Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change


{kind=link}
{kind=link}
Abstract
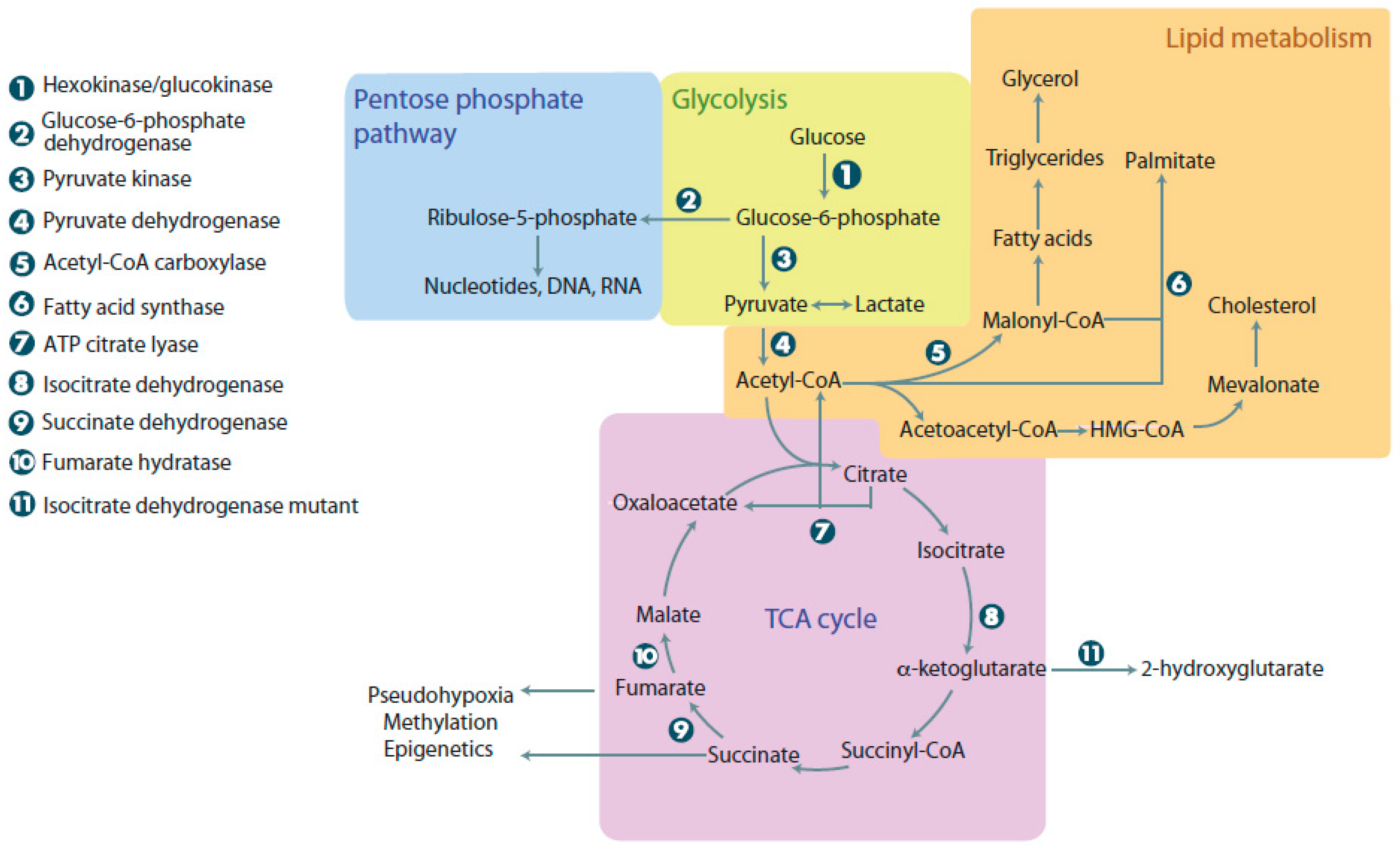
:1. Aerobic Glycolysis in Normal and Cancer Cells
2. Advantages of Metabolic Reprogramming in Cancer Cells
2.1. Lipid Synthesis
2.2. Nucleotide Synthesis
2.3. 1C Metabolism
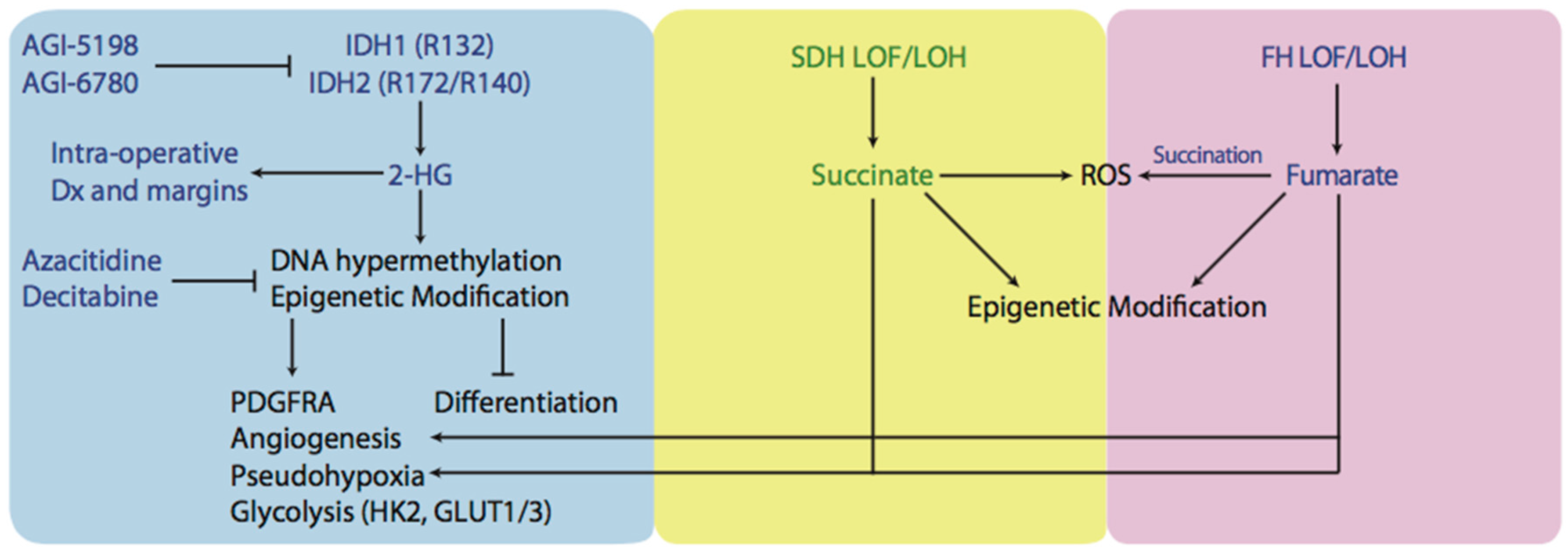
3. Production of Oncometabolites and Effects on Normal Metabolism: 2-Hydroxyglutarate, Succinate, and Fumarate
3.1. Mutations Affecting Isocitrate Dehydrogenase
3.2. Mutations Affecting Succinate Dehydrogenase and Fumarate Hydratase
4. The Link between Oncometabolites and Mitochondrial Dynamics
5. Novel Therapies Targeting Metabolic Aberrations in Cancer Cells
6. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Cura, A.J.; Carruthers, A. Role of monosaccharide transport proteins in carbohydrate assimilation, distribution, metabolism, and homeostasis. Compr. Physiol. 2012, 2, 863–914. [Google Scholar] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Enriquez, J.A.; Perez-Martos, A.; Montoya, J.; Fernandez-Silva, P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion 2011, 11, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Farrar, G.J.; Chadderton, N.; Kena, P.F.; Millington-Ward, S. Mitochondrial disorders: Aetiologies, models systems, and candidate therapies. Trends Genet. 2013, 29, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Darlington, C.D. The plasmagene theory of the origin of cancer. Br. J. Cancer 1948, 2, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Woods, M.W.; DuBuy, H.G. Cytoplasmic diseases and cancer. Science 1945, 102, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Döppler, H.; DelGiorno K, E.; Zhang, L.; Leitges, M.; Crawford, C.H.; Murphy, M.P.; Storz, P. Mutant Kras-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signalling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Makinoshima, H.; Takita, M.; Saruwatari, K.; Umemura, S.; Obata, Y.; Ishii, G.; Matsumoto, S.; Sugiyama, E.; Ochiai, A.; Abe, R.; et al. Signaling through the Phosphatidylinositol 3-Kinase (PI3K)/Mammalian Target of Rapamycin (mTOR) Axis Is Responsible for Aerobic Glycolysis mediated by Glucose Transporter in Epidermal Growth Factor Receptor (EGFR)-mutated Lung Adenocarcinoma. J. Biol. Chem. 2015, 290, 17495–17504. [Google Scholar] [CrossRef] [PubMed]
- Losman, J.A.; Looper, R.E.; Koivunen, P.; Lee, S.; Schneider, R.K.; McMahon, C.; Cowley, G.S.; Root, D.E.; Ebert, B.L.; Kaelin, W.G., Jr. (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 2013, 339, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Pollard, P.J.; Tomlinson, I. SDH mutations in cancer. Biochim. Biophys. Acta 2011, 1807, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Picaud, S.; Kavanagh, K.L.; Yue, W.W.; Lee, W.H.; Muller-Knapp, S.; Gileadi, O.; Sacchettini, J.; Oppermann, U. Structural basis of fumarate hydratase deficienct. J. Inherit. Metab. Dis. 2011, 34, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Kurelac, I.; Romeo, C.; Gasparre, G. Mitochondrial metabolism and cancer. Mitochondrion 2010, 11, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Nayak, B.; Shim, E.H.; Livi, C.B.; Block, K.; Sudarshan, S. The oncometabolite fumarate promotes pseudohypoxia through noncanonical activation of NF-κB signaling. J. Biol. Chem. 2014, 289, 24691–24699. