Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins?


 ,
,  and
and 
Abstract
:1. The Biological Significance of Vitamin E
2. Absorption and Distribution of Vitamin E
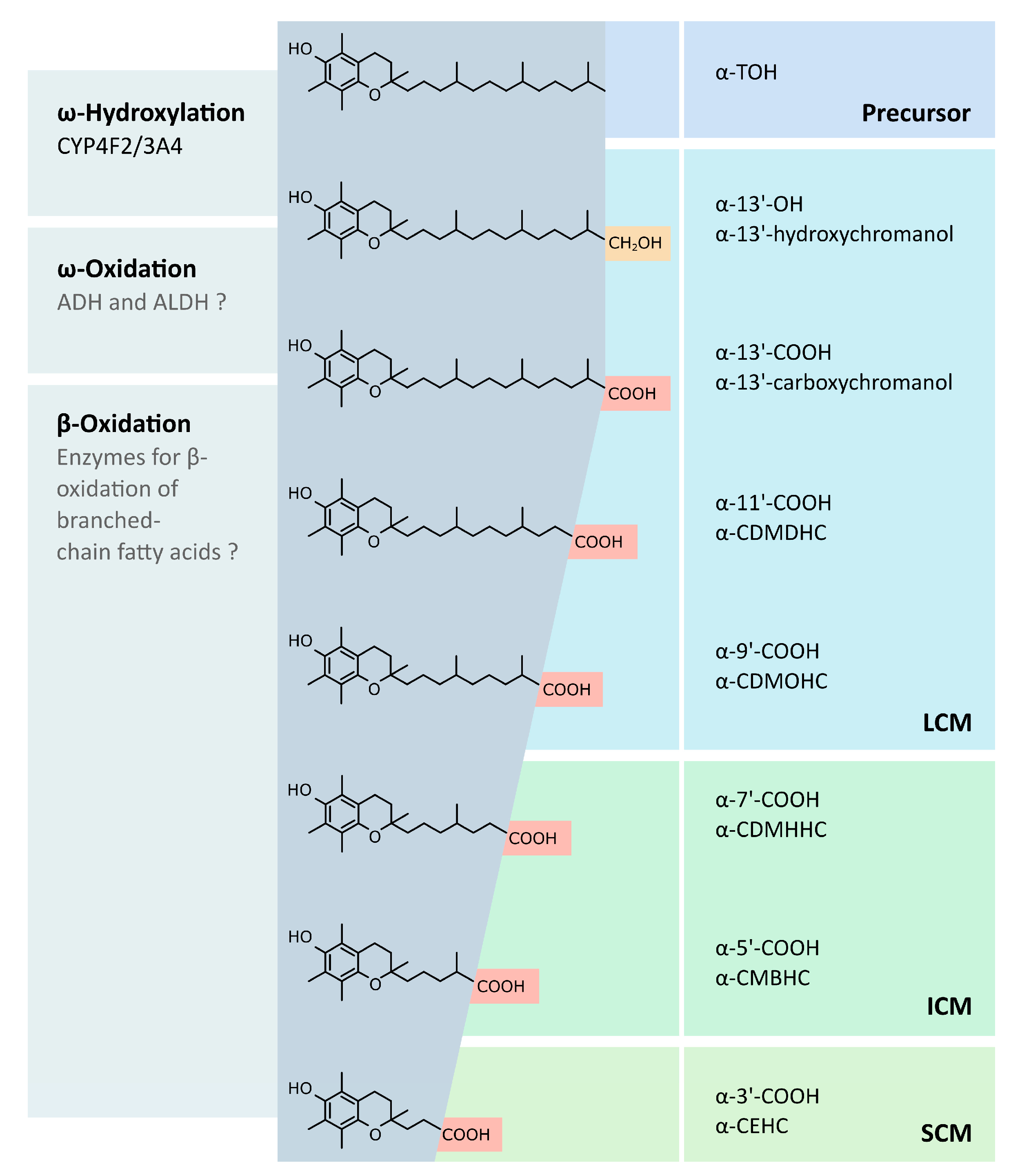
3. Metabolism of Vitamin E
- (i).
- Under physiological conditions, retinyl esters (in the intestinal lumen) and carotenoids (in enterocytes) are converted into retinol before or during their intestinal absorption, respectively. Inside the enterocytes, retinol is re-esterified by lecithin-retinol acyl transferase or acyl-CoA:retinol-acyltransferase and packed into chylomicrons for transport. The retinyl esters are transferred to the liver and stored in hepatic parenchymal and non-parenchymal cells. Vitamin A is mobilized from liver stores by the retinol-binding protein, a specific transporter allowing the transport of retinol in blood circulation [50]. These results suggest that vitamin A has an active (retinol) and a storage form (retinyl ester). In addition, the oxidation of retinol leads to the formation of retinal, another active form of vitamin A, which is primarily bound to opsins in the photoreceptors of the retina [51]. More current research indicates that all-trans retinoic acid (ATRA), 9-cis-RA, and all-trans-4-oxo-RA are the vitamin A metabolites with the highest biological activity. These active vitamin A metabolites serve as ligands for nuclear receptors, called retinoic acid receptors (RARs) [52] and retinoid receptors (RXRs) [53], which act as ligand-activated transcription factors controlling the expression of their respective target genes. Therefore, hepatic retinol is transferred to extrahepatic tissues and metabolized to retinoic acid by different enzymatic systems. Lampen and co-workers found that ATRA is also formed in the small intestine via direct oxidation of vitamin A. Based on this result, they hypothesized that biologically active retinoids are formed in the gastrointestinal tract and act as retinoid-receptor ligands controlling various processes in the intestinal mucosa via RAR [53].
- (ii).
