The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling


Abstract
:1. Introduction
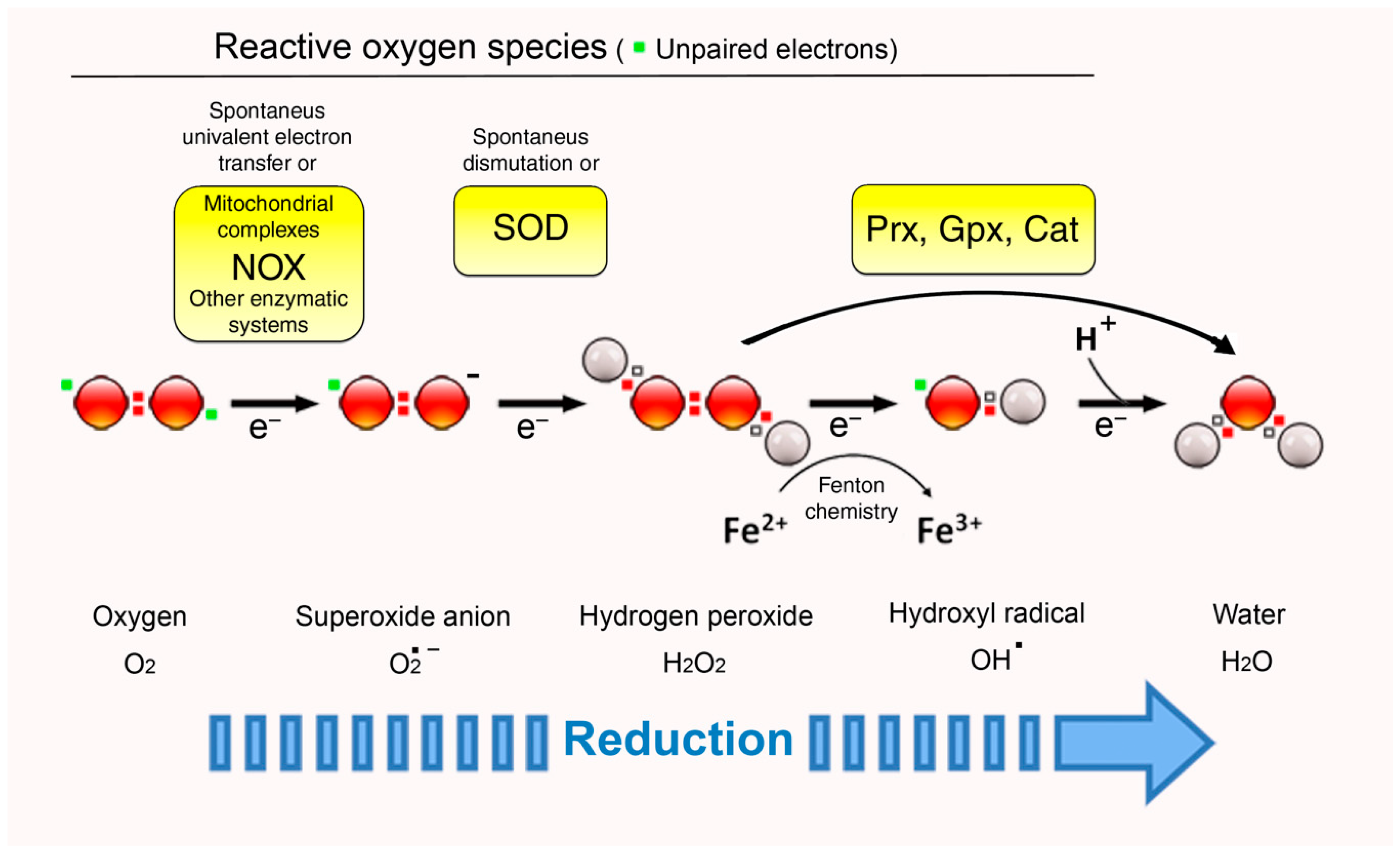
2. ROS: Signals, Second Messengers, or Simply Foes?
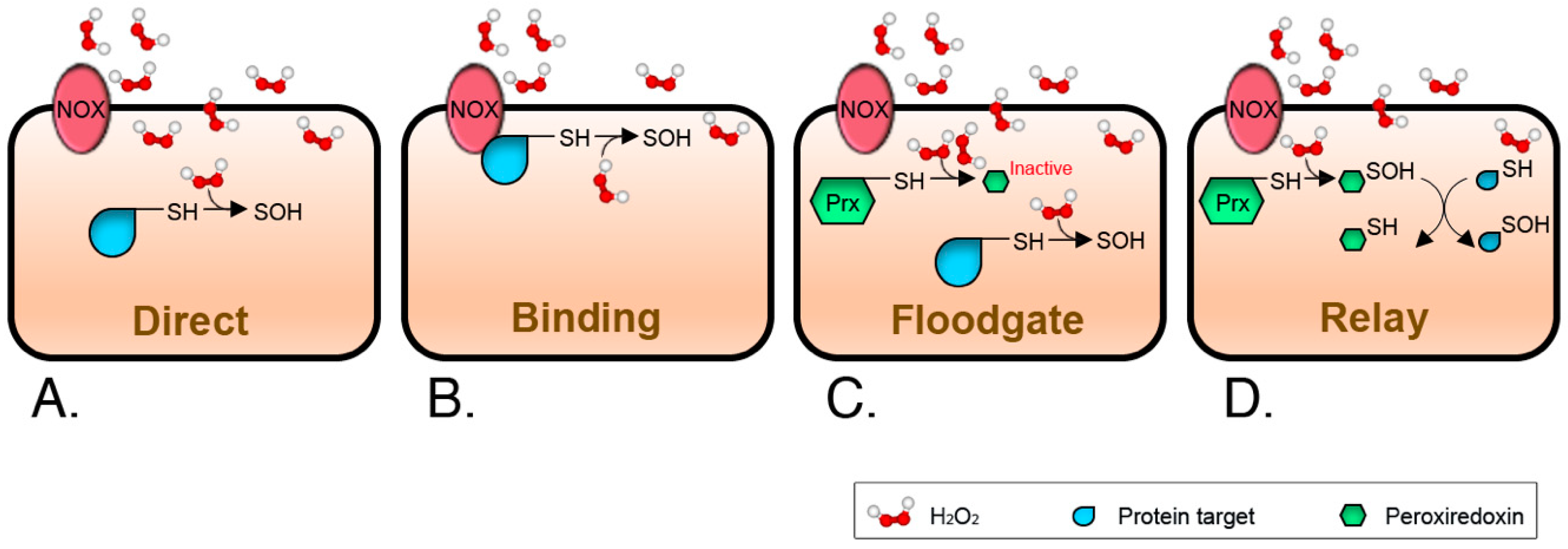
3. Signal Thiol Oxidations Mediated by Hydrogen Peroxide
4. The Plasma Membrane as a Platform for Redox Signal Transmission
4.1. The Plasma Membrane: More Than Lipids
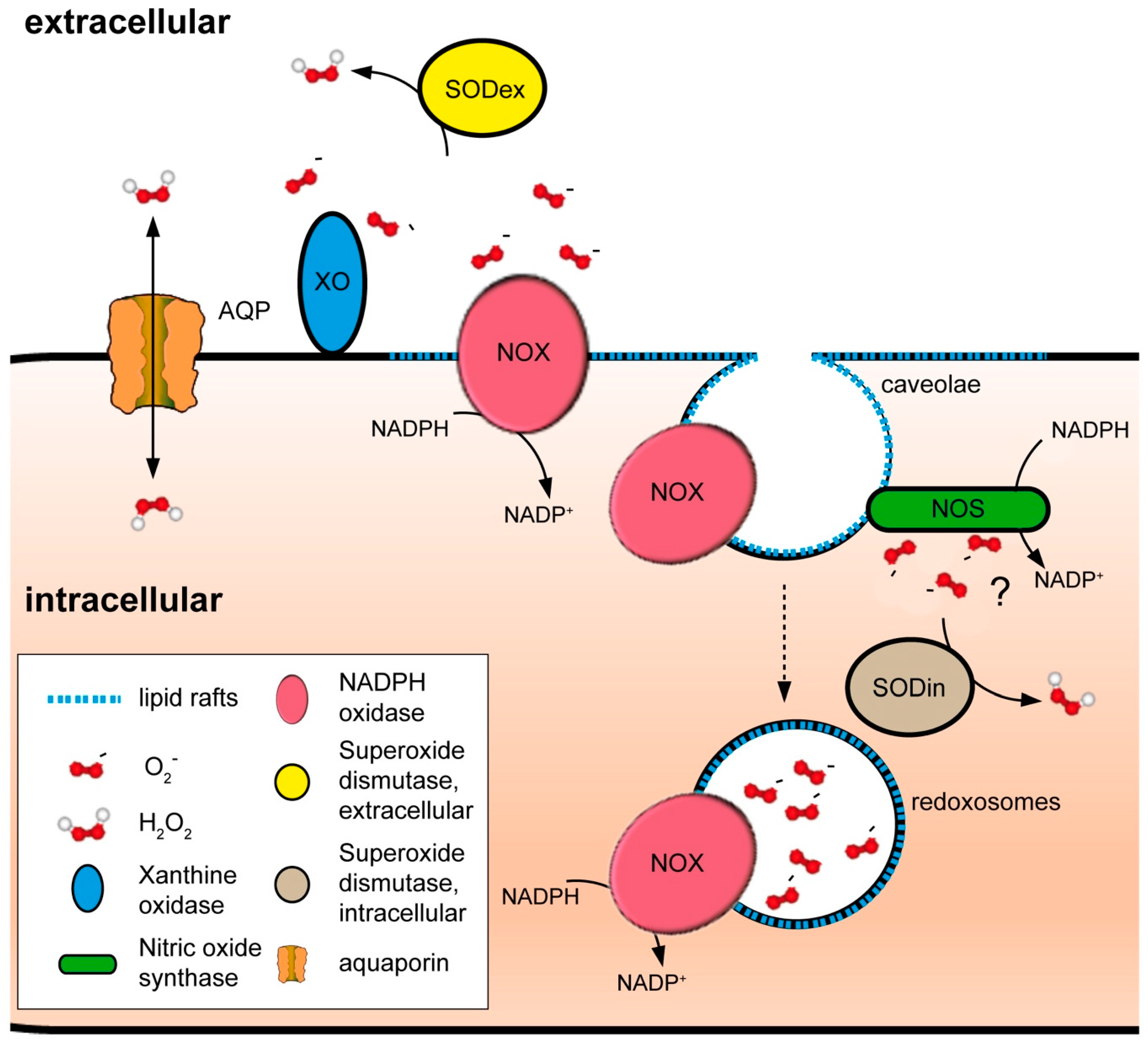
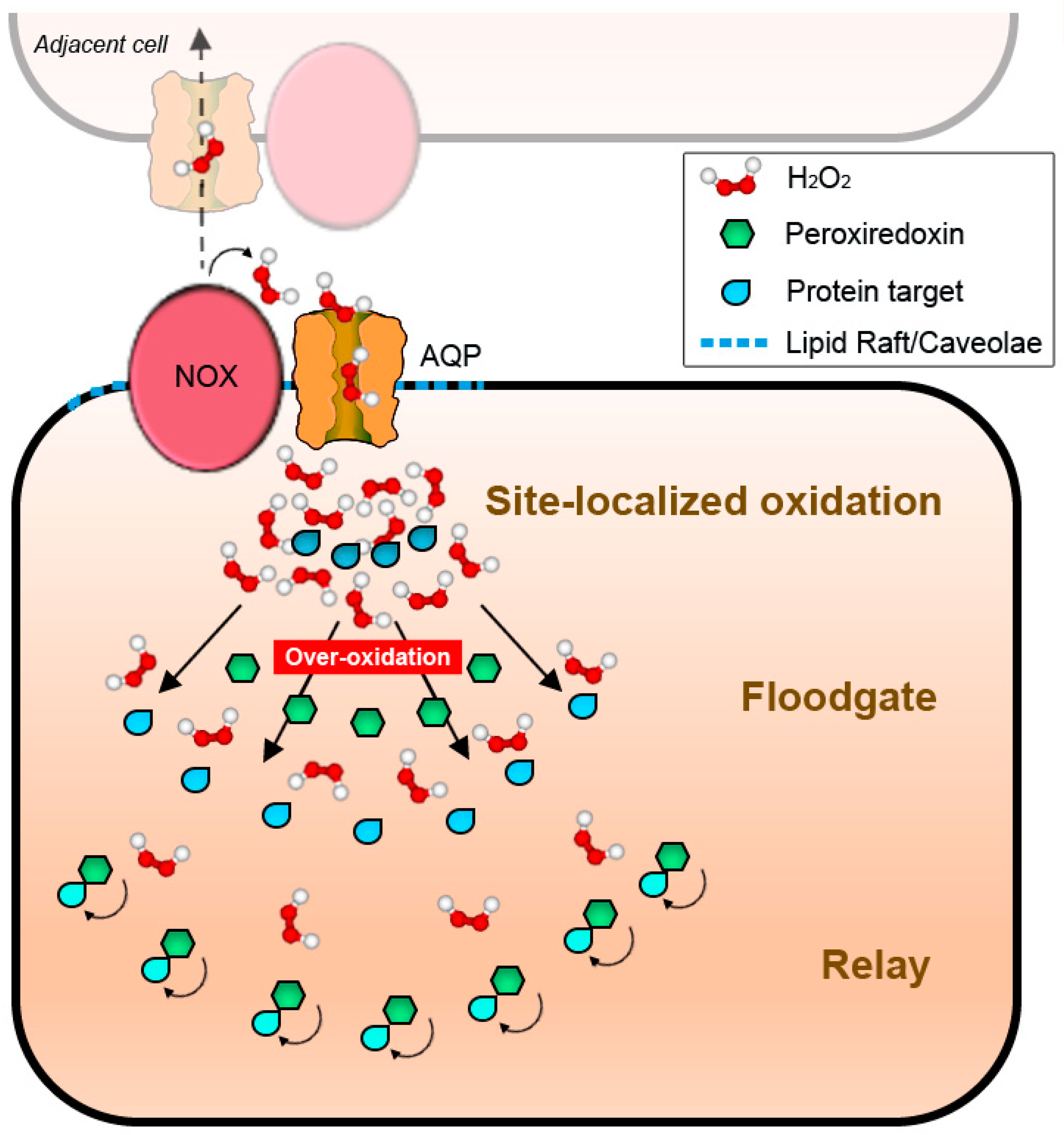
4.2. NADPH Oxidases and Peroxiporins as a Generator–Facilitator System on the Plasma Membrane
4.3. Other Sources of ROS at the Plasma Membrane
5. Dysregulation of Redox Signal Transmission through the Plasma Membrane: Impact on Disease
5.1. Immune System Diseases, NADPH Oxidases, and Peroxiporins
5.2. Inflammatory Bowel Disease, NADPH Oxidases, and Peroxiporins
5.3. Neurodegenerative Diseases, NADPH Oxidases, and Peroxiporins
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rall, T.W.; Sutherland, E.W.; Berthet, J. The relationship of epinephrine and glucagon to liver phosphorylase. J. Biol. Chem. 1957, 224, 459–468. [Google Scholar]
- Bassler, J.; Schultz, J.E.; Lupas, A.N. Adenylate cyclases: Receivers, transducers, and generators of signals. Cell Signal. 2018, 46, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol. Cell. Proteom. MCP 2013, 12, 3498–3508. [Google Scholar] [CrossRef] [PubMed]
- Cary, S.P.; Winger, J.A.; Derbyshire, E.R.; Marletta, M.A. Nitric oxide signaling: No longer simply on or off. Trends Biochem. Sci. 2006, 31, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Landry, W.D.; Cotter, T.G. ROS signalling, NADPH oxidases and cancer. Biochem. Soc. Trans. 2014, 42, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. (Eds.) Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Superoxide and iron: Partners in crime. IUBMB Life 1999, 48, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.G.; Banerjee, R. Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanas’ev, I. Mechanisms of superoxide signaling in epigenetic processes: Relation to aging and cancer. Aging Dis. 2015, 6, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fridovich, I. Superoxide dismutase: A comparison of rate constants. Arch. Biochem. Biophys. 1973, 158, 396–400. [Google Scholar] [CrossRef]
- Hancock, J.T.; Whiteman, M. Cellular Redox Environment and Its Influence on Redox Signaling Molecules. React. Oxyg Spec. 2018, 5, 78–85. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Yim, M.B.; Chock, P.B.; Stadtman, E.R. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1990, 87, 5006–5010. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [PubMed]
- Medraño-Fernandez, I.; Bestetti, S.; Bertolotti, M.; Bienert, G.P.; Bottino, C.; Laforenza, U.; Rubartelli, A.; Sitia, R. Stress Regulates Aquaporin-8 Permeability to Impact Cell Growth and Survival. Antioxid. Redox Signal. 2016, 24, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imlay, J.A. Pathways of oxidative damage. Ann. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Berkowitz, G. Hydrogen peroxide, a messenger with too many roles? Redox Rep. Commun. Free Radic. Res. 2001, 6, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, A.V.; Matveyeva, N.P.; Yermakov, I.P. Reactive oxygen species are involved in regulation of pollen wall cytomechanics. Plant Biol. 2014, 16, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nagy, K.; Pasti, G.; Bene, L.; Zs-Nagy, I. Induction of granulocytic maturation in HL-60 human leukemia cells by free radicals: A hypothesis of cell differentiation involving hydroxyl radicals. Free Radic. Res. Commun. 1993, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Andriollo, M.; Guiraud, P.; Belhoussine, R.; Jeannesson, P. Oxidative stress involvement in chemically induced differentiation of K562 cells. Free Radic. Biol. Med. 2000, 28, 18–27. [Google Scholar] [CrossRef]
- Nagy, I.Z. On the true role of oxygen free radicals in the living state, aging, and degenerative disorders. Ann. N. Y. Acad. Sci. 2001, 928, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Putker, M.; Vos, H.R.; van Dorenmalen, K.; de Ruiter, H.; Duran, A.G.; Snel, B.; Burgering, B.M.T.; Vermeulen, M.; Dansen, T.B. Evolutionary Acquisition of Cysteines Determines FOXO Paralog-Specific Redox Signaling. Antioxid. Redox Signal. 2015, 22, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.M.; Gladyshev, V.N. Proteomics: Mapping reactive cysteines. Nat. Chem. Biol. 2011, 7, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Ann. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Hecht, H.J.; Flohe, L. Peroxiredoxins. Biol. Chem. 2002, 383, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Babbush, R.; Lebrero, M.C.; Trujillo, M.; Radi, R.; Estrin, D.A. Molecular basis of the mechanism of thiol oxidation by hydrogen peroxide in aqueous solution: Challenging the SN2 paradigm. Chem. Res. Toxicol. 2012, 25, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Allison, W.S. Formation and reactions of sulfenic acids in proteins. Acc. Chem. Res. 1976, 9, 293–299. [Google Scholar] [CrossRef]
- Lo Conte, M.; Carroll, K.S. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.R. Chemistry of sulphenic acids and esters. In Sulfenic Acids and Derivatives; Patai, S., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1990; pp. 361–402. [Google Scholar]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Winter, J. The physiological role of reversible methionine oxidation. Biochim. Biophys. Acta 2014, 1844, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. Off. J. Jpn. Circ. Soc. 2008, 72, 1–16. [Google Scholar] [CrossRef]
