Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
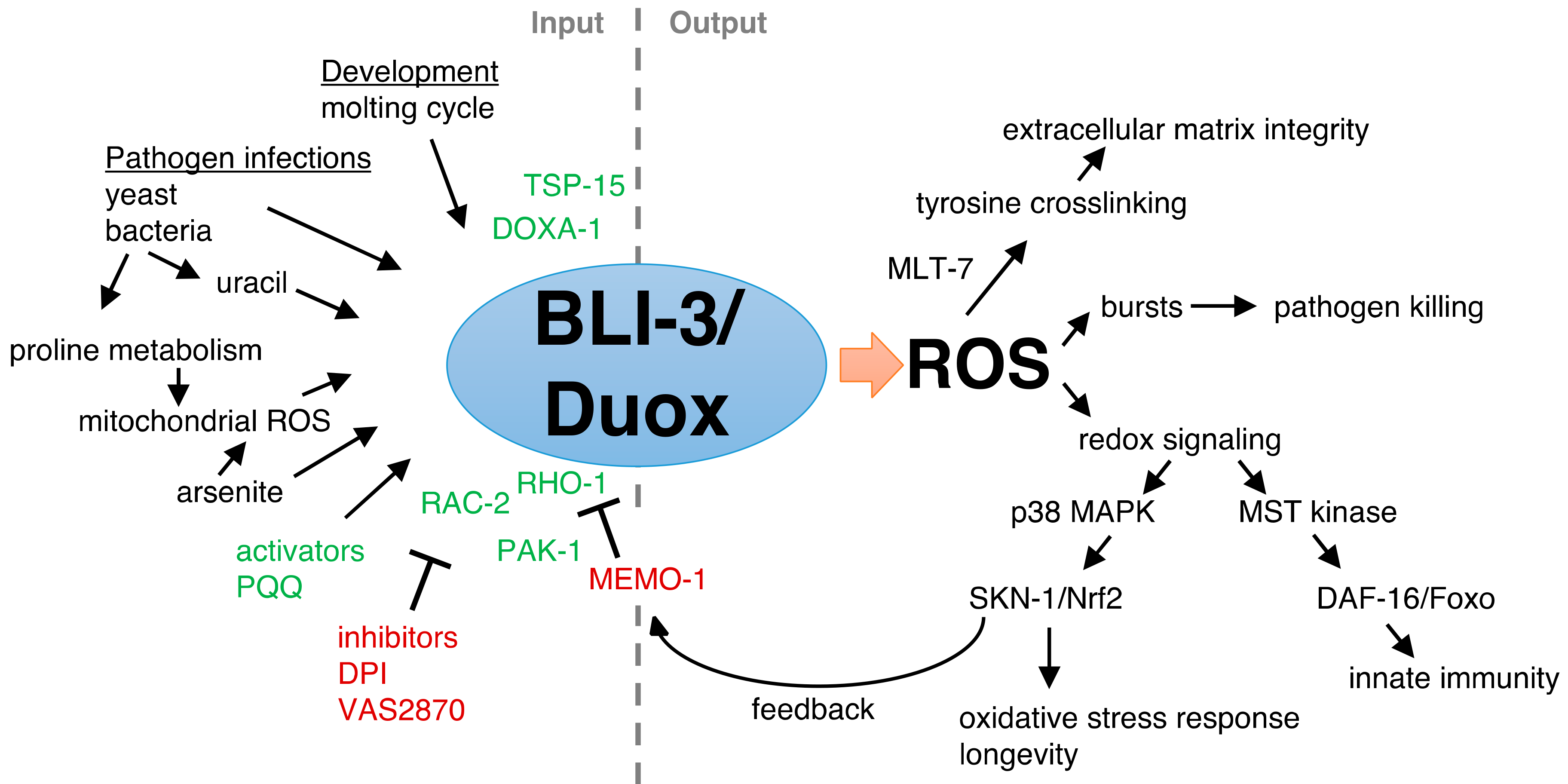
2. Insights of Duox Functions from the Model Organism C. elegans
3. Tissue Distribution of Duox in C. elegans
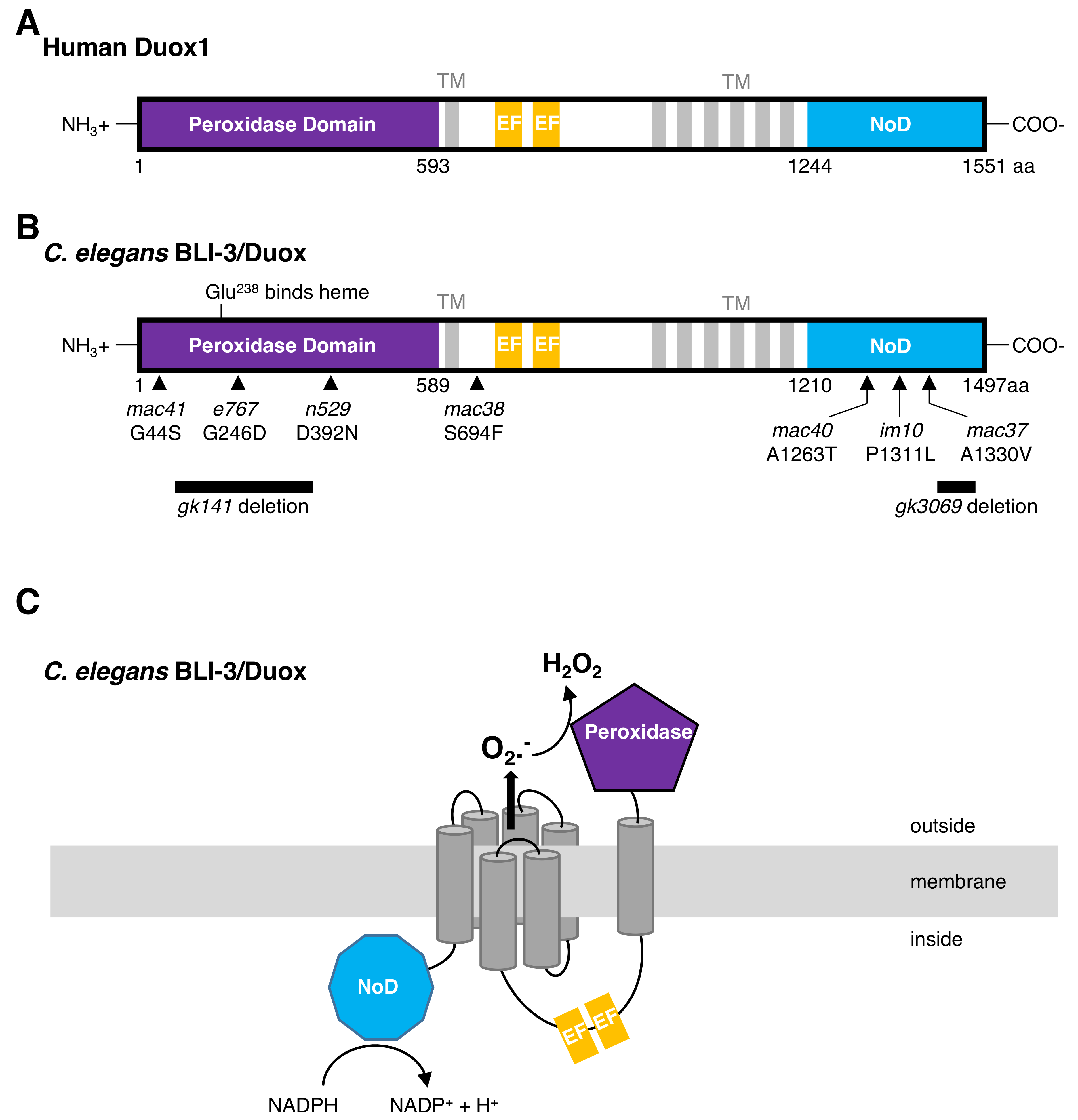
4. The Maturation Complex of Duox in C. elegans
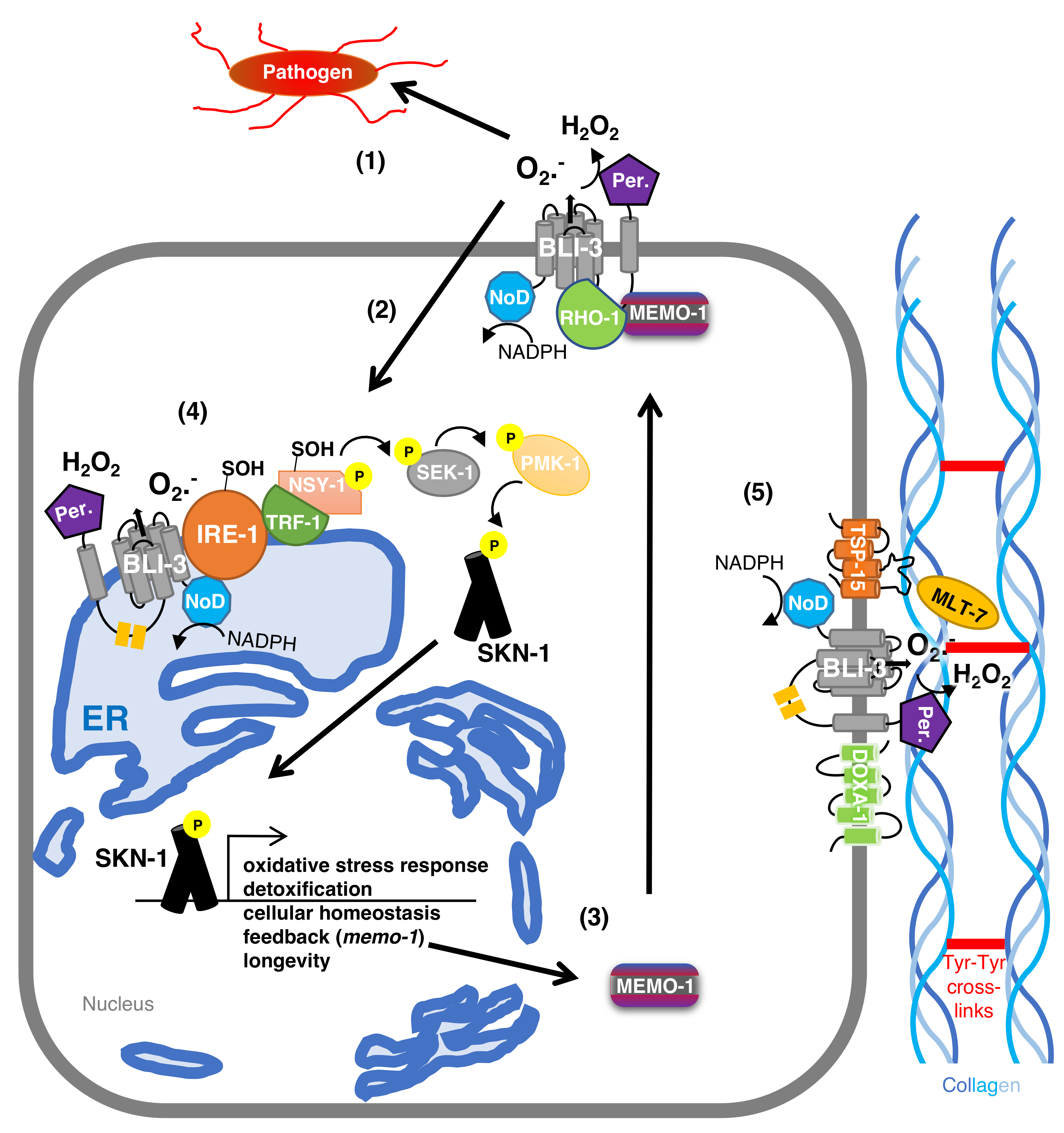
5. BLI-3-Generated Hydrogen Peroxide Catalyzes Collagen Crosslinking
6. The Protective Role of BLI-3 in Pathogen Defense
7. BLI-3-Generated ROS as a Signaling Molecule to Activate the Oxidative Stress Response
8. BLI-3-Generated ROS Redox Signaling to SKN-1 to Promote Longevity
9. The Role of NADPH Oxidases during Mammalian Aging
10. Implication of NADPH Oxidases in Human Aging
11. Conclusive Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristow, M. Unraveling the truth about antioxidants: Mitohormesis explains ROS-induced health benefits. Nat. Med. 2014, 20, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst. Rev. 2012, 336, CD007176. [Google Scholar]
- Pan, Y.; Schroeder, E.A.; Ocampo, A.; Barrientos, A.; Shadel, G.S. Regulation of Yeast Chronological Life Spanby TORC1 via Adaptive Mitochondrial ROS Signaling. Cell Metab. 2011, 13, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, A.; Weinberger, M.; Silva, A.; Sampaio-Marques, B.; Almeida, B.; Leão, C.; Costa, V.; Rodrigues, F.; Burhans, W.C.; Ludovico, P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc. Natl. Acad. Sci. USA 2010, 107, 15123–15128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, E.A.; Raimundo, N.; Shadel, G.S. Epigenetic Silencing Mediates Mitochondria Stress-Induced Longevity. Cell Metab. 2013, 17, 954–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht, S.C.; Barata, A.G.; Grosshans, J.; Teleman, A.A.; Dick, T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Doonan, R.; McElwee, J.J.; Matthijssens, F.; Walker, G.A.; Houthoofd, K.; Back, P.; Matscheski, A.; Vanfleteren, J.R.; Gems, D. Against the oxidative damage theory of aging: Superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008, 22, 3236–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Schmeisser, S.; Priebe, S.; Groth, M.; Monajembashi, S.; Hemmerich, P.; Guthke, R.; Platzer, M.; Ristow, M. Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab. 2013, 2, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Hwang, A.B.; Kenyon, C.J. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Hourihan, J.M.; Bland, M.S.; Obieglo, C.; Katic, I.; Moronetti Mazzeo, L.E.; Alcedo, J.; Blackwell, T.K.; Hynes, N.E. NADPH oxidase-mediated redox signaling promotes oxidative stress resistance and longevity through memo-1 in C. elegans. Elife 2017, 6, 819. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hekimi, S. Early mitochondrial dysfunction in long-lived Mclk1+/− mice. J. Biol. Chem. 2008, 283, 26217–26227. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jiang, N.; Hughes, B.; Bigras, E.; Shoubridge, E.; Hekimi, S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: Loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005, 19, 2424–2434. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Vizuete, A.; Veal, E.A. Caenorhabditis elegans as a model for understanding ROS function in physiology and disease. Redox Biol. 2017, 11, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noël-Hudson, M.S.; Dème, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cDNAs. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.E.; van Trotsenburg, A.S.P.; Baas, F.; de Vijlder, J.J.M.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Heinecke, J.W.; Shapiro, B.M. The respiratory burst oxidase of fertilization. A physiological target for regulation by protein kinase C. J. Biol. Chem. 1992, 267, 7959–7962. [Google Scholar] [PubMed]
- Anh, N.T.T.; Nishitani, M.; Harada, S.; Yamaguchi, M.; Kamei, K. Essential role of Duox in stabilization of Drosophila wing. J. Biol. Chem. 2011, 286, 33244–33251. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Molina-Cruz, A.; Gupta, L.; Rodrigues, J.; Barillas-Mury, C. A peroxidase/dual oxidase system modulates midgut epithelial immunity in Anopheles gambiae. Science 2010, 327, 1644–1648. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.-H. Tissue distribution and putative physiological function of NOX family NADPH oxidases. Jpn. J. Infect. Dis. 2004, 57, S28–S29. [Google Scholar] [PubMed]
- Ha, E.-M.; Oh, C.-T.; Bae, Y.S.; Lee, W.-J. A direct role for dual oxidase in Drosophila gut immunity. Science 2005, 310, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Lee, W.-J. Role of DUOX in gut inflammation: Lessons from Drosophila model of gut-microbiota interactions. Front. Cell. Infect. Microbiol. 2014, 3, 116. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.V.; Crawford, K.C.; Pullin, L.M.; Hall, C.J.; Crosier, K.E.; Crosier, P.S. Dual oxidase in the intestinal epithelium of zebrafish larvae has anti-bacterial properties. Biochem. Biophys. Res. Commun. 2010, 400, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Edens, W.A.; Sharling, L.; Cheng, G.; Shapira, R.; Kinkade, J.M.; Lee, T.; Edens, H.A.; Tang, X.; Sullards, C.; Flaherty, D.B.; et al. Tyrosine cross-linking of extracellular matrix is catalyzed by Duox, a multidomain oxidase/peroxidase with homology to the phagocyte oxidase subunit gp91phox. J. Cell Biol. 2001, 154, 879–891. [Google Scholar] [CrossRef] [PubMed]