- The human metabolism of vitamin D is primarily located in liver and kidney. Metabolism of vitamin D2 and D3 starts with the formation of 25-OHD, the major circulating vitamin D metabolite, by vitamin D-25 hydroxylase. Afterwards, 25-OHD is transferred to the kidney and further catabolized by 25-OHD-1α-hydroxylase to 1,25-dihydroxyvitamin D2/3. These molecules serve as ligands for the vitamin D receptor (VDR), a transcription factor expressed in various tissues. Vitamin D receptor binds to specific regions in the promoter regions of genes, the so-called vitamin D responsive elements, thus controlling the expression of respective target genes. Therefore, 1,25-dihydroxyvitamin D is the active metabolic form of vitamin D [54,55].
- (iii).
- Phylloquinone (vitamin K1) and menaquinone (vitamin K2) are summarized by the term vitamin K. Phylloquinone is synthesized in plants, while menaquinone is derived from animal and bacterial origins [30,56]. Both compounds share a 2-methyl-1,4-naphthoquinone structure, called menadione, and a side chain at the 3′-position. The side chain of phylloquinone is composed of three isopentyl units and one isopentenyl unit, while the side chain of menaquinone contains a variable number of only isopentenyl units (2–13) [30]. The metabolism of vitamin K is localized in the liver and has not been studied in detail so far [57]. Nevertheless, the metabolic pathway of phylloquinone and menaquinone degradation likely follows that of vitamin E. Hence, the degradation starts with an initial ω-oxidation, which is mediated by CYP. While the ω-oxidation of vitamin E is catalyzed primarily by CYP4F2, CYP3A4 has been described as the possible mediator for the ω-oxidation of vitamin K. Next, the following degradation of the side chain of vitamin K occurs via β-oxidation [30,56,58]. A 5-carbon carboxylic acid metabolite termed K acid 2 has been identified as the end-product of either phylloquinone or menaquinone metabolism and is excreted via urine and bile [30,58]. In addition to their metabolic degradation, it has been suggested that phylloquinones could also be converted to menaquinones [59,60]. For this, phylloquinone is likely transformed to the intermediate menadione by removing its side chain, which is subsequently replaced by a newly synthesized isopentenyl side chain to form menaquinone [30]. While menaquinone is considered as the physiologically active form of vitamin K in humans [56], almost nothing is known about a possible biological activity of the vitamin K metabolites. Further studies are needed to unravel whether vitamin K must be included into the general concept of a metabolic pre-activation of lipid-soluble vitamins.
In Vivo Verification of Systemic LCM Availability
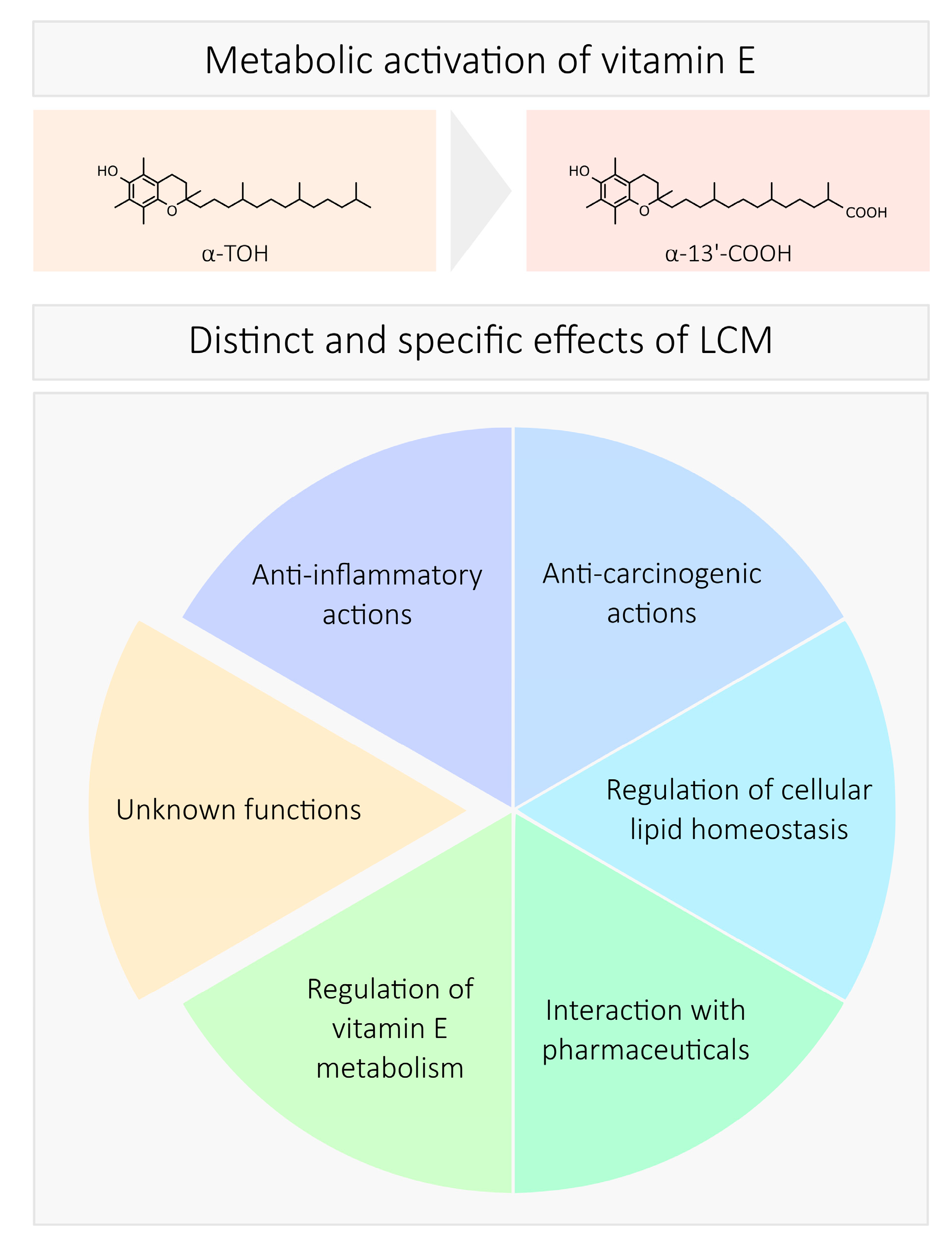
4. Biological Activity
4.1. Anti-Inflammatory Actions
4.2. Cancerogenesis and Chemoprevention
4.3. Cellular Lipid Homeostasis
4.4. Interaction with Pharmaceuticals
4.5. Regulation of LCM Formation
5. Structure-Specific Effects
6. Receptors of Vitamin Metabolites
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Horn, M.; Gunn, P.; van Emon, M.; Lemenager, R.; Burgess, J.; Pyatt, N.A.; Lake, S.L. Effects of natural (RRR alpha-tocopherol acetate) or synthetic (all-rac-alpha-tocopherol acetate) vitamin E supplementation on reproductive efficiency in beef cows. J. Anim. Sci. 2010, 88, 3121–3127. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.; Schubert, M.; Schmölz, L.; Birringer, M.; Wallert, M.; Lorkowski, S. Garcinoic Acid: A Promising Bioactive Natural Product for Better Understanding the Physiological Functions of Tocopherol Metabolites. Stud. Nat. Prod. Chem. 2016, 51, 435–481. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E.; et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Dysken, M.W.; Guarino, P.D.; Vertrees, J.E.; Asthana, S.; Sano, M.; Llorente, M.; Pallaki, M.; Love, S.; Schellenberg, G.D.; McCarten, J.R.; et al. Vitamin E and memantine in Alzheimer’s disease: Clinical trial methods and baseline data. Alzheimer Dement. J. Alzheimer Assoc. 2014, 10, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Jishage, K.; Arita, M.; Igarashi, K.; Iwata, T.; Watanabe, M.; Ogawa, M.; Ueda, O.; Kamada, N.; Inoue, K.; Arai, H.; et al. Alpha-tocopherol transfer protein is important for the normal development of placental labyrinthine trophoblasts in mice. J. Biol. Chem. 2001, 276, 1669–1672. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Yoshida, Y.; Ishida, N.; Hagihara, Y.; Iwahashi, H.; Tamai, H.; Niki, E. α-Tocopherol suppresses lipid peroxidation and behavioral and cognitive impairments in the Ts65Dn mouse model of Down syndrome. Free Radic. Biol. Med. 2011, 50, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Czeizel, A.E.; Dudás, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Chandler, A.L.; Hobbs, C.A.; Mosley, B.S.; Berry, R.J.; Canfield, M.A.; Qi, Y.P.; Siega-Riz, A.M.; Shaw, G.M. Neural tube defects and maternal intake of micronutrients related to one-carbon metabolism or antioxidant activity. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med. 2009, 46, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Wallert, M.; Schmölz, L.; Galli, F.; Birringer, M.; Lorkowski, S. Regulatory metabolites of vitamin E and their putative relevance for atherogenesis. Redox Biol. 2014, 2, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q. Natural forms of vitamin E: Metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic. Biol. Med. 2014, 72, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Rigotti, A. Absorption, transport, and tissue delivery of vitamin E. Mol. Asp. Med. 2007, 28, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Bjørneboe, A.; Bjørneboe, G.E.; Drevon, C.A. Absorption, transport and distribution of vitamin E. J. Nutr. 1990, 120, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Borel, P.; Pasquier, B.; Armand, M.; Tyssandier, V.; Grolier, P.; Alexandre-Gouabau, M.C.; Andre, M.; Senft, M.; Peyrot, J.; Jaussan, V.; et al. Processing of vitamin A and E in the human gastrointestinal tract. Am. J. Physiol.-Gastrointest. Liver Physiol. 2001, 280, G95–G103. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E. Intestinal absorption of vitamin D: From the meal to the enterocyte. Food Funct. 2015, 6, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Bieri, J.G.; Wu, A.L.; Tolliver, T.J. Reduced intestinal absorption of vitamin E by low dietary levels of retinoic acid in rats. J. Nutr. 1981, 111, 458–467. [Google Scholar] [PubMed]
- Richelle, M.; Enslen, M.; Hager, C.; Groux, M.; Tavazzi, I.; Godin, J.-P.; Berger, A.; Métairon, S.; Quaile, S.; Piguet-Welsch, C.; et al. Both free and esterified plant sterols reduce cholesterol absorption and the bioavailability of beta-carotene and alpha-tocopherol in normocholesterolemic humans. Am. J. Clin. Nutr. 2004, 80, 171–177. [Google Scholar] [PubMed]
- Doi, K.; Matsuura, M.; Kawara, A.; Tanaka, T.; Baba, S. Influence of dietary fiber (konjac mannan) on absorption of vitamin B12 and vitamin E. Tohoku J. Exp. Med. 1983, 141, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Ingold, K.U.; Foster, D.O.; Cheng, S.C.; Webb, A.; Hughes, L.; Lusztyk, E. Comparison of free alpha-tocopherol and alpha-tocopheryl acetate as sources of vitamin E in rats and humans. Lipids 1988, 23, 834–840. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Li, L.; van Bennekum, A.M.; Potter, S.H.; Harrison, E.H.; Blaner, W.S.; Breslow, J.L.; Fisher, E.A. Intestinal absorption of dietary cholesteryl ester is decreased but retinyl ester absorption is normal in carboxyl ester lipase knockout mice. Biochemistry 1999, 38, 4143–4149. [Google Scholar] [CrossRef] [PubMed]
- Maislos, M.; Shany, S. Bile salt deficiency and the absorption of vitamin D metabolites. In vivo study in the rat. Isr. J. Med. Sci. 1987, 23, 1114–1117. [Google Scholar] [PubMed]
- Reboul, E.; Klein, A.; Bietrix, F.; Gleize, B.; Malezet-Desmoulins, C.; Schneider, M.; Margotat, A.; Lagrost, L.; Collet, X.; Borel, P. Scavenger receptor class B type I (SR-BI) is involved in vitamin E transport across the enterocyte. J. Biol. Chem. 2006, 281, 4739–4745. [Google Scholar] [CrossRef] [PubMed]
- Hacquebard, M.; Carpentier, Y.A. Vitamin E: Absorption, plasma transport and cell uptake. Curr. Opin. Clin. Nutr. Metab. Care 2005, 8, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Yamanashi, Y.; Takada, T.; Kurauchi, R.; Tanaka, Y.; Komine, T.; Suzuki, H. Transporters for the Intestinal Absorption of Cholesterol, Vitamin E, and Vitamin K. J. Atheroscler. Thromb. 2017, 24, 347–359. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E. Absorption of vitamin A and carotenoids by the enterocyte: Focus on transport proteins. Nutrients 2013, 5, 3563–3581. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Burton, G.W.; Ingold, K.U.; Kayden, H.J. RRR- and SRR-alpha-tocopherols are secreted without discrimination in human chylomicrons, but RRR-alpha-tocopherol is preferentially secreted in very low density lipoproteins. J. Lipid Res. 1990, 31, 675–685. [Google Scholar] [PubMed]
- Traber, M.G.; Burton, G.W.; Hughes, L.; Ingold, K.U.; Hidaka, H.; Malloy, M.; Kane, J.; Hyams, J.; Kayden, H.J. Discrimination between forms of vitamin E by humans with and without genetic abnormalities of lipoprotein metabolism. J. Lipid Res. 1992, 33, 1171–1182. [Google Scholar] [PubMed]