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, T.W.; Lane, A.N.; Higashi, R.M.; Farag, M.A.; Gao, H.; Bousamra, M.; Miller, D.M. Altered regulation of metabolic pathways is human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 2009, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Comerford, S.A.; Huang, Z.; Du, X.; Wang, Y.; Cai, L.; Witkiewicz, A.K.; Walters, H.; Tantawy, M.N.; Fu, A.; Manning, H.C.; et al. Acetate dependence of tumors. Cell 2014, 159, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Jenni, S.; Ban, N. Architecture of mammalian fatty acid synthase at 4.5 Å resolution. Science 2006, 311, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Asturias, F.J.; Chadick, J.Z.; Cheung, I.K.; Stark, H.; Witkowski, A.; Joshi, A.K.; Smith, S. Structure and molecular organization of mammalian fatty acid synthase. Nat. Struct. Mol. Biol. 2005, 12, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Chirala, S.S.; Wakil, S.J. Structure and function of animal fatty acid synthase. Lipids 2004, 39, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.; Hoffmann, G.E.; Schreiber, R.; Andres, H.; Fuchs, E.; Körber, E.; Kolb, H.J. Fatty-acid biosynthesis in man, a pathway of minor importance. Purification, optimal assay conditions, and organ distribution of fatty-acid synthase. Biol. Chem. Hoppe Seyler 1986, 367, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Pizer, E.S.; Kurman, R.J.; Pasternack, G.R.; Kuhajda, F.P. Expression of fatty acid synthase is closely linked to proliferation and stromal decidualization in cycling endometrium. Int. J. Gynecol. Pathol. 1997, 16, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kusakabe, T.; Maeda, M.; Hoshi, N.; Sugino, T.; Watanabe, K.; Fukuda, T.; Suzuki, T. Fatty acid synthase is expressed mainly in adult hormone-sensitive cells or cells with high lipid metabolism and in proliferating fetal cells. J. Histochem. Cytochem. 2000, 48, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Wagle, S.; Bui, A.; Ballard, P.L.; Shuman, H.; Gonzales, J.; Gonzales, L.W. Hormonal regulation and cellular localization of fatty acid synthase in human fetal lung. Am. J. Physiol. 1999, 277 Pt 1, L381–L390. [Google Scholar] [CrossRef] [PubMed]
- Szutowicz, A.; Kwiatkowski, J.; Angielski, S. Lipogenetic and glycolytic enzyme activities in carcinoma and nonmalignant diseases of the human breast. Br. J. Cancer 1979, 39, 681–687. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue IV: A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Costello, L.C.; Franklin, R. Tumor cell metabolism: The marriage of molecular genetics and proteomics with cellular intermediary metabolism; proceed with caution! Mol. Cancer 2006, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R. ‘Why do tumour cells glycolyse?’: From glycolysis through citrate to lipogenesis. Mol. Cell. Biochem. 2005, 280, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Q.; Zhao, X.Y.; Wang, H.Y.; Liang, Y. Increased fatty acid synthase as a potential therapeutic target in multiple myeloma. J. Zhejiang Univ. Sci. B 2008, 9, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Migita, T.; Narita, T.; Nomura, K.; Miyagi, E.; Inazuka, F.; Matsuura, M.; Ushijima, M.; Mashima, T.; Seimiya, H.; Satoh, Y.; et al. ATP citrate lyase: Activation and therapeutic implications in non-small cell lung cancer. Cancer Res. 2008, 68, 8547–8554. [Google Scholar] [CrossRef] [PubMed]