- DeYulia, G.J., Jr.; Carcamo, J.M.; Borquez-Ojeda, O.; Shelton, C.C.; Golde, D.W. Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 5044–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R.; Rubbo, H.; Bush, K.; Freeman, B.A. Xanthine oxidase binding to glycosaminoglycans: Kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch. Biochem. Biophys. 1997, 339, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C.; Barber, C.M.; Lynch, K.R. Mutation of N-myristoylation site converts endothelial cell nitric oxide synthase from a membrane to a cytosolic protein. Circ. Res. 1993, 72, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Januel, C.; El Hentati, F.Z.; Carreras, M.; Arthur, J.R.; Calzada, C.; Lagarde, M.; Vericel, E. Phospholipid-hydroperoxide glutathione peroxidase (GPx-4) localization in resting platelets, and compartmental change during platelet activation. Biochim. Biophys. Acta 2006, 1761, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int.J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.E. Catalase: An old enzyme with a new role? Can. J. Biochem. Cell Biol. 1984, 62, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.Y.; Engelhardt, J.F. The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies. Biochemistry 2014, 53, 1551–1564. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J.; Nicholson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tillman, T.S.; Cascio, M. Effects of membrane lipids on ion channel structure and function. Cell Biochem. Biophys. 2003, 38, 161–190. [Google Scholar] [CrossRef]
- Caliceti, C.; Zambonin, L.; Rizzo, B.; Fiorentini, D.; Vieceli Dalla Sega, F.; Hrelia, S.; Prata, C. Role of plasma membrane caveolae/lipid rafts in VEGF-induced redox signaling in human leukemia cells. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, Y.; Yi, F. Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid. Redox Signal. 2007, 9, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Zhang, Y.; Li, X.; Gulbins, E. Ceramide: A novel player in reactive oxygen species-induced signaling? Antioxid. Redox Signal. 2007, 9, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Kim, Y.S.; Liu, Z. Lipid rafts and oxidative stress-induced cell death. Antioxid. Redox Signal. 2007, 9, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Volonte, D.; Engelman, J.A.; Galbiati, F.; Mehta, P.; Zhang, X.L.; Scherer, P.E.; Lisanti, M.P. Crowded little caves: Structure and function of caveolae. Cell Signal. 1998, 10, 457–463. [Google Scholar] [CrossRef]
- Parton, R.G. Caveolae and caveolins. Curr. Opin. Cell Biol. 1996, 8, 542–548. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Mastick, C.C. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal. 2003, 15, 289–298. [Google Scholar] [CrossRef]
- Li, S.; Seitz, R.; Lisanti, M.P. Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J. Biol. Chem. 1996, 271, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.B.; Li, S.M.; Qian, X.X.; Moon, C.; Zheng, J. Tyrosine phosphorylation of caveolin 1 by oxidative stress is reversible and dependent on the c-src tyrosine kinase but not mitogen-activated protein kinase pathways in placental artery endothelial cells. Biol. Reprod. 2005, 73, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Insel, P.A. Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxid. Redox Signal. 2009, 11, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Grande-García, A.; Echarri, A.; de Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; del Pozo, M.A. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Volonte, D.; Zhang, K.; Lisanti, M.P.; Galbiati, F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol. Biol. Cell 2002, 13, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Rungtabnapa, P.; Nimmannit, U.; Halim, H.; Rojanasakul, Y.; Chanvorachote, P. Hydrogen peroxide inhibits non-small cell lung cancer cell anoikis through the inhibition of caveolin-1 degradation. Am. J. Physiol. Cell Physiol. 2011, 300, C235–C245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.S.; Ko, Y.G.; Seo, J.S. Caveolin internalization by heat shock or hyperosmotic shock. Exp. Cell Res. 2000, 255, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhou, F.; Katirai, F.; Li, P.L. Lipid raft redox signaling: Molecular mechanisms in health and disease. Antioxid. Redox Signal. 2011, 15, 1043–1083. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F. Signaling components of redox active endosomes: The redoxosomes. Antioxid. Redox Signal. 2009, 11, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Filali, M.; Huss, G.J.; Stanic, B.; Chamseddine, A.; Barna, T.J.; Lamb, F.S. Cytokine activation of nuclear factor ĸB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 2007, 101, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.; Marden, J.J.; Banfi, B.; Engelhardt, J.F. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem. J. 2008, 411, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Tosoni, D.; Comai, R.; Rabellino, A.; Segat, D.; Caneva, F.; Luzzi, P.; Di Fiore, P.P.; Tacchetti, C. Relationships between EGFR signaling-competent and endocytosis-competent membrane microdomains. Mol. Biol. Cell 2005, 16, 2704–2718. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.V.; Lamaze, C.; Schmid, S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996, 274, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pennock, S.; Chen, X.; Wang, Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol. Cell. Biol. 2002, 22, 7279–7290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pennock, S.D.; Chen, X.; Kazlauskas, A.; Wang, Z. Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J. Biol. Chem. 2004, 279, 8038–8046. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Farrow, M.A.; Chumbler, N.M.; Lapierre, L.A.; Franklin, J.L.; Rutherford, S.A.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 18674–18679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Fan, L.; Chen, T.; Li, R.; Li, X.; He, Q.; Botella, M.A.; Lin, J. Clathrin and membrane microdomains cooperatively regulate RbohD dynamics and activity in Arabidopsis. Plant. Cell 2014, 26, 1729–1745. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Phosphoinositide recognition domains. Traffic 2003, 4, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Lambeth, J.D. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 2004, 279, 4737–4742. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Virbasius, J.V.; Song, X.; Pomerleau, D.P.; Zhou, G.W. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem. 2002, 277, 4512–4518. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Lambeth, J.D. Phosphatidylinositol (4,5)-bisphosphate modulates Nox5 localization via an N-terminal polybasic region. Mol. Biol. Cell 2008, 19, 4020–4031. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J. Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, L.; Jope, R.S. Cholinergic stimulation of AP-1 and NF kappa B transcription factors is differentially sensitive to oxidative stress in SH-SY5Y neuroblastoma: Relationship to phosphoinositide hydrolysis. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 5914–5922. [Google Scholar] [CrossRef]
- van den Bout, I.; Jones, D.R.; Shah, Z.H.; Halstead, J.R.; Keune, W.J.; Mohammed, S.; D’Santos, C.S.; Divecha, N. Collaboration of AMPK and PKC to induce phosphorylation of Ser413 on PIP5K1B resulting in decreased kinase activity and reduced PtdIns(4,5)P2 synthesis in response to oxidative stress and energy restriction. Biochem. J. 2013, 455, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.P. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J. Biol. Chem. 2011, 286, 32444–32453. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Wied, S.; Walz, N.; Jung, C. Translocation of glycosylphosphatidylinositol-anchored proteins from plasma membrane microdomains to lipid droplets in rat adipocytes is induced by palmitate, H2O2, and the sulfonylurea drug glimepiride. Mol. Pharmacol. 2008, 73, 1513–1529. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegold, A.A.; Shatwell, K.P.; Segal, A.W.; Klausner, R.D.; Dancis, A. Intramembrane bis-heme motif for transmembrane electron transport conserved in a yeast iron reductase and the human NADPH oxidase. J. Biol. Chem. 1996, 271, 31021–31024. [Google Scholar] [CrossRef] [PubMed]
- Stasia, M.J. CYBA encoding p22phox, the cytochrome b558 alpha polypeptide: Gene structure, expression, role and physiopathology. Gene 2016, 586, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Sugawara, Y.; Wen, K.; Giulivi, C. Generation of oxygen free radicals in thyroid cells and inhibition of thyroid peroxidase. Exp. Biol. Med. 2002, 227, 141–146. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Ann. Rev.Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells Devoted Mol. Cell. Mech. 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, D.; Diaz, B.; Taulet, N.; Fowler, B.; Courtneidge, S.A.; Bokoch, G.M. Novel p47phox-related organizers regulate NADPH oxidase 1 (Nox1) activity and localization. Sci. Signal. 2010, 2, ra54. [Google Scholar]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function—Role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.L.; Brockman, D.; Campos, B.; Myatt, L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: Involvement in preeclampsia. Placenta 2006, 27, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Murillo, I.; Henderson, L.M. Expression of GP91phox/Nox2 in COS-7 cells: Cellular localization of the protein and the detection of outward proton currents. Biochem. J. 2005, 385, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cruz, A.; Vilchis-Landeros, M.M.; Guinzberg, R.; Villalobos-Molina, R.; Pina, E. Nox2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free Radic. Res. 2011, 45, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef] [PubMed]
- Paffenholz, R.; Bergstrom, R.A.; Pasutto, F.; Wabnitz, P.; Munroe, R.J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J.; et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004, 18, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Chun, Y.S.; Park, K.S.; Kim, S.J.; Choi, S.O.; Kim, H.L.; Park, J.W. Identification of subdomains in NADPH oxidase-4 critical for the oxygen-dependent regulation of TASK-1 K+ channels. Am. J. Physiol. Cell Physiol. 2009, 297, C855–C864. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid. Redox Signal. 2009, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed]
- Forteza, R.; Salathe, M.; Miot, F.; Conner, G.E. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhardt, F.; van Deurs, B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004, 23, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Rizzo, V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H954–H962. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Diebold, B.A. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 2002, 100, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.; DeLeo, F.R.; Gallois, A.; Toyoshima, S.; Suzuki, K.; Nauseef, W.M. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood 1999, 93, 3521–3530. [Google Scholar] [PubMed]