- Shearer, M.J.; Newman, P. Metabolism and cell biology of vitamin K. Thromb. Haemost. 2008. [Google Scholar] [CrossRef]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [PubMed]
- Kiyose, C.; Muramatsu, R.; Kameyama, Y.; Ueda, T.; Igarashi, O. Biodiscrimination of alpha-tocopherol stereoisomers in humans after oral administration. Am. J. Clin. Nutr. 1997, 65, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Weiser, H.; Riss, G.; Kormann, A.W. Selective biodiscrimination of alpha-tocopherol stereoisomers. Similar enrichment of all 2R forms in rat tissues after oral all-rac-alpha-tocopheryl acetate. Ann. N. Y. Acad. Sci. 1992, 669, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Kayden, H.J. Alpha-tocopherol as compared with gamma-tocopherol is preferentially secreted in human lipoproteins. Ann. N. Y. Acad. Sci. 1989, 570, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Arita, M.; Nomura, K.; Arai, H.; Inoue, K. alpha-tocopherol transfer protein stimulates the secretion of alpha-tocopherol from a cultured liver cell line through a brefeldin A-insensitive pathway. Proc. Natl. Acad. Sci. USA 1997, 94, 12437–12441. [Google Scholar] [CrossRef] [PubMed]
- Oram, J.F.; Vaughan, A.M.; Stocker, R. ATP-binding cassette transporter A1 mediates cellular secretion of alpha-tocopherol. J. Biol. Chem. 2001, 276, 39898–39902. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.J.; Shields, J.; Horton, R.A.; Brown, M.K.; Reed, D.J. Biliary secretion of alpha-tocopherol and the role of the mdr2 P-glycoprotein in rats and mice. Arch. Biochem. Biophys. 1998, 350, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Lemaire-Ewing, S.; Desrumaux, C.; Néel, D.; Lagrost, L. Vitamin E transport, membrane incorporation and cell metabolism: Is alpha-tocopherol in lipid rafts an oar in the lifeboat? Mol. Nutr. Food Res. 2010, 54, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Schmölz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of vitamin E metabolism. World J. Biol. Chem. 2016, 7, 14–43. [Google Scholar] [CrossRef] [PubMed]
- Abe, C.; Uchida, T.; Ohta, M.; Ichikawa, T.; Yamashita, K.; Ikeda, S. Cytochrome P450-dependent metabolism of vitamin E isoforms is a critical determinant of their tissue concentrations in rats. Lipids 2007, 42, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Bardowell, S.A.; Ding, X.; Parker, R.S. Disruption of P450-mediated vitamin E hydroxylase activities alters vitamin E status in tocopherol supplemented mice and reveals extra-hepatic vitamin E metabolism. J. Lipid Res. 2012, 53, 2667–2676. [Google Scholar] [CrossRef] [PubMed]
- Grebenstein, N.; Schumacher, M.; Graeve, L.; Frank, J. α-Tocopherol transfer protein is not required for the discrimination against γ-tocopherol in vivo but protects it from side-chain degradation in vitro. Mol. Nutr. Food Res. 2014, 58, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Chiku, S.; Hamamura, K.; Nakamura, T. Novel urinary metabolite of d-delta-tocopherol in rats. J. Lipid Res. 1984, 25, 40–48. [Google Scholar] [PubMed]
- Swanson, J.E.; Ben, R.N.; Burton, G.W.; Parker, R.S. Urinary excretion of 2,7,8-trimethyl-2-(beta-carboxyethyl)-6-hydroxychroman is a major route of elimination of gamma-tocopherol in humans. J. Lipid Res. 1999, 40, 665–671. [Google Scholar] [PubMed]
- Sontag, T.J.; Parker, R.S. Cytochrome P450 omega-hydroxylase pathway of tocopherol catabolism. Novel mechanism of regulation of vitamin E status. J. Biol. Chem. 2002, 277, 25290–25296. [Google Scholar] [CrossRef] [PubMed]
- Birringer, M.; Pfluger, P.; Kluth, D.; Landes, N.; Brigelius-Flohé, R. Identities and differences in the metabolism of tocotrienols and tocopherols in HepG2 cells. J. Nutr. 2002, 132, 3113–3118. [Google Scholar] [PubMed]
- Parker, R.S.; Sontag, T.J.; Swanson, J.E. Cytochrome P4503A-dependent metabolism of tocopherols and inhibition by sesamin. Biochem. Biophys. Res. Commun. 2000, 277, 531–534. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Slanař, O.; Krausz, K.W.; Kang, D.W.; Patterson, A.D.; Kim, J.-H.; Luecke, H.; Gonzalez, F.J.; Idle, J.R. Novel metabolites and roles for α-tocopherol in humans and mice discovered by mass spectrometry-based metabolomics. Am. J. Clin. Nutr. 2012, 96, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lee, M.-J.; Cheung, C.; Ju, J.-H.; Chen, Y.-K.; Liu, B.; Hu, L.-Q.; Yang, C.S. Analysis of multiple metabolites of tocopherols and tocotrienols in mice and humans. J. Agric. Food Chem. 2010, 58, 4844–4852. [Google Scholar] [CrossRef] [PubMed]
- Goodman, D.S. Overview of current knowledge of metabolism of vitamin A and carotenoids. J. Natl. Cancer Inst. 1984, 73, 1375–1379. [Google Scholar] [PubMed]