- Yancy, H.F.; Mason, J.A.; Peters, S.; Thompson, C.E., 3rd; Littleton, G.K.; Jett, M.; Day, A.A. Metastatic progression and gene expression between breast cancer cell lines from African American and Caucasian women. J. Carcinog. 2007, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Hasegawa, K.; Ohashi, K.; Matsuzaka, T.; Najima, Y.; Sekiya, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Co-ordinate activation of lipogenic enzymes in hepatocellular carcinoma. Eur. J. Cancer 2005, 41, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Varis, A.; Wolf, M.; Monni, O.; Vakkari, M.L.; Kokkola, A.; Moskaluk, C.; Frierson, H., Jr.; Powell, S.M.; Knuutila, S.; Kallioniemi, A.; et al. Targets of gene amplification and overexpression at 17q in gastric cancer. Cancer Res. 2002, 62, 2625–2629. [Google Scholar] [PubMed]
- Turyn, J.; Schlichtholz, B.; Dettlaff-Pokora, A.; Presler, M.; Goyke, E.; Matuszewski, M.; Kmiec, Z.; Krajka, K.; Swierczynski, J. Increased activity of glycerol 3-phosphate dehydrogenase and other lipogenic enzymes in human bladder cancer. Horm. Metab. Res. 2003, 35, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Halliday, K.R.; Fenoglio-Preiser, C.; Sillerud, L.O. Differentiation of human tumors from nonmalignant tissue by natural-abundance 13C NMR spectroscopy. Magn. Reson. Med. 1988, 7, 384–411. [Google Scholar] [CrossRef] [PubMed]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Tong, L. Acetyl-coenzyme A carboxylase: Crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life Sci. 2005, 62, 1784–1803. [Google Scholar] [CrossRef] [PubMed]
- Milgraum, L.Z.; Witters, L.A.; Pasternack, G.R.; Kuhajda, F.P. Enzymes of the fatty acid synthesis pathway are highly expressed in in situ breast carcinoma. Clin. Cancer Res. 1997, 3, 2115–2120. [Google Scholar] [PubMed]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Magnard, C.; Bachelier, R.; Vincent, A.; Jaquinod, M.; Kieffer, S.; Lenoir, G.M.; Venezia, N.D. BRCA1 interacts with acetyl-CoA carboxylase through its tandem of BRCT domains. Oncogene 2002, 21, 6729–6739. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Dizin, E.; Ray, H.; Luquain, C.; Lefai, E.; Foufelle, F.; Billaud, M.; Lenoir, G.M.; Venezia, N.D. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. J. Biol. Chem. 2006, 281, 3172–3181. [Google Scholar] [CrossRef] [PubMed]
- Chajes, V.; Cambot, M.; Moreau, K.; Lenoir, G.M.; Joulin, V. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res. 2006, 66, 5287–5294. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; De Schrijver, E.; Verhoeven, G.; Swinnen, J.V. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res. 2005, 65, 6719–6725. [Google Scholar] [CrossRef] [PubMed]
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218. [Google Scholar] [PubMed]