- McCallum, K.C.; Garsin, D.A. The Role of Reactive Oxygen Species in Modulating the Caenorhabditis elegans Immune Response. PLoS Pathog. 2016, 12, e1005923. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Sasakura, H.; Moribe, H.; Nakano, M.; Ikemoto, K.; Takeuchi, K.; Mori, I. Lifespan extension by peroxidase and dual oxidase-mediated ROS signaling through pyrroloquinoline quinone in C. elegans. J. Cell Sci. 2017, 130, 2631–2643. [Google Scholar] [CrossRef] [PubMed]
- Moribe, H.; Konakawa, R.; Koga, D.; Ushiki, T.; Nakamura, K.; Mekada, E. Tetraspanin is required for generation of reactive oxygen species by the dual oxidase system in Caenorhabditis elegans. PLoS Genet. 2012, 8, e1002957. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Ortiz de Montellano, P.R. Caenorhabditis elegans and human dual oxidase 1 (DUOX1) “peroxidase” domains: Insights into heme binding and catalytic activity. J. Biol. Chem. 2009, 284, 18634–18643. [Google Scholar] [CrossRef] [PubMed]
- Van der Hoeven, R.; Cruz, M.R.; Chávez, V.; Garsin, D.A. Localization of the Dual Oxidase BLI-3 and Characterization of Its NADPH Oxidase Domain during Infection of Caenorhabditis elegans. PLoS ONE 2015, 10, e0124091. [Google Scholar] [CrossRef] [PubMed]
- Chávez, V.; Mohri-Shiomi, A.; Garsin, D.A. Ce-Duox1/BLI-3 generates reactive oxygen species as a protective innate immune mechanism in Caenorhabditis elegans. Infect. Immun. 2009, 77, 4983–4989. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, L.T.; Pons, C.; Arda, H.E.; Giese, G.E.; Myers, C.L.; Walhout, A.J.M. Transcription Factor Activity Mapping of a Tissue-Specific in vivo Gene Regulatory Network. Cell Syst. 2015, 1, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Barreiro, O.; Gordon-Alonso, M.; Sala-Valdés, M.; Sánchez-Madrid, F. Tetraspanin-enriched microdomains: A functional unit in cell plasma membranes. Trends Cell Biol. 2009, 19, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef] [PubMed]
- The cuticle. Available online: www.wormbook.org/chapters/www_cuticle/cuticle.html (accessed on 9 September 2018).