- Zhong, M.; Kawaguchi, R.; Kassai, M.; Sun, H. Retina, retinol, retinal and the natural history of vitamin A as a light sensor. Nutrients 2012, 4, 2069–2096. [Google Scholar] [CrossRef] [PubMed]
- Petkovich, M.; Brand, N.J.; Krust, A.; Chambon, P. A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 1987, 330, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Lampen, A.; Meyer, S.; Arnhold, T.; Nau, H. Metabolism of vitamin A and its active metabolite all-trans-retinoic acid in small intestinal enterocytes. J. Pharmacol. Exp. Ther. 2000, 295, 979–985. [Google Scholar] [PubMed]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Urrutia-Pereira, M.; Solé, D. Vitamin D deficiency in pregnancy and its impact on the fetus, the newborn and in childhood. Rev. Paul. Pediatr. 2015, 33, 104–113. [Google Scholar] [CrossRef]
- Landes, N.; Birringer, M.; Brigelius-Flohé, R. Homologous metabolic and gene activating routes for vitamins E and K. Mol. Asp. Med. 2003, 24, 337–344. [Google Scholar] [CrossRef]
- Hodges, S.J.; Pitsillides, A.A.; Ytrebø, L.M.; Soper, R. Anti-inflammatory actions of vitamin K. In Vitamin K2—Vital for Health and Wellbeing; Gordeladze, J.O., Ed.; InTech: Rijeka, Croatia, 2017. [Google Scholar]
- Traber, M.G. Vitamin E and K interactions—A 50-year-old problem. Nutr. Rev. 2008, 66, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, R.; Komai, M.; Kojima, K.; Furukawa, Y.; Kimura, S. Menaquinone-4 accumulation in various tissues after an oral administration of phylloquinone in Wistar rats. J. Nutr. Sci. Vitaminol. 1997, 43, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Okano, T.; Shimomura, Y.; Yamane, M.; Suhara, Y.; Kamao, M.; Sugiura, M.; Nakagawa, K. Conversion of phylloquinone (Vitamin K1) into menaquinone-4 (Vitamin K2) in mice: Two possible routes for menaquinone-4 accumulation in cerebra of mice. J. Biol. Chem. 2008, 283, 11270–11279. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Kelly, F.J.; Salonen, J.T.; Neuzil, J.; Zingg, J.-M.; Azzi, A. The European perspective on vitamin E: Current knowledge and future research. Am. J. Clin. Nutr. 2002, 76, 703–716. [Google Scholar] [PubMed]
- Azzi, A.; Ricciarelli, R.; Zingg, J.-M. Non-antioxidant molecular functions of α-tocopherol (vitamin E). FEBS Lett. 2002, 519, 8–10. [Google Scholar] [CrossRef]
- Wallert, M.; Mosig, S.; Rennert, K.; Funke, H.; Ristow, M.; Pellegrino, R.M.; Cruciani, G.; Galli, F.; Lorkowski, S.; Birringer, M. Long-chain metabolites of α-tocopherol occur in human serum and inhibit macrophage foam cell formation in vitro. Free Radic. Biol. Med. 2014, 68, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Schmölz, L.; Wallert, M.; Rozzino, N.; Cignarella, A.; Galli, F.; Glei, M.; Werz, O.; Koeberle, A.; Birringer, M.; Lorkowski, S. Structure-Function Relationship Studies in vitro Reveal Distinct and Specific Effects of Long-Chain Metabolites of Vitamin E. Mol. Nutr. Food Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Torquato, P.; Ripa, O.; Giusepponi, D.; Galarini, R.; Bartolini, D.; Wallert, M.; Pellegrino, R.; Cruciani, G.; Lorkowski, S.; Birringer, M.; et al. Analytical strategies to assess the functional metabolome of vitamin E. J. Pharm. Biomed. Anal. 2016, 124, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Giusepponi, D.; Torquato, P.; Bartolini, D.; Piroddi, M.; Birringer, M.; Lorkowski, S.; Libetta, C.; Cruciani, G.; Moretti, S.; Saluti, G.; et al. Determination of tocopherols and their metabolites by liquid-chromatography coupled with tandem mass spectrometry in human plasma and serum. Talanta 2017, 170, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Munteanu, A.; Zingg, J.-M.; Azzi, A. Anti-atherosclerotic effects of vitamin E—Myth or reality? J. Cell. Mol. Med. 2004, 8, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Traber, M.G.; Atkinson, J. Vitamin E, antioxidant and nothing more. Free Radic. Biol. Med. 2007, 43, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Davies, K.J.A. Is vitamin E an antioxidant, a regulator of signal transduction and gene expression, or a ‘junk’ food? Comments on the two accompanying papers: “Molecular mechanism of alpha-tocopherol action” by A. Azzi and “Vitamin E, antioxidant and nothing more” by M. Traber and J. Atkinson. Free Radic. Biol. Med. 2007, 43, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yin, X.; Lill, M.A.; Danielson, M.L.; Freiser, H.; Huang, J. Long-chain carboxychromanols, metabolites of vitamin E, are potent inhibitors of cyclooxygenases. Proc. Natl. Acad. Sci. USA 2008, 105, 20464–20469. [Google Scholar] [CrossRef] [PubMed]