- Wang, C.; Xu, C.; Sun, M.; Luo, D.; Liao, D.F.; Cao, D. Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 2009, 385, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Beckers, A.; Organe, S.; Timmermans, L.; Scheys, K.; Peeters, A.; Brusselmans, K.; Verhoeven, G.; Swinnen, J.V. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 2007, 67, 8180–8187. [Google Scholar] [CrossRef] [PubMed]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2016, 90, 927–963. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro-Oncology 2016, 18, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 Regulates Biosynthesis through Direct Inactivation of Glucose-6-Phosphate Dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 2014, 5, 3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, A.; Tedeschi, P.M.; Bertino, J.R. Overexpression of the mitochondrial folate and glycine-serine pathway: A new determinant of methotrexate selectivity in tumors. Cancer Res. 2013, 73, 478–482. [Google Scholar] [CrossRef] [PubMed]
- Tibbetts, A.S.; Appling, D.R. Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 2010, 30, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fan, J.; Venneti, S.; Wan, Y.W.; Pawel, B.R.; Zhang, J.; Finley, L.W.; Lu, C.; Lindsten, T.; Cross, J.R.; et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014, 4, 1406–1417. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Fiske, B.P.; Birsoy, K.; Freinkman, E.; Kami, K.; Possemato, R.L.; Chudnovsky, Y.; Pacold, M.E.; Chen, W.W.; Cantor, J.R.; et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 2015, 520, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of Cytosolic One-Carbon Flux Compensates for Loss of the Mitochondrial Folate Pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Bolusani, S.; Young, B.A.; Cole, N.A.; Tibbetts, A.S.; Momb, J.; Bryant, J.D.; Solmonson, A.; Appling, D.R. Mammalian MTHFD2L encodes a mitochondrial methylenetetrahydrofolate dehydrogenase isozyme expressed in adult tissues. J. Biol. Chem. 2011, 286, 5166–5174. [Google Scholar] [CrossRef] [PubMed]
- Meiser, J.; Tumanov, S.; Maddocks, O.; Labuschagne, C.F.; Athineos, D.; Van Den Broek, N.; Mackay, G.M.; Gottlieb, E.; Blyth, K.; Vousden, K.; et al. Serine one-carbon catabolism with formate overflow. Sci. Adv. 2016, 2, e1601273. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Parker, S.J.; Fiske, B.P.; McCloskey, D.; Gui, D.Y.; Green, C.R.; Vokes, N.I.; Feist, A.M.; Vander Heiden, M.G.; Metallo, C.M. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 2014, 55, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Soga, T.; Pollard, P.J. Oncometabolites: Linking altered metabolism with cancer. J. Clin. Investig. 2013, 123, 3652–3658. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Pollard, P.J.; Gottlieb, E. Inborn and acquired metabolic defects in cancer. J. Mol. Med. (Berl.) 2011, 89, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R.; Ding, L.; Dooling, D.J.; Larson, D.E.; McLellan, M.D.; Chen, K.; Koboldt, D.C.; Fulton, R.S.; Delehaunty, K.D.; McGrath, S.D.; et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009, 361, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.B. Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 2009, 360, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O'Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Borger, D.R.; Tanabe, K.K.; Fan, K.C.; Lopez, H.U.; Fantin, V.R.; Straley, K.S.; Schenkein, D.P.; Hezel, A.F.; Ancukiewicz, M.; Liebman, H.M.; et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012, 17, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Oermann, E.K.; Wu, J.; Guan, K.L.; Xiong, Y. Alterations of metabolic genes and metabolites in cancer. Semin. Cell Dev. Biol. 2012, 23, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ye, D.; Guan, K.L.; Xiong, Y. IDH1 and IDH2 mutations in tumorigenesis: Mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012, 18, 5562–5571. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2010, 465, 966. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.; Cairns, R.A.; Minden, M.D.; Driggers, E.M.; Bittinger, M.A.; Jang, H.G.; Jang, H.G.; Sasaki, M.; Jin, S.; Schenkein, D.P.; Su, S.M.; et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J. Exp. Med. 2010, 207, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Subramanian, C.; Jackowski, S.; Rock, C.O. Cancer-associated isocitrate dehydrogenase mutations inactivate NADPH-dependent reductive carboxylation. J. Biol. Chem. 2012, 287, 14615–14620. [Google Scholar] [CrossRef] [PubMed]
- Jochmanova, I.; Pacak, K. Pheochromocytoma: The First Metabolic Endocrine Cancer. Clin. Cancer Res. 2016, 22, 5001–5011. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Burger, P.; Kleihues, P. Definition of primary and secondary glioblastoma—Response. Clin. Cancer Res. 2014, 20, 2013. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Drier, Y.; Liau, B.B.; Gillespie, S.M.; Venteicher, A.S.; Stemmer-Rachamimov, A.O.; Suva, M.L.; Bernstein, B.E. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 2016, 529, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Lee, S.; Duncan, C.G.; Lopez, G.; Lu, G.; Ramkissoon, S.; Losman, J.A.; Joensuu, P.; Bergmann, U.; Gross, S.; et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 2012, 483, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Knobbe, C.B.; Itsumi, M.; Elia, A.J.; Harris, I.S.; Chio, I.I.; Cairns, R.A.; McCracken, S.; Wakeham, A.; Haight, J.; et al. D-2-hydroxyglutarate produced by mutant IDH1 perturbs collagen maturation and basement membrane function. Genes Dev. 2012, 26, 2038–2049. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Knobbe, C.B.; Munger, J.C.; Lind, E.F.; Brenner, D.; Brustle, A.; Harris, I.S.; Holmes, R.; Wakeham, A.; Haight, J.; et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature 2012, 488, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zha, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Mapstone, T.B. Malignant glioma physiology: Cellular response to hypoxia and its role in tumor progression. Ann. Intern. Med. 2003, 138, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.P.; Chou, W.C.; Buckstein, R.; Cermak, J.; et al. Multicenter, randomized, open-label, phase III trial of decitabine versus patient choice, with physician advice, of either supportive care or low-dose cytarabine for the treatment of older patients with newly diagnosed acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 2670–2677. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs. conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, A.; Salmasi, V.; Turcan, S.; Fabius, A.W.; Baia, G.S.; Eberhart, C.G.; Weingart, J.D.; Gallia, G.L.; Baylin, S.B.; Chan, T.A.; et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget 2013, 4, 1737–1747. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Fabius, A.W.; Borodovsky, A.; Pedraza, A.; Brennan, C.; Huse, J.; Viale, A.; Riggins, G.J.; Chan, T.A.; et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget 2013, 4, 1729–1736. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Travins, J.; DeLaBarre, B.; Penard-Lacronique, V.; Schalm, S.; Hansen, E.; Straley, K.; Kernytsky, A.; Liu, W.; Gliser, C.; et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science 2013, 340, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Kernytsky, A.; Wang, F.; Hansen, E.; Schalm, S.; Straley, K.; Gliser, C.; Yang, H.; Travins, J.; Murray, S.; Dorsch, M.; et al. IDH2 mutation-induced histone and DNA hypermethylation is progressively reversed by small-molecule inhibition. Blood 2015, 125, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Quivoron, C.; David, M.; Straley, K.; Travins, J.; Kim, H.; Chen, Y.; Zhu, D.; Saada, V.; Bawa, O.; Opolon, P.; et al. AG-221, an oral, selective, first-in-class, potent IDH2-R140Q mutant inhibitor, induces differentiation in a Xenotransplant Model. Blood 2014, 124, 3735. [Google Scholar]
- Hansen, E.; Quivoron, C.; Straley, K.; Lemieux, R.M.; Popovici-Muller, J.; Sadrzadeh, H.; Fathi, A.T.; Gliser, C.; David, M.; Saada, V.; et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated Ex Vivo. Blood 2014, 124, 3734. [Google Scholar]
- Mondesir, J.; Willekens, C.; Touat, M.; de Botton, S. IDH1 and IDH2 mutations as novel therapeutic targets: Current perspectives. J. Blood Med. 2016, 7, 171–180. [Google Scholar] [PubMed]
- Miettinen, M.; Lasota, J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—A review. Int. J. Biochem. Cell Biol. 2014, 53, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Lehtonen, H.J.; Kiuru, M.; Ylisaukko-Oja, S.K.; Salovaara, R.; Herva, R.; Koivisto, P.A.; Vierimaa, O.; Aittomaki, K.; Pukkala, E.; Launonen, V.; et al. Increased risk of cancer in patients with fumarate hydratase germline mutation. J. Med. Genet. 2006, 43, 523–526. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P. Warburg tumours and the mechanisms of mitochondrial tumour suppressor genes. Barking up the right tree? Curr. Opin. Genet. Dev. 2010, 20, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Rieubland, C.; Kerlan, V.; Plouin, P.F.; Rotig, A.; Jeunemaitre, X. Functional consequences of a SDHB gene mutation in an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab. 2002, 87, 4771–4774. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Roqueplo, A.P.; Favier, J.; Rustin, P.; Mourad, J.J.; Plouin, P.F.; Corvol, P.; Rotig, A.; Jeunemaitre, X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am. J. Hum. Genet. 2001, 69, 1186–1197. [Google Scholar] [CrossRef] [PubMed]
- Pollard, P.J.; Briere, J.J.; Alam, N.A.; Barwell, J.; Barclay, E.; Wortham, N.C.; Hunt, T.; Mitchell, M.; Olpin, S.; Moat, S.J.; et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet. 2005, 14, 2231–2239. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar] [PubMed]