- Pierini, L.M.; Eddy, R.J.; Fuortes, M.; Seveau, S.; Casulo, C.; Maxfield, F.R. Membrane lipid organization is critical for human neutrophil polarization. J. Biol. Chem. 2003, 278, 10831–10841. [Google Scholar] [CrossRef] [PubMed]
- Tall, E.G.; Spector, I.; Pentyala, S.N.; Bitter, I.; Rebecchi, M.J. Dynamics of phosphatidylinositol 4,5-bisphosphate in actin-rich structures. Curr. Biol. 2000, 10, 743–746. [Google Scholar] [CrossRef]
- Grasberger, H.; De Deken, X.; Miot, F.; Pohlenz, J.; Refetoff, S. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol. Endocrinol. 2007, 21, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, H.; Refetoff, S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, M.; Anderson, R.G. Calcium signal transduction from caveolae. Cell Calcium 1999, 26, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Weerth, S.H.; Holtzclaw, L.A.; Russell, J.T. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium 2007, 41, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Kim, B.; You, H.; Lee, W.J. Uracil-induced signaling pathways for DUOX-dependent gut immunity. Fly 2015, 9, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, D.E.; van der Vliet, A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2016, 8, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Mistry, R.K.; Brewer, A.C. Redox regulation of gasotransmission in the vascular system: A focus on angiogenesis. Free Radic. Biol. Med. 2017, 108, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Mkaddem, S.B.; Vandewalle, A. NOX enzymes and Toll-like receptor signaling. Sem. Immunopathol. 2008, 30, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Smith, S.W.; Anderson, B.D. Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J. Pharm. Sci. 2005, 94, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Wadley, A.J.; Aldred, S.; Coles, S.J. An unexplored role for Peroxiredoxin in exercise-induced redox signalling? Redox Biol. 2016, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins at a glance. J. Cell Sci. 2011, 124, 2107–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Bestetti, S.; Garcia-Manteiga, J.M.; Medrano-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine kinase signal modulation: A matter of H2O2 membrane permeability? Antioxid. Redox Signal. 2013, 19, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Steudle, E. Oxidative gating of water channels (aquaporins) in corn roots. Plant Cell Environ. 2006, 29, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampilogova, Y.N.; Zhestkova, I.M.; Trofimova, M.S. Redox modulation of osmotic water permeability in plasma membranes isolated from roots and shoots of pea seedlings. Russ. J. Plant. Physiol. 2006, 53, 622–628. [Google Scholar] [CrossRef]
- Henzler, T.; Ye, Q.; Steudle, E. Oxidative gating of water channels (aquaporins) in Chara by hydroxyl radicals. Plant. Cell Environ. 2004, 27, 1184–1195. [Google Scholar] [CrossRef]
- Dynowski, M.; Schaaf, G.; Loque, D.; Moran, O.; Ludewig, U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem. J. 2008, 414, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Cavez, D.; Besserer, A.; Berny, M.C.; Gilis, D.; Rooman, M.; Chaumont, F. A conserved cysteine residue is involved in disulfide bond formation between plant plasma membrane aquaporin monomers. Biochem. J. 2012, 445, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursiac, Y.; Boudet, J.; Postaire, O.; Luu, D.T.; Tournaire-Roux, C.; Maurel, C. Stimulus-induced downregulation of root water transport involves reactive oxygen species-activated cell signalling and plasma membrane intrinsic protein internalization. Plant J. Cell Mol. Biol. 2008, 56, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestetti, S.; Medraño-Fernandez, I.; Galli, M.; Ghitti, M.; Bienert, G.P.; Musco, G.; Orsi, A.; Rubartelli, A.; Sitia, R. A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 2018, 4, eaar5770. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; Verkman, A.S. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-κB signalling in keratinocytes and development of psoriasis. Nat. Commun. 2015, 6, 7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, M.; Farinelli, G.; Galli, M.; Aiuti, A.; Sitia, R. AQP8 transports Nox2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukocyte Biol. 2016, 100, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, A.; Tietz, P.; Jefferson, J.; Pagano, R.; LaRusso, N.F. Isolation and characterization of lipid microdomains from apical and basolateral plasma membranes of rat hepatocytes. Hepatology 2006, 43, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy-Scaria, A.M.; Dietzen, D.J.; Kwong, J.; Link, D.C.; Lublin, D.M. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J. Cell Biol. 1994, 126, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Leu, S.; Cheong, A.; Zhang, H.; Baibakov, B.; Shih, C.; Birnbaum, M.J.; Donowitz, M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: Role in absorption and differentiation. Gastroenterology 2004, 126, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Park, S.J.; Joe, E.H.; Jou, I. Raft-mediated Src homology 2 domain-containing proteintyrosine phosphatase 2 (SHP-2) regulation in microglia. J. Biol. Chem. 2006, 281, 11872–11878. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Mazzinghi, B.; Camici, G.; Manao, G.; Ramponi, G. Some protein tyrosine phosphatases target in part to lipid rafts and interact with caveolin-1. Biochem. Biophys. Res. Commun. 2002, 296, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Jin, S.; Zhang, F.; Xia, M.; Bao, J.X.; Hu, J.; Poklis, J.L.; Li, P.L. Formation of lipid raft redox signalling platforms in glomerular endothelial cells: An early event of homocysteine-induced glomerular injury. J. Cell. Mol. Med. 2009, 13, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kuhn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [PubMed]