- Ciffolilli, S.; Wallert, M.; Bartolini, D.; Krauth, V.; Werz, O.; Piroddi, M.; Sebastiani, B.; Torquato, P.; Lorkowski, S.; Birringer, M.; et al. Human serum determination and in vitro anti-inflammatory activity of the vitamin E metabolite α-(13′-hydroxy)-6-hydroxychroman. Free Radic. Biol. Med. 2015, 89, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Park, N.-Y.; Rostgaard-Hansen, A.L.; Huang, J.; Jiang, Q. Vitamin E metabolite 13′-carboxychromanols inhibit pro-inflammatory enzymes, induce apoptosis and autophagy in human cancer cells by modulating sphingolipids and suppress colon tumor development in mice. Free Radic. Biol. Med. 2016, 95, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yin, X.; Jiang, Q. Natural forms of vitamin E and 13′-carboxychromanol, a long-chain vitamin E metabolite, inhibit leukotriene generation from stimulated neutrophils by blocking calcium influx and suppressing 5-lipoxygenase activity, respectively. J. Immunol. 2011, 186, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Wallert, M.; Schmolz, L.; Koeberle, A.; Krauth, V.; Glei, M.; Galli, F.; Werz, O.; Birringer, M.; Lorkowski, S. Alpha-Tocopherol long-chain metabolite alpha-13′-COOH affects the inflammatory response of lipopolysaccharide-activated murine RAW264.7 macrophages. Mol. Nutr. Food Res. 2015, 59, 1524–1534. [Google Scholar] [CrossRef] [PubMed]
- Schmölz, L.; Wallert, M.; Lorkowski, S. Optimized incubation regime for nitric oxide measurements in murine macrophages using the Griess assay. J. Immunol. Methods 2017. [Google Scholar] [CrossRef] [PubMed]
- Mazzini, F.; Betti, M.; Netscher, T.; Galli, F.; Salvadori, P. Configuration of the vitamin E analogue garcinoic acid extracted from Garcinia Kola seeds. Chirality 2009, 21, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Birringer, M.; Lington, D.; Vertuani, S.; Manfredini, S.; Scharlau, D.; Glei, M.; Ristow, M. Proapoptotic effects of long-chain vitamin E metabolites in HepG2 cells are mediated by oxidative stress. Free Radic. Biol. Med. 2010, 49, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Podszun, M.C.; Jakobi, M.; Birringer, M.; Weiss, J.; Frank, J. The long chain α-tocopherol metabolite α-13′-COOH and γ-tocotrienol induce P-glycoprotein expression and activity by activation of the pregnane X receptor in the intestinal cell line LS 180. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Torquato, P.; Bartolini, D.; Giusepponi, D.; Saluti, G.; Russo, A.; Barola, C.; Birringer, M.; Galarini, R.; Galli, F. a-13′-OH is the main product of a-tocopherol metabolism and influences CYP4F2 and PPARg: Gene expression in HepG2 human hepatocarcinoma cells. Free Radic. Biol. Med. 2016, 96, S19–S20. [Google Scholar] [CrossRef]
- Reddi, K.; Henderson, B.; Meghji, S.; Wilson, M.; Poole, S.; Hopper, C.; Harris, M.; Hodges, S.J. Interleukin 6 production by lipopolysaccharide-stimulated human fibroblasts is potently inhibited by naphthoquinone (vitamin K) compounds. Cytokine 1995, 7, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Soper, R.J.; Oguz, C.; Emery, R.; Pitsillides, A.A.; Hodges, S.J. Vitamin K catabolite inhibition of ovariectomy-induced bone loss: Structure-activity relationship considerations. Mol. Nutr. Food Res. 2014, 58, 1658–1666. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Shimizu, A.; Takeda, N.; Oguchi, K.; Katsurai, T.; Shirakawa, H.; Komai, M.; Kagechika, H. Systematic synthesis and anti-inflammatory activity of ω-carboxylated menaquinone derivatives—Investigations on identified and putative vitamin K2 metabolites. Bioorg. Med. Chem. 2015, 23, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Harrington, D.J.; Soper, R.; Edwards, C.; Savidge, G.F.; Hodges, S.J.; Shearer, M.J. Determination of the urinary aglycone metabolites of vitamin K by HPLC with redox-mode electrochemical detection. J. Lipid Res. 2005, 46, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Conte, C.; Floridi, A.; Aisa, C.; Piroddi, M.; Floridi, A.; Galli, F. Gamma-tocotrienol metabolism and antiproliferative effect in prostate cancer cells. Ann. N. Y. Acad. Sci. 2004, 1031, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Stabile, A.M.; Betti, M.; Conte, C.; Pistilli, A.; Rende, M.; Floridi, A.; Azzi, A. The effect of alpha- and gamma-tocopherol and their carboxyethyl hydroxychroman metabolites on prostate cancer cell proliferation. Arch. Biochem. Biophys. 2004, 423, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Kennel, K.A.; Drake, M.T. Vitamin D in the cancer patient. Curr. Opin. Support. Palliat. Care 2013, 7, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Vanoirbeek, E.; Krishnan, A.; Eelen, G.; Verlinden, L.; Bouillon, R.; Feldman, D.; Verstuyf, A. The anti-cancer and anti-inflammatory actions of 1,25(OH)2D3. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-H.; Gudas, L.J. Retinoids, retinoic acid receptors, and cancer. Ann. Rev. Pathol. 2011, 6, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-Y.; Fujii, S.; Shimizu, A.; Kagechika, H.; Kojima, S. Carboxylic Derivatives of Vitamin K2 Inhibit Hepatocellular Carcinoma Cell Growth through Caspase/Transglutaminase-Related Signaling Pathways. J. Nutr. Sci. Vitaminol. 2015, 61, 285–290. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.R. Vitamin A and Retinoids as Mitochondrial Toxicants. Oxid. Med. Cell. Longev. 2015, 2015, 140267. [Google Scholar] [CrossRef] [PubMed]