- Sudarshan, S.; Sourbier, C.; Kong, H.S.; Block, K.; Valera Romero, V.A.; Yang, Y.; Galindo, C.; Mollapour, M.; Scroggins, B.; Goode, N.; et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol. Cell. Biol. 2009, 29, 4080–4090. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Launonen, V.; Tomlinson, I.P. The FH mutation database: An online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med. Genet. 2008, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Blatnik, M.; Thorpe, S.R.; Baynes, J.W. Succination of proteins by fumarate: Mechanism of inactivation of glyceraldehyde-3-phosphate dehydrogenase in diabetes. Ann. N. Y. Acad. Sci. 2008, 1126, 272–275. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Martinez-Garcia, E.; Nguyen, H.; Mullen, A.R.; Dufour, E.; Sudarshan, S.; Licht, J.D.; Deberardinis, R.J.; Chandel, N.S. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 2013, 51, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Hatipoglu, E.; O’Flaherty, L.; Ternette, N.; Sahgal, N.; Lockstone, H.; Baban, D.; Nye, E.; Stamp, G.W.; Wolhuter, K.; et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011, 20, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Ternette, N.; Yang, M.; Laroyia, M.; Kitagawa, M.; O’Flaherty, L.; Wolhulter, K.; Igarashi, K.; Saito, K.; Kato, K.; Fischer, R.; et al. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 2013, 3, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Ooi, A.; Wong, J.C.; Petillo, D.; Roossien, D.; Perrier-Trudova, V.; Whitten, D.; Min, B.W.; Tan, M.H.; Zhang, Z.; Yang, X.J.; et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011, 20, 511–523. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Wentzel, J.F.; Lewies, A.; Bronkhorst, A.J.; van Dyk, E.; du Plessis, L.H.; Pretorius, P.J. Exposure to high levels of fumarate and succinate leads to apoptotic cytotoxicity and altered global DNA methylation profiles in vitro. Biochimie 2017, 135, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.; Scorrano, L.; Campello, S. Changing perspective on oncometabolites: From metabolic signature of cancer to tumorigenic and immunosuppressive agents. Oncotarget 2016, 7, 46692–46706. [Google Scholar] [CrossRef] [PubMed]
- Janin, M.; Esteller, M. Oncometabolite Accumulation and Epithelial-to-Mesenchymal Transition: The Turn of Fumarate. Cell Metab. 2016, 24, 529–530. [Google Scholar] [CrossRef] [PubMed]
- Colvin, H.; Nishida, N.; Konno, M.; Haraguchi, N.; Takahashi, H.; Nishimura, J.; Hata, T.; Kawamoto, K.; Asai, A.; Tsunekuni, K.; et al. Oncometabolite D-2-Hydroxyglurate Directly Induces Epithelial-Mesenchymal Transition and is Associated with Distant Metastasis in Colorectal Cancer. Sci. Rep. 2016, 6, 36289. [Google Scholar] [CrossRef] [PubMed]
- Porporato, P.E.; Payen, V.L.; Perez-Escuredo, J.; De Saedeleer, C.J.; Danhier, P.; Copetti, T.; Dhup, S.; Tardy, M.; Vazeille, T.; Bouzin, C.; et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014, 8, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-da-Silva, A.; Valacca, C.; Rios, E.; Populo, H.; Soares, P.; Sobrinho-Simoes, M.; Scorrano, L.; Maximo, V.; Campello, S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.Y.; Zhang, J.F.; Yang, Z.J.; Jiang, L.P.; Wei, Y.F.; Lai, Q.N.; Wang, J.B.; Xin, H.B.; Han, X.J. Involvement of Drp1 in hypoxia-induced migration of human glioblastoma U251 cells. Oncol. Rep. 2014, 32, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Campello, S.; Lacalle, R.A.; Bettella, M.; Manes, S.; Scorrano, L.; Viola, A. Orchestration