- Kage-Nakadai, E.; Imae, R.; Suehiro, Y.; Yoshina, S.; Hori, S.; Mitani, S. A conditional knockout toolkit for Caenorhabditis elegans based on the Cre/loxP recombination. PLoS ONE 2014, 9, e114680. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Brandman, R.; Ortiz de Montellano, P.R. Perturbed heme binding is responsible for the blistering phenotype associated with mutations in the Caenorhabditis elegans dual oxidase 1 (DUOX1) peroxidase domain. J. Biol. Chem. 2010, 285, 40991–41000. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Cox, G.N.; Kusch, M.; Edgar, R.S. Cuticle of Caenorhabditis elegans: Its isolation and partial characterization. J. Cell Biol. 1981, 90, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Norman, K.R.; Moerman, D.G. The let-268 locus of Caenorhabditis elegans encodes a procollagen lysyl hydroxylase that is essential for type IV collagen secretion. Dev. Biol. 2000, 227, 690–705. [Google Scholar] [CrossRef] [PubMed]
- Thein, M.C.; Winter, A.D.; Stepek, G.; McCormack, G.; Stapleton, G.; Johnstone, I.L.; Page, A.P. Combined extracellular matrix cross-linking activity of the peroxidase MLT-7 and the dual oxidase BLI-3 is critical for post-embryonic viability in Caenorhabditis elegans. J. Biol. Chem. 2009, 284, 17549–17563. [Google Scholar] [CrossRef] [PubMed]
- Benedetto, A.; Au, C.; Avila, D.S.; Milatovic, D.; Aschner, M. Extracellular dopamine potentiates Mn-induced oxidative stress, lifespan reduction, and dopaminergic neurodegeneration in a BLI-3-dependent manner in Caenorhabditis elegans. PLoS Genet. 2010, 6, e1001084. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Luo, J.; Li, Y.; Ma, L. The BLI-3/TSP-15/DOXA-1 dual oxidase complex is required for iodide toxicity in Caenorhabditis elegans. G3 2014, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Degroot, L.J.; Niepomniszcze, H. Biosynthesis of thyroid hormone: Basic and clinical aspects. Metab. Clin. Exp. 1977, 26, 665–718. [Google Scholar] [CrossRef]
- Xiao, N.; Venton, B.J. Characterization of dopamine releasable and reserve pools in Drosophila larvae using ATP/P2X2-mediated stimulation. J. Neurochem. 2015, 134, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Hoeven, R.V.D.; McCallum, K.C.; Cruz, M.R.; Garsin, D.A. Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog. 2011, 7, e1002453. [Google Scholar]
- Tang, H.; Pang, S. Proline Catabolism Modulates Innate Immunity in Caenorhabditis elegans. Cell Rep. 2016, 17, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Yun, M.; Politz, S.M.; Rao, R.P. A pathogenesis assay using Saccharomyces cerevisiae and Caenorhabditis elegans reveals novel roles for yeast AP-1, Yap1, and host dual oxidase BLI-3 in fungal pathogenesis. Eukaryot. Cell 2009, 8, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Pastor, K.; Gonzalez, A.Y.; Lorenz, M.C.; Rao, R.P. The role of Candida albicans AP-1 protein against host derived ROS in in vivo models of infection. Virulence 2013, 4, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Pujol, N.; Cypowyj, S.; Ziegler, K.; Millet, A.; Astrain, A.; Goncharov, A.; Jin, Y.; Chisholm, A.D.; Ewbank, J.J. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. CURBIO 2008, 18, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.-G.; Tu, Q.; Niu, J.; Ji, X.-L.; Zhang, K.-Q. The DAF-16/FOXO transcription factor functions as a regulator of epidermal innate immunity. PLoS Pathog. 2013, 9, e1003660. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-A.; Kim, S.-H.; Kim, E.-K.; Ha, E.-M.; You, H.; Kim, B.; Kim, M.-J.; Kwon, Y.; Ryu, J.-H.; Lee, W.-J. Bacterial-derived uracil as a modulator of mucosal immunity and gut-microbe homeostasis in Drosophila. Cell 2013, 153, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Tiller, G.R.; Garsin, D.A. The SKPO-1 peroxidase functions in the hypodermis to protect Caenorhabditis elegans from bacterial infection. Genetics 2014, 197, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.-C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Glutathione metabolism and its selective modification. J. Biol. Chem. 1988, 263, 17205–17208. [Google Scholar] [PubMed]
- Romero-Aristizabal, C.; Marks, D.S.; Fontana, W.; Apfeld, J. Regulated spatial organization and sensitivity of cytosolic protein oxidation in Caenorhabditis elegans. Nat. Commun. 2014, 5, 5020. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.K.; Steinbaugh, M.J.; Hourihan, J.M.; Ewald, C.Y.; Isik, M. SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegans. Free Radic. Biol. Med. 2015, 88, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-J.; Sun, Z.; Chen, W.; Li, Y.; Villeneuve, N.F.; Zhang, D.D. Activation of Nrf2 by arsenite and monomethylarsonous acid is independent of Keap1-C151: Enhanced Keap1-Cul3 interaction. Toxicol. Appl. Pharmacol. 2008, 230, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Teuscher, A.C.; Ewald, C.Y. Overcoming Autofluorescence to Assess GFP Expression during Normal Physiology and Aging in Caenorhabditis elegans. BIO-PROTOCOL 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Hourihan, J.M.; Blackwell, T.K. Oxidative Stress Assays (arsenite and tBHP) in Caenorhabditis elegans. BIO-PROTOCOL 2017, 7, e2365. [Google Scholar] [CrossRef] [PubMed]