- Uray, I.P.; Dmitrovsky, E.; Brown, P.H. Retinoids and rexinoids in cancer prevention: From laboratory to clinic. Semin. Oncol. 2016, 43, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T.; Liebisch, G.; Moehle, C.; Schifferer, R.; Dayoub, R.; Heiduczek, S.; Grandl, M.; Dada, A.; Schmitz, G. Gene expression profiling identifies retinoids as potent inducers of macrophage lipid efflux. Biochim. Biophys. Acta 2005, 1740, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Azzi, A.; Birringer, M.; Cook-Mills, J.M.; Eggersdorfer, M.; Frank, J.; Cruciani, G.; Lorkowski, S.; Özer, N.K. Vitamin E: Emerging aspects and new directions. Free Radic. Biol. Med. 2017, 102, 16–36. [Google Scholar] [CrossRef] [PubMed]
- Argmann, C.A.; Sawyez, C.G.; McNeil, C.J.; Hegele, R.A.; Huff, M.W. Activation of peroxisome proliferator-activated receptor gamma and retinoid X receptor results in net depletion of cellular cholesteryl esters in macrophages exposed to oxidized lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Sidell, N. Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology 2002, 106, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Wuttge, D.M. Induction of CD36 by all-trans retinoic acid: Retinoic acid receptor signaling in the pathogenesis of atherosclerosis. FASEB J. 2001. [Google Scholar] [CrossRef]
- Barber, N.; Belov, L.; Christopherson, R.I. All-trans retinoic acid induces different immunophenotypic changes on human HL60 and NB4 myeloid leukaemias. Leuk. Res. 2008, 32, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Endemann, G.; Stanton, L.W.; Madden, K.S.; Bryant, C.M.; White, R.T.; Protter, A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993, 268, 11811–11816. [Google Scholar] [PubMed]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar] [PubMed]
- Tontonoz, P.; Nagy, L.; Alvarez, J.G.; Thomazy, V.A.; Evans, R.M. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 1998, 93, 241–252. [Google Scholar] [CrossRef]
- Schrijvers, D.M.; De Meyer, G.R.Y.; Herman, A.G.; Martinet, W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc. Res. 2007, 73, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Weng, S.; Felton, S.K.; Bhandare, S.; Riek, A.; Butler, B.; Proctor, B.M.; Petty, M.; Chen, Z.; Schechtman, K.B.; et al. 1,25(OH)2 vitamin D inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation 2009, 120, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Riek, A.E.; Oh, J.; Bernal-Mizrachi, C. Vitamin D regulates macrophage cholesterol metabolism in diabetes. J. Steroid Biochem. Mol. Biol. 2010, 121, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; Feo, V.D.; Zia-Ul-Haq, M. Natural products as alternative choices for P-Glycoprotein (P-gp) inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remião, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Henry, H.L. Regulation of vitamin D metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Mustacich, D.J.; Leonard, S.W.; Patel, N.K.; Traber, M.G. Alpha-tocopherol beta-oxidation localized to rat liver mitochondria. Free Radic. Biol. Med. 2010, 48, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Benbrook, D.; Lernhardt, E.; Pfahl, M. A new retinoic acid receptor identified from a hepatocellular carcinoma. Nature 1988, 333, 669–672. [Google Scholar] [CrossRef] [PubMed]
- Brand, N.; Petkovich, M.; Krust, A.; Chambon, P.; de Thé, H.; Marchio, A.; Tiollais, P.; Dejean, A. Identification of a second human retinoic acid receptor. Nature 1988, 332, 850–853. [Google Scholar] [CrossRef] [PubMed]
- Giguere, V.; Ong, E.S.; Segui, P.; Evans, R.M. Identification of a receptor for the morphogen retinoic acid. Nature 1987, 330, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Zelent, A.; Krust, A.; Petkovich, M.; Kastner, P.; Chambon, P. Cloning of murine alpha and beta retinoic acid receptors and a novel receptor gamma predominantly expressed in skin. Nature 1989, 339, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Brumbaugh, P.F.; Hughes, M.R.; Haussler, M.R. Cytoplasmic and nuclear binding components for 1alpha25-dihydroxyvitamin D3 in chick parathyroid glands. Proc. Natl. Acad. Sci. USA 1975, 72, 4871–4875. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.C.; Norman, A.W. Studies on calciferol metabolism. 8. Evidence for a cytoplasmic receptor for 1,25-dihydroxy-vitamin D3 in the intestinal mucosa. J. Biol. Chem. 1973, 248, 5967–5975. [Google Scholar] [PubMed]