- Perrone, G.; Zagami, M.; Altomare, V.; Battista, C.; Morini, S.; Rabitti, C. Cox-2 localization within plasma membrane caveolae-like structures in human lobular intraepithelial neoplasia of the breast. Virchows Arch. 2007, 451, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Feron, O. Nitric oxide synthases: Which, where, how, and why? J. Clin. Investig. 1997, 100, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Braam, B.; Verhaar, M.C. Understanding eNOS for pharmacological modulation of endothelial function: A translational view. Curr. Pharm. Des. 2007, 13, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Dudzinski, D.M.; Igarashi, J.; Greif, D.; Michel, T. The regulation and pharmacology of endothelial nitric oxide synthase. Ann. Rev. Pharmacol. Toxicol. 2006, 46, 235–276. [Google Scholar] [CrossRef] [PubMed]
- Bouloumie, A.; Bauersachs, J.; Linz, W.; Scholkens, B.A.; Wiemer, G.; Fleming, I.; Busse, R. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 1997, 30, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, B.; John, M.; Klatt, P.; Bohme, E.; Mayer, B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 1992, 281 Pt 3, 627–630. [Google Scholar] [CrossRef]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Roe, N.D.; Ren, J. Nitric oxide synthase uncoupling: A therapeutic target in cardiovascular diseases. Vasc. Pharmacol. 2012, 57, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kou, R.; Lin, A.J.; Golan, D.E.; Michel, T. Subcellular targeting and agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. J. Biol. Chem. 2002, 277, 39554–39560. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W. Regulation of endothelial nitric oxide synthase: Location, location, location. Ann. Rev. Physiol. 2002, 64, 749–774. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.C.; Duncan, J.A.; Yamashita, S.; Michel, T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca2+-calmodulin. J. Biol. Chem. 1999, 274, 33148–33154. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Feron, O.; Sase, K.; Prabhakar, P.; Michel, T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J. Biol. Chem. 1997, 272, 25907–25912. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, P.; Cheng, V.; Michel, T. A chimeric transmembrane domain directs endothelial nitric-oxide synthase palmitoylation and targeting to plasmalemmal caveolae. J. Biol. Chem. 2000, 275, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Bender, F.C.; Courjault-Gautier, F.; Bron, C.; Quest, A.F. Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14334–14339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharavi, N.M.; Baker, N.A.; Mouillesseaux, K.P.; Yeung, W.; Honda, H.M.; Hsieh, X.; Yeh, M.; Smart, E.J.; Berliner, J.A. Role of endothelial nitric oxide synthase in the regulation of SREBP activation by oxidized phospholipids. Circ. Res. 2006, 98, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Thomas, D.D.; Miranda, K.M.; Wink, D.A. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc. Natl. Acad. Sci. USA 2002, 99, 11127–11132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, D.M.; Black, S.M.; Todor, H.; Schmidt-Ullrich, R.K.; Dawson, K.S.; Mikkelsen, R.B. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J. Biol. Chem. 2005, 280, 14453–14461. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of ROS in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: How, where, and what for? Free Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Decoursey, T.E.; Ligeti, E. Regulation and termination of NADPH oxidase activity. Cell. Mol. Life Sci. 2005, 62, 2173–2193. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Ann. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Bustamante, J.; Kannengiesser, C.; de Boer, M.; van Leeuwen, K.; Koker, M.Y.; Wolach, B.; Roesler, J.; et al. Hematologically important mutations: The autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 2010, 44, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Roesler, J.; Lopez, J.A.; Ariga, T.; Avcin, T.; de Boer, M.; Bustamante, J.; Condino-Neto, A.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol. Dis. 2010, 45, 246–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; deBoer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.A.; Tao, W.; Yang, F.; Kim, C.; Gu, Y.; Mansfield, P.; Levine, J.E.; Petryniak, B.; Derrow, C.W.; Harris, C.; et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 2000, 96, 1646–1654. [Google Scholar] [PubMed]