- An, J.H.; Blackwell, T.K. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev. 2003, 17, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B. The molecular hug between the ER and the mitochondria. Curr. Opin. Cell Biol. 2013, 25, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Durai, S.; Singh, N.; Kundu, S.; Balamurugan, K. Proteomic investigation of Vibrio alginolyticus challenged Caenorhabditis elegans revealed regulation of cellular homeostasis proteins and their role in supporting innate immune system. Proteomics 2014, 14, 1820–1832. [Google Scholar] [CrossRef] [PubMed]
- Glover-Cutter, K.M.; Lin, S.; Blackwell, T.K. Integration of the unfolded protein and oxidative stress responses through SKN-1/Nrf. PLoS Genet. 2013, 9, e1003701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol, N.; Link, E.M.; Liu, L.X.; Kurz, C.L.; Alloing, G.; Tan, M.W.; Ray, K.P.; Solari, R.; Johnson, C.D.; Ewbank, J.J. A reverse genetic analysis of components of the Toll signaling pathway in Caenorhabditis elegans. CURBIO 2001, 11, 809–821. [Google Scholar]
- Kim, D.H.; Feinbaum, R.; Alloing, G.; Emerson, F.E.; Garsin, D.A.; Inoue, H.; Tanaka-Hino, M.; Hisamoto, N.; Matsumoto, K.; Tan, M.-W.; et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 2002, 297, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Kato, K.; Hayakawa, R.; Hisamoto, N.; Matsumoto, K.; Takeda, K.; Ichijo, H. Regulation of anoxic death in Caenorhabditis elegans by mammalian apoptosis signal-regulating kinase (ASK) family proteins. Genetics 2011, 187, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Hordijk, P.L. Regulation of NADPH oxidases: The role of Rac proteins. Circ. Res. 2006, 98, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Landis, J.N.; Porter Abate, J.; Murphy, C.T.; Blackwell, T.K. Dauer-independent insulin/IGF-1-signalling implicates collagen remodelling in longevity. Nature 2015, 519, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Won, H.Y.; Bae, M.A.; Hong, J.-H.; Hwang, E.S. Spontaneous and aging-dependent development of arthritis in NADPH oxidase 2 deficiency through altered differentiation of CD11b+ and Th/Treg cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9548–9553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.-R.; Lazarenko, O.P.; Blackburn, M.L.; Mercer, K.E.; Badger, T.M.; Ronis, M.J.J. p47phox-Nox2-dependent ROS Signaling Inhibits Early Bone Development in Mice but Protects against Skeletal Aging. J. Biol. Chem. 2015, 290, 14692–14704. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-H.; Lee, J. A Mini-Review of the NADPH oxidases in Vascular Dementia: Correlation with NOXs and Risk Factors for VaD. Int. J. Mol. Sci. 2017, 18, 2500. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.W.; Wang, J.; Zhang, Q.; Wang, R.; Dhandapani, K.M.; Vadlamudi, R.K.; Brann, D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017, 12, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Zhang, J.; Walker, S.J.; Dworakowski, R.; Lakatta, E.G.; Shah, A.M. Involvement of NADPH oxidase in age-associated cardiac remodeling. J. Mol. Cell. Cardiol. 2010, 48, 765–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Kwak, H.-B.; Hord, J.; Kim, J.-H.; Lawler, J.M. Exercise training attenuates age-dependent elevation of angiotensin II type 1 receptor and Nox2 signaling in the rat heart. Exp. Gerontol. 2015, 70, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Mahfouf, W.; Serrano-Sanchez, M.; Raad, H.; Harfouche, G.; Bonneu, M.; Claverol, S.; Mazurier, F.; Rossignol, R.; Taieb, A.; et al. Premature skin aging features rescued by inhibition of NADPH oxidase activity in XPC-deficient mice. J. Investig. Dermatol. 2015, 135, 1108–1118. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Matsushima, S.; Kuroda, J.; Zablocki, D.; Kitazono, T.; Sadoshima, J. The NADPH oxidase Nox4 and aging in the heart. Aging 2010, 2, 1012–1016. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar] [CrossRef] [PubMed]
- Ago, T.; Kuroda, J.; Pain, J.; Fu, C.; Li, H.; Sadoshima, J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ. Res. 2010, 106, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Lener, B.; Kozieł, R.; Pircher, H.; Hütter, E.; Greussing, R.; Herndler-Brandstetter, D.; Hermann, M.; Unterluggauer, H.; Jansen-Dürr, P. The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem. J. 2009, 423, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezende, F.; Schürmann, C.; Schütz, S.; Harenkamp, S.; Herrmann, E.; Seimetz, M.; Weissmann, N.; Schröder, K. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox Biol. 2017, 11, 312–314. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, A.; Hadzić, N.; Morgan, G.; Strobel, S.; Levinsky, R.J. Prognosis of chronic granulomatous disease. Arch. Dis. Child. 1990, 65, 942–945. [Google Scholar] [CrossRef] [PubMed]
- Kayaaltı, Z.; Kaya, D.; Bacaksız, A.; Söylemez, E.; Söylemezoğlu, T. An association between polymorphism of the NADH/NADPH oxidase p22phox (phagocyte oxidase) subunit and aging in Turkish population. Aging Clin. Exp. Res. 2013, 25, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-I.; Na, J.-E.; Kang, S.Y.; Cho, Y.-S.; Choi, D.-J.; Kim, C.-H.; Kim, H.-S.; Oh, B.-H.; Choi, Y.-H.; Kwon, I.S.; et al. Impact of NAD(P)H oxidase p22 phox gene polymorphism on vascular aging in Korean centenarian and nonagenarian. Int. J. Cardiol. 