- Baker, A.R.; McDonnell, D.P.; Hughes, M.; Crisp, T.M.; Mangelsdorf, D.J.; Haussler, M.R.; Pike, J.W.; Shine, J.; O’Malley, B.W. Cloning and expression of full-length cDNA encoding human vitamin D receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 3294–3298. [Google Scholar] [CrossRef] [PubMed]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Krishnan, A.V.; Malloy, P.J.; Eccleshall, T.R.; Zhao, X.Y.; Feldman, D. The vitamin D receptor gene start codon polymorphism: A functional analysis of FokI variants. J. Bone Miner. Res. 1998, 13, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Allenby, G.; Janocha, R.; Kazmer, S.; Speck, J.; Grippo, J.F.; Levin, A.A. Binding of 9-cis-retinoic acid and all-trans-retinoic acid to retinoic acid receptors alpha, beta, and gamma. Retinoic acid receptor gamma binds all-trans-retinoic acid preferentially over 9-cis-retinoic acid. J. Biol. Chem. 1994, 269, 16689–16695. [Google Scholar] [PubMed]
- Heyman, R.A.; Mangelsdorf, D.J.; Dyck, J.A.; Stein, R.B.; Eichele, G.; Evans, R.M.; Thaller, C. 9-cis-retinoic acid is a high affinity ligand for the retinoid X receptor. Cell 1992, 68, 397–406. [Google Scholar] [CrossRef]
- Wolf, G. Is 9-cis-retinoic acid the endogenous ligand for the retinoic acid-X receptor? Nutr. Rev. 2006, 64, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Landes, N.; Pfluger, P.; Kluth, D.; Birringer, M.; Rühl, R.; Böl, G.-F.; Glatt, H.; Brigelius-Flohé, R. Vitamin E activates gene expression via the pregnane X receptor. Biochem. Pharmacol. 2003, 65, 269–273. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Tabb, M.M.; Sun, A.; Zhou, C.; Grün, F.; Errandi, J.; Romero, K.; Pham, H.; Inoue, S.; Mallick, S.; Lin, M.; et al. Vitamin K2 regulation of bone homeostasis is mediated by the steroid and xenobiotic receptor SXR. J. Biol. Chem. 2003, 278, 43919–43927. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Horie-Inoue, K.; Ikeda, K.; Blumberg, B.; Inoue, S. Steroid and xenobiotic receptor SXR mediates vitamin K2-activated transcription of extracellular matrix-related genes and collagen accumulation in osteoblastic cells. J. Biol. Chem. 2006, 281, 16927–16934. [Google Scholar] [CrossRef] [PubMed]
- Suhara, Y.; Watanabe, M.; Nakagawa, K.; Wada, A.; Ito, Y.; Takeda, K.; Takahashi, K.; Okano, T. Synthesis of novel vitamin K2 analogues with modification at the ω-terminal position and their biological evaluation as potent steroid and xenobiotic receptor (SXR) agonists. J. Med. Chem. 2011, 54, 4269–4273. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Targets | Cells | Effects | Substances | Refs. |
|---|---|---|---|---|
| COX2 | A549 cells | Reduced activity in arachidonic acid-pre-induced cells | γ-13′-COOH | [70] |
| δ-13′-COOH | [70,72] | |||
| δ-9′-COOH | [70] | |||
| Isolated enzyme | Inhibition of activity | δ-13′-COOH | [70] | |
| δ-9′-COOH | ||||
| RAW264.7 | Inhibition of LPS-stimulated mRNA and protein expression, as well as reduced PG release | α-13′-OH | [71] | |
| α-13′-COOH | [74] | |||
| iNos | RAW264.7 | Inhibition of LPS-stimulated mRNA and protein expression, as well as reduced release of nitric oxide | α-13′-OH | [64,71,74,75] |
| α-13′-COOH | ||||
| δ-13′-OH | ||||
| δ-13′-COOH | ||||
| 5-LO | Isolated enzyme | Inhibition of activity | δ-13′-COOH | [72,73] |
| HL-60 neutrophils | Reduced activity and LT release in pre-induced cells | δ-13′-COOH | [73] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schubert, M.; Kluge, S.; Schmölz, L.; Wallert, M.; Galli, F.; Birringer, M.; Lorkowski, S. Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins? Antioxidants 2018, 7, 10. https://doi.org/10.3390/antiox7010010
Schubert M, Kluge S, Schmölz L, Wallert M, Galli F, Birringer M, Lorkowski S. Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins? Antioxidants. 2018; 7(1):10. https://doi.org/10.3390/antiox7010010
Chicago/Turabian StyleSchubert, Martin, Stefan Kluge, Lisa Schmölz, Maria Wallert, Francesco Galli, Marc Birringer, and Stefan Lorkowski. 2018. "Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins?" Antioxidants 7, no. 1: 10. https://doi.org/10.3390/antiox7010010
APA StyleSchubert, M., Kluge, S., Schmölz, L., Wallert, M., Galli, F., Birringer, M., & Lorkowski, S. (2018). Long-Chain Metabolites of Vitamin E: Metabolic Activation as a General Concept for Lipid-Soluble Vitamins? Antioxidants, 7(1), 10. https://doi.org/10.3390/antiox7010010