- van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Espanol, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic granulomatous disease: The European experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.M. Chronic granulomatous disease. Hematol./Oncol. Clin. N. Am. 2013, 27, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Baehner, R.L.; Karnovsky, M.L. Deficiency of reduced nicotinamide-adenine dinucleotide oxidase in chronic granulomatous disease. Science 1968, 162, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Goldblatt, D.; Buddle, J.; Morton, L.; Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukocyte Biol. 2003, 73, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; MacDonald, K.L.; Brown, K.L.; Mydel, P.; Collins, L.V.; Hancock, R.E.; Speert, D.P. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-κB. Eur. J. Immunol. 2007, 37, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.L.; Bylund, J.; MacDonald, K.L.; Song-Zhao, G.X.; Elliott, M.R.; Falsafi, R.; Hancock, R.E.; Speert, D.P. ROS-deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin. Immunol. 2008, 129, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, L.; Dahlgren, C.; Karlsson, A.; Brown, K.L.; Bylund, J. Phagocyte-derived reactive oxygen species as suppressors of inflammatory disease. Arthritis Rheum. 2008, 58, 2931–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, D.; Lehmann, N.; Hoffmann, F.; Jansson, A.; Hector, A.; Notheis, G.; Roos, D.; Belohradsky, B.H.; Wintergerst, U. Dysregulation of innate immune receptors on neutrophils in chronic granulomatous disease. J. Allergy Clin. Immunol. 2008, 121, 375.e379–382.e379. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukocyte Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.M.; Kashgarian, M.; Shlomchik, M.J. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci. Transl. Med. 2012, 4, 157ra141. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Bäcklund, J.; Bauer, K.; Gelderman, K.A.; Holmdahl, R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J. Immunol. 2007, 179, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Kelkka, T.; Kienhöfer, D.; Hoffmann, M.; Linja, M.; Wing, K.; Sareila, O.; Hultqvist, M.; Laajala, E.; Chen, Z.; Vasconcelos, J.; et al. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid. Redox Signal. 2014, 21, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.J.; Deffit, S.N.; McLetchie, S.; Perez, L.; Walline, C.C.; Blum, J.S. A role for NADPH oxidase in antigen presentation. Front. Immunol. 2013, 4, 295. [Google Scholar] [CrossRef] [PubMed]
- Schappi, M.G.; Jaquet, V.; Belli, D.C.; Krause, K.H. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Sem. Immunopathol. 2008, 30, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Jancic, C.; Savina, A.; Wasmeier, C.; Tolmachova, T.; El-Benna, J.; Dang, P.M.; Pascolo, S.; Gougerot-Pocidalo, M.A.; Raposo, G.; Seabra, M.C.; et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 2007, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Dumenil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. Nox2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Kotsias, F.; Hoffmann, E.; Amigorena, S.; Savina, A. Reactive oxygen species production in the phagosome: Impact on antigen presentation in dendritic cells. Antioxid. Redox Signal. 2013, 18, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, A.; Ganguly, A.; Mu, L.; Davis, S.P.; Stenner, M.D.; Lam, R.; Munro, F.; Namet, I.; Alghamdi, E.; Furstenhaupt, T.; et al. Redirecting soluble antigen for MHC class I cross-presentation during phagocytosis. Eur. J. Immunol. 2015, 45, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, J.M.; Balce, D.R.; Chaudhuri, S.; Allan, E.R.; Yates, R.M. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J. 2012, 31, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Allan, E.R.; Tailor, P.; Balce, D.R.; Pirzadeh, P.; McKenna, N.T.; Renaux, B.; Warren, A.L.; Jirik, F.R.; Yates, R.M. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J. Immunol. 2014, 192, 4989–5001. [Google Scholar] [CrossRef] [PubMed]
- Cotugno, N.; Finocchi, A.; Cagigi, A.; Di Matteo, G.; Chiriaco, M.; Di Cesare, S.; Rossi, P.; Aiuti, A.; Palma, P.; Douagi, I. Defective B-cell proliferation and maintenance of long-term memory in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2015, 135, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleesing, J.J.; Souto-Carneiro, M.M.; Savage, W.J.; Brown, M.R.; Martinez, C.; Yavuz, S.; Brenner, S.; Siegel, R.M.; Horwitz, M.E.; Lipsky, P.E.; et al. Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J. Immunol. 2006, 176, 7096–7103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Feng, X.; He, C.; Gao, H.; Yang, L.; Ma, Q.; Guo, L.; Qiao, Y.; Yang, H.; Ma, T. Defective macrophage function in aquaporin-3 deficiency. FASEB J. 2011, 25, 4233–4239. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, C.; Asplund, A.; Catrina, A.; Nielsen, S.; Rutzler, M. A Systematic Characterization of Aquaporin-9 Expression in Human Normal and Pathological Tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2016, 64, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.; Rousseau, R.; Soria, J.C.; Hoque, M.O.; Lee, J.; Jang, S.J.; Trink, B.; Sidransky, D.; Mao, L. Aquaporin expression in human lymphocytes and dendritic cells. Am. J. Hematol. 2004, 75, 128–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loitto, V.M.; Forslund, T.; Sundqvist, T.; Magnusson, K.E.; Gustafsson, M. Neutrophil leukocyte motility requires directed water influx. J. Leukocyte Biol. 2002, 71, 212–222. [Google Scholar] [PubMed]