2007, 123, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.; Ferreira-Sae, M.C.; Ronchi, J.A.; Pio-Magalhães, J.A.; Cipolli, J.A.; Matos-Souza, J.R.; Mill, J.G.; Vercesi, A.E.; Krieger, J.E.; Franchini, K.G.; et al. The C242T polymorphism of the p22-phox gene (CYBA) is associated with higher left ventricular mass in Brazilian hypertensive patients. BMC Med. Genet. 2011, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Ferrell, R.E.; Park, J.-J.; Hagberg, J.M.; Phares, D.A.; Jones, J.M.; Brown, M.D. NADPH oxidase p22phox gene variants are associated with systemic oxidative stress biomarker responses to exercise training. J. Appl. Physiol. 2005, 99, 1905–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muthusamy, V.R.; Kannan, S.; Sadhaasivam, K.; Gounder, S.S.; Davidson, C.J.; Boeheme, C.; Hoidal, J.R.; Wang, L.; Rajasekaran, N.S. Acute exercise stress activates Nrf2/ARE signaling and promotes antioxidant mechanisms in the myocardium. Free Radic. Biol. Med. 2012, 52, 366–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal. 2014, 20, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C. Serendipity in search for longevity from experiences of Hansen people. Transl. Med. Aging 2017, 1, 12–17. [Google Scholar] [CrossRef]
- Cho, S.C.; Rhim, J.-H.; Son, Y.H.; Lee, S.J.; Park, S.C. Suppression of ROS generation by 4,4-diaminodiphenylsulfone in non-phagocytic human diploid fibroblasts. Exp. Mol. Med. 2010, 42, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.C.; Park, M.C.; Keam, B.; Choi, J.M.; Cho, Y.; Hyun, S.; Park, S.C.; Lee, J. DDS, 4,4′-diaminodiphenylsulfone, extends organismic lifespan. Proc. Natl. Acad. Sci. USA 2010, 107, 19326–19331. [Google Scholar] [CrossRef] [PubMed]
- Shore, D.E.; Ruvkun, G. A cytoprotective perspective on longevity regulation. Trends Cell Biol. 2013, 23, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ewald, C.Y. Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging. Antioxidants 2018, 7, 130. https://doi.org/10.3390/antiox7100130
Ewald CY. Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging. Antioxidants. 2018; 7(10):130. https://doi.org/10.3390/antiox7100130
Chicago/Turabian StyleEwald, Collin Y. 2018. "Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging" Antioxidants 7, no. 10: 130. https://doi.org/10.3390/antiox7100130
APA StyleEwald, C. Y. (2018). Redox Signaling of NADPH Oxidases Regulates Oxidative Stress Responses, Immunity and Aging. Antioxidants, 7(10), 130. https://doi.org/10.3390/antiox7100130