scholar] [CrossRef] [PubMed]
- Jung, O.; Schreiber, J.G.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. Gp91phox-Containing NADPH Oxidase Mediates Endothelial Dysfunction in Renovascular Hypertension. Circulation 2004, 109, 1795–1801. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.; Clempus, R.; Lassegue, B.; Cheng, G.; McCoy, J.; Dikalov, S.; San Martin, A.; Lyle, A.; Weber, D.S.; Weiss, D.; et al. Nox1 Overexpression Potentiates Angiotensin II-Induced Hypertension and Vascular Smooth Muscle Hypertrophy in Transgenic Mice. Circulation 2005, 112, 2668–2676. [Google Scholar] [CrossRef] [PubMed]
- Bendall, J.K.; Rinze, R.; Adlam, D.; Tatham, A.L.; de Bono, J.; Wilson, N.; Volpi, E.; Channon, K.M. Endothelial Nox2 Overexpression Potentiates Vascular Oxidative Stress and Hemodynamic Response to Angiotensin II: Studies in Endothelial-Targeted Nox2 Transgenic Mice. Circ. Res. 2007, 100, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH Oxidase Enhances Vasodilatation and Reduces Blood Pressure in Vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Kang, J.; Hu, K.; Tang, S.; Zhou, X.; Xu, L.; Li, Y.; Yu, S. The Role of the Nox4-Derived ROS-Mediated RhoA/Rho Kinase Pathway in Rat Hypertension Induced by Chronic Intermittent Hypoxia. Sleep Breath. 2017. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr.; Yang, C.; Zheleznova, N.N.; Staruschenko, A.; Kurth, T.; Rein, L.; Kumar, V.; Sadovnikov, K.; Dayton, A.; Hoffman, M.; et al. Evidence of the Importance of Nox4 in Production of Hypertension in Dahl Salt-Sensitive Rats. Hypertension 2016, 67, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chandrashekar, K.; Lu, Y.; Duan, Y.; Qu, P.; Wei, J.; Juncos, L.A.; Liu, R. Enhanced Expression and Activity of Nox2 and Nox4 in the Macula Densa in ANG II-Induced Hypertensive Mice. Am. J. Physiol. Renal Physiol. 2014, 306, F344–F350. [Google Scholar] [CrossRef] [PubMed]
- Barman, S.A.; Chen, F.; Su, Y.; Dimitropoulou, C.; Wang, Y.; Catravas, J.D.; Han, W.; Orfi, L.; Szantai-Kis, C.; Keri, G.; et al. NADPH Oxidase 4 Is Expressed in Pulmonary Artery Adventitia and Contributes to Hypertensive Vascular Remodeling. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1704–1715. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Pignatelli, P.; Pignata, C.; Plebani, A.; Rossi, P.; Sanguigni, V.; Carnevale, R.; Soresina, A.; Finocchi, A.; Cirillo, E.; et al. Reduced Atherosclerotic Burden in Subjects With Genetically Determined Low Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; San Martin, A.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of Vascular Smooth Muscle NADPH Oxidase 1 (Nox1) Contribution to Injury-Induced Neointimal Formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide Production and Expression of Nox Family Proteins in Human Atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kirk, E.A.; Dinauer, M.C.; Rosen, H.; Chait, A.; Heinecke, J.W.; LeBoeuf, R.C. Impaired Superoxide Production Due to a Deficiency in Phagocyte NADPH Oxidase Fails to Inhibit Atherosclerosis in Mice. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Hsich, E.; Segal, B.H.; Pagano, P.J.; Rey, F.E.; Paigen, B.; Deleonardis, J.; Hoyt, R.F.; Holland, S.M.; Finkel, T. Vascular Effects Following Homozygous Disruption of P47(Phox) : An Essential Component of NADPH Oxidase. Circulation 2000, 101, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Barry-Lane, P.A.; Patterson, C.; van der, M.M.; Hu, Z.; Holland, S.M.; Yeh, E.T.; Runge, M.S. P47phox Is Required for Atherosclerotic Lesion Progression in ApoE(-/-) Mice. J. Clin. Investig. 2001, 108, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.F.; Liu, J.T.; Wang, Y.; Xu, A.; Vanhoutte, P.M. Toll-Like Receptor 4 Mutation Protects Obese Mice Against Endothelial Dysfunction by Decreasing NADPH Oxidase Isoforms 1 and 4. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Ling, D.; Tu, L.; van Wijngaarden, P.; Dusting, G.J.; Liu, G.S. Gene Therapy for Diabetic Retinopathy: Are We Ready to Make the Leap From Bench to Bedside? Pharmacol. Ther. 2017, 173, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Fassina, G.; Morini, M.; Aluigi, M.G.; Masiello, L.; Fontanini, G.; D’Agostini, F.; De Flora, S.; Noonan, D.M.; Albini, A. N-Acetylcysteine Inhibits Endothelial Cell Invasion and Angiogenesis. Lab. Investig. 