- Moniaga, C.S.; Watanabe, S.; Honda, T.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9-expressing neutrophils are required for the establishment of contact hypersensitivity. Sci. Rep. 2015, 5, 15319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Arseneau, K.O.; Cominelli, F. Inflammatory bowel disease. Immunol. Lett. 2014, 161, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyette, P.; Labbé, C.; Trinh, T.T.; Xavier, R.J.; Rioux, J.D. Molecular pathogenesis of inflammatory bowel disease: Genotypes, phenotypes and personalized medicine. Ann. Med. 2007, 39, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Lee, P.; Ceteci, F.; Nixon, C.; Blyth, K.; Sansom, O.J.; Vousden, K.H. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 2016, 30, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Alzoghaibi, M.A. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J. Gastroenterol. 2013, 19, 6540–6547. [Google Scholar] [CrossRef] [PubMed]
- Pravda, J. Radical induction theory of ulcerative colitis. World J. Gastroenterol. 2005, 11, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Lekstrom, K.; Brenner, S.; Hewitt, S.M.; Dana, R.; Malech, H.L.; Leto, T.L. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J. Immunol. 2003, 171, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Bäckhed, F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 2015, 8, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; Ben Mkaddem, S.; Pedruzzi, E.; Guichard, C.; Treton, X.; Ducroc, R.; Freund, J.N.; Cazals-Hatem, D.; Bouhnik, Y.; Woerther, P.L.; et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol. Cell. Biol. 2010, 30, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Ann. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, E.M.; Oh, C.T.; Bae, Y.S.; Lee, W.J. A direct role for dual oxidase in Drosophila gut immunity. Science 2005, 310, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Misaka, T.; Matsumoto, I.; Watanabe, H.; Abe, K. Aquaporin-9 is expressed in a mucus-secreting goblet cell subset in the small intestine. FEBS Lett. 2003, 540, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Differential expression of aquaporin 8 in human colonic epithelial cells and colorectal tumors. BMC Physiol. 2001, 1, 1. [Google Scholar] [CrossRef]
- Yang, B.; Song, Y.; Zhao, D.; Verkman, A.S. Phenotype analysis of aquaporin-8 null mice. Am. J. Physiol. Cell Physiol. 2005, 288, C1161–C1170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, J.; Wang, J.; Shen, X.; Sun, J. Aquaporin 3 and 8 are down-regulated in TNBS-induced rat colitis. Biochem. Biophys. Res. Commun. 2014, 443, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.A.; Wallace, L.E.; Wong, J.F.; O’Loughlin, E.V.; Urbanski, S.J.; Gall, D.G.; MacNaughton, W.K.; Beck, P.L. Aquaporin expression is downregulated in a murine model of colitis and in patients with ulcerative colitis, Crohn’s disease and infectious colitis. Cell Tissue Res. 2004, 318, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zahn, A.; Moehle, C.; Langmann, T.; Ehehalt, R.; Autschbach, F.; Stremmel, W.; Schmitz, G. Aquaporin-8 expression is reduced in ileum and induced in colon of patients with ulcerative colitis. World J. Gastroenterol. 2007, 13, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.X.; Dong, P.P.; Peng, R.; Li, J.; Zhang, D.Y.; Wang, J.Y.; Shen, X.Z.; Dong, L.; Sun, J.Y. Expression, localization and possible functions of aquaporins 3 and 8 in rat digestive system. Biotech. Histochem. Off. Publ. Biol. Stain Comm. 2016, 91, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Kon, R.; Sugiyama, K. Aquaporins in the Colon as a New Therapeutic Target in Diarrhea and Constipation. Int. J. Mol. Sci. 2016, 17, 1172. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Suzuki, T.; Koyama, H.; Tanaka, S.; Takata, K. Water channel protein AQP3 is present in epithelia exposed to the environment of possible water loss. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1999, 47, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Zhao, D.; Verkman, A.S. Impaired enterocyte proliferation in aquaporin-3 deficiency in mouse models of colitis. Gut 2007, 56, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xu, Y.; Chen, Z.; Xu, Z.; Xu, H. Knockdown of aquaporin 3 is involved in intestinal barrier integrity impairment. FEBS Lett. 2011, 585, 3113–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, S.; Jaquet, V.; Sorce, S.; Dubois-Dauphin, M.; Hultqvist, M.; Backdahl, L.; Holmdahl, R.; Colaianna, M.; Cuomo, V.; Trabace, L.; et al. NADPH oxidase elevations in pyramidal neurons drive psychosocial stress-induced neuropathology. Transl. Psychiatry 2012, 2, e111. [Google Scholar] [CrossRef] [PubMed]
- Kallenborn-Gerhardt, W.; Schröder, K.; Del Turco, D.; Lu, R.; Kynast, K.; Kosowski, J.; Niederberger, E.; Shah, A.M.; Brandes, R.P.; Geisslinger, G.; et al. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 10136–10145. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain J. Neurol. 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a Nox1-dependent NADPH oxidase. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, P.M.; Satriotomo, I.; Windelborn, J.A.; Mitchell, G.S. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J. Physiol. 2009, 587, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.Y.; Quinn, M.T.; Klann, E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Pao, M.; Holland, S.M.; Klann, E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J. Neurochem. 2005, 94, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Yoneda, K.; Asai, K.; Sobue, K.; Tada, T.; Fujita, Y.; Katsuya, H.; Fujita, M.; Aihara, N.; Mase, M.; et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Brain Res. Mol. Brain Res. 2001, 90, 26–38. [Google Scholar] [CrossRef]