of lymphocyte chemotaxis by mitochondrial dynamics. J. Exp. Med. 2006, 203, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.F.; Mariotti, F.R.; Maximo, V.; Campello, S. Mitochondria dynamism: Of shape, transport and cell migration. Cell. Mol. Life Sci. 2014, 71, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Corrado, M.; Scorrano, L.; Campello, S. Mitochondrial dynamics in cancer and neurodegenerative and neuroinflammatory diseases. Int. J. Cell Biol. 2012, 2012, 729290. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Hackenbrock, C.R. Ultrastructural bases for metabolically linked mechanical activity in mitochondria. I. Reversible ultrastructural changes with change in metabolic steady state in isolated liver mitochondria. J. Cell Biol. 1966, 30, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- SongTao, Q.; Lei, Y.; Si, G.; YanQing, D.; HuiXia, H.; XueLin, Z.; LanXiao, W.; Fei, Y. IDH mutations predict longer survival and response to temozolomide in secondary glioblastoma. Cancer Sci. 2012, 103, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Briere, J.J.; Favier, J.; Benit, P.; El Ghouzzi, V.; Lorenzato, A.; Rabier, D.; Di Renzo, M.F.; Gimenez-Roqueplo, A.P.; Rustin, P. Mitochondrial succinate is instrumental for HIF1alpha nuclear translocation in SDHA-mutant fibroblasts under normoxic conditions. Hum. Mol. Genet. 2005, 14, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
- Letouze, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Obara, N.; Ema, M.; Horie, M.; Naka, A.; Takahashi, S.; Imagawa, S. Antitumor effects of 2-oxoglutarate through inhibition of angiogenesis in a murine tumor model. Cancer Sci. 2009, 100, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, T.; Chesnelong, C.; Pankov, A.; Jalbert, L.E.; Hong, C.; Hayes, J.; Smirnov, I.V.; Marshall, R.; Souza, C.F.; Shen, Y.; et al. Clonal expansion and epigenetic reprogramming following deletion or amplification of mutant IDH1. Proc. Natl. Acad. Sci. USA 2017, 114, 10743–10748. [Google Scholar] [CrossRef] [PubMed]
- Rohle, D.; Popovici-Muller, J.; Palaskas, N.; Turcan, S.; Grommes, C.; Campos, C.; Tsoi, J.; Clark, O.; Oldrini, B.; Komisopoulou, E.; et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science 2013, 340, 626–630. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Eberlin, L.S.; Norton, I.; Calligaris, D.; Feldman, D.R.; Ide, J.L.; Liu, X.; Wiley, J.S.; Vestal, M.L.; Ramkissoon, S.H.; et al. Intraoperative mass spectrometry mapping of an onco-metabolite to guide brain tumor surgery. Proc. Natl. Acad. Sci. USA 2014, 111, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants 2018, 7, 16. https://doi.org/10.3390/antiox7010016
Zhou Z, Ibekwe E, Chornenkyy Y. Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants. 2018; 7(1):16. https://doi.org/10.3390/antiox7010016
Chicago/Turabian StyleZhou, Zhengqiu, Elochukwu Ibekwe, and Yevgen Chornenkyy. 2018. "Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change" Antioxidants 7, no. 1: 16. https://doi.org/10.3390/antiox7010016
APA StyleZhou, Z., Ibekwe, E., & Chornenkyy, Y. (2018). Metabolic Alterations in Cancer Cells and the Emerging Role of Oncometabolites as Drivers of Neoplastic Change. Antioxidants, 7(1), 16. https://doi.org/10.3390/antiox7010016