1999, 79, 1151–1159. [Google Scholar] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting Cancer Cells by ROS-Mediated Mechanisms: A Radical Therapeutic Approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Myung, S.K.; Ju, W.; Cho, B.; Oh, S.W.; Park, S.M.; Koo, B.K.; Park, B.J. Efficacy of Vitamin and Antioxidant Supplements in Prevention of Cardiovascular Disease: Systematic Review and Meta-Analysis of Randomised Controlled Trials. BMJ 2013, 346, f10. [Google Scholar] [CrossRef] [PubMed]
- Rodiño-Janeiro, B.K.; Paradela-Dobarro, B.; Castiñeiras-Landeira, M.I.; Raposeiras-Roubín, S.; González-Juanatey, J.R.; Alvarez, E. Current Status of NADPH Oxidase Research in Cardiovascular Pharmacology. Vasc. Health Risk Manag. 2013, 9, 401–428. [Google Scholar] [PubMed]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin Is Not an Inhibitor of Vascular NADPH Oxidases but an Antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Sharifpanah, F.; De Silva, S.; Bekhite, M.M.; Hurtado-Oliveros, J.; Preissner, K.T.; Wartenberg, M.; Sauer, H. Stimulation of Vasculogenesis and Leukopoiesis of Embryonic Stem Cells by Extracellular Transfer RNA and Ribosomal RNA. Free Radic. Biol. Med. 2015, 89, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Pi, X.; Xie, L.; Portbury, A.L.; Kumar, S.; Lockyer, P.; Li, X.; Patterson, C. NADPH Oxidase-Generated Reactive Oxygen Species Are Required for Stromal Cell-Derived Factor-1alpha-Stimulated Angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Tang, Y.; Cai, X.; Peng, X.; Liu, X.; Zhang, L.; Xiang, Y.; Wang, D.; Wang, X.; Pan, T. Celastrol Inhibits Vasculogenesis by Suppressing the VEGF-Induced Functional Activity of Bone Marrow-Derived Endothelial Progenitor Cells. Biochem. Biophys. Res. Commun. 2012, 423, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Zhu, L.; Zhang, Y.; Zhang, J.; Yao, L.; Liang, D.; Wang, L. Celastrol Nanomicelles Attenuate Cytokine Secretion in Macrophages and Inhibit Macrophage-Induced Corneal Neovascularization in Rats. Int. J. Nanomed. 2016, 11, 6135–6148. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Inhibitor | Main Target | Other Targets | Side Effects | Reports in Angiogenesis |
|---|---|---|---|---|
| GKT136901 | Nox1, Nox4 and Nox5 | Nox2 | Peroxynitrite scavenger | Inhibition of tumor angiogenesis in mouse models [135] |
| GKT137831 | Nox1, Nox4 and Nox5 | Nox2 | None tested | |
| ML171 | Nox1 | Nox2, Nox3, Nox4 and XO | Inhibitor of serotonin and adrenergic receptors | Inhibition of vasculogenesis in embryoid bodies [211] |
| VAS2870 | Nox2 | Nox4 and Nox5 | Thioalkylate RyR1 and GSH | Inhibition of vasculogenesis in embryoid bodies [211] |
| VAS3947 | Nox1, Nox2 and Nox4 | Nox4 and Nox5 | ||
| S17834 | Nox * | AMPK activation | ||
| Fulvene-5 | Nox2 and Nox4 | None tested | None tested | |
| Triphenylmethane derivates | Nox2 and Nox4 | None tested | None tested | |
| Ebselen | Nox1, Nox2 and Nox5 | Peroxynitrite scavenger in vitro, and eNOS inhibitor | Inhibits in vitro ECs migration induced by SCDF-1a [212] | |
| Celastrol | Nox1 and Nox2 | Nox4 and Nox5 | Inhibitor of topoisomerase II and proteasome | Inhibition of tumor VEGF-induced BM-EPC- supported vasculogenesis in vitro [213] Inhibition of macrophage-induced corneal neovascularization in rats [214] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prieto-Bermejo, R.; Hernández-Hernández, A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants 2017, 6, 32. https://doi.org/10.3390/antiox6020032
Prieto-Bermejo R, Hernández-Hernández A. The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants. 2017; 6(2):32. https://doi.org/10.3390/antiox6020032
Chicago/Turabian StylePrieto-Bermejo, Rodrigo, and Angel Hernández-Hernández. 2017. "The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis" Antioxidants 6, no. 2: 32. https://doi.org/10.3390/antiox6020032
APA StylePrieto-Bermejo, R., & Hernández-Hernández, A. (2017). The Importance of NADPH Oxidases and Redox Signaling in Angiogenesis. Antioxidants, 6(2), 32. https://doi.org/10.3390/antiox6020032