- Yang, M.; Gao, F.; Liu, H.; Yu, W.H.; Sun, S.Q. Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat brains after permanent focal cerebral ischemia. Brain Res. 2009, 1290, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.J.; Wang, K.J.; Gan, S.W.; Xu, J.; Xu, S.Y.; Sun, S.Q. Expression of aquaporin8 in human astrocytomas: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2013, 440, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Tanaka, Y.; Matsuzaki, T.; Morishita, Y.; Ishibashi, K. Aquaporin-11 (AQP11) Expression in the Mouse Brain. Int. J. Mol. Sci. 2016, 17, 861. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Tao, S.; Liu, Z.; Zheng, W.; Yu, D. Down-regulation of AQP8 suppresses glioma cells growth and invasion/migration via cell cycle pathway. Int. J. Clin Exp. Pathol. 2016, 9, 1240–1248. [Google Scholar]
- Jelen, S.; Parm Ulhøi, B.; Larsen, A.; Frøkiaer, J.; Nielsen, S.; Rützler, M. AQP9 expression in glioblastoma multiforme tumors is limited to a small population of astrocytic cells and CD15+/CalB+ leukocytes. PLoS ONE 2013, 8, e75764. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol. 2006, 26, 5908–5920. [Google Scholar] [CrossRef] [PubMed]
- Pao, M.; Wiggs, E.A.; Anastacio, M.M.; Hyun, J.; DeCarlo, E.S.; Miller, J.T.; Anderson, V.L.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Cognitive function in patients with chronic granulomatous disease: A preliminary report. Psychosomatics 2004, 45, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Pra, I.; Rossi, F. beta-amyloid activates the O−2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef] [PubMed]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, E.; Hashimoto, Y.; Kanekura, K.; Niikura, T.; Aiso, S.; Nishimoto, I. Characterization of the toxic mechanism triggered by Alzheimer’s amyloid-β peptides via p75 neurotrophin receptor in neuronal hybrid cells. J. Neurosci. Res. 2003, 73, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Cooper, C.; Liu, B.; Wilson, B.; Hong, J.S. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 2002, 83, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zheng, W.; Xin, N.; Xie, J.W.; Wang, T.; Wang, Z.Y. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 2012, 61, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Qin, L.; Gao, H.M.; Wilson, B.; Ali, S.F.; Hong, J.S.; Liu, B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: Role of NADPH oxidase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Li, G.; Qin, L.; Wu, X.; Pei, Z.; Wang, T.; Wilson, B.; Yang, J.; Hong, J.S. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: Substance P vs. dynorphin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhou, Y.; Hong, J.S.; Zhang, J. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 533–542. [Google Scholar]
- Hu, X.; Zhang, D.; Pang, H.; Caudle, W.M.; Li, Y.; Gao, H.; Liu, Y.; Qian, L.; Wilson, B.; Di Monte, D.A.; et al. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J. Immunol. 2008, 181, 7194–7204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Choi, D.H.; Block, M.L.; Lorenzl, S.; Yang, L.; Kim, Y.J.; Sugama, S.; Cho, B.P.; Hwang, O.; Browne, S.E.; et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Re, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristóvão, A.C.; Choi, D.H.; Baltazar, G.; Beal, M.F.; Kim, Y.S. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid. Redox Signal. 2009, 11, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef] [PubMed]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Wu, P.F.; Long, L.H.; Yu, D.F.; Wu, W.N.; Hu, Z.L.; Fu, H.; Xie, N.; Jin, Y.; Ni, L.; et al. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell 2010, 9, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Muñoz, M.; Garzón, J. Nitric oxide and zinc-mediated protein assemblies involved in mu opioid receptor signaling. Mol. Neurobiol. 2013, 48, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Calero, C.I.; Vickers, E.; Moraga Cid, G.; Aguayo, L.G.; von Gersdorff, H.; Calvo, D.J. Allosteric modulation of retinal GABA receptors by ascorbic acid. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isoform | Regulatory Subunits | Major Distribution Sites Reported | References |
|---|---|---|---|
| Nox1 | p22phox NOXA1 NOXO1 Rac | Vascular smooth muscle cells (VSMC), colon, endothelium, placenta, central nervous system (CNS) | [115,116,117,118,119] |
| Nox2 | p22phox p67phox p47phox p40phox Rac | Phagocytes, ovary, central nervous system (CNS), cardiomyocytes, hepatocytes, endothelium, gastrointestinal tract, hematopoietic stem cells | [13,118,120,121,122,123] |
| Nox3 | p22phox NOXA1 NOXO1 Rac? | Inner ear | [124,125] |
| Nox4 | p22phox | Ubiquitous, including: Endothelial cells, vascular smooth muscle cells (VSMC), kidney, lungs, hematopoietic stem cells, placenta, central nervous system (CNS) | [123,126] |
| Nox5 | Ca2+ (as activator) | Lymphoid tissue, testis, ovary, spleen | [100,101] |
| DUOX1 | Ca2+ (as activator) DUOXA1 (as maturation factor) | Respiratory epithelium, thyroid, gastrointestinal epithelia, salivary and rectal glands | [127,128,129,130,131] |
| DUOX2 | Ca2+ (as activator) DUOXA2 (as maturation factor) | Respiratory epithelium, thyroid, gastrointestinal epithelia, salivary and rectal glands | [127,128,129,130,131] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. https://doi.org/10.3390/antiox7110168
Nordzieke DE, Medraño-Fernandez I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants. 2018; 7(11):168. https://doi.org/10.3390/antiox7110168
Chicago/Turabian StyleNordzieke, Daniela E., and Iria Medraño-Fernandez. 2018. "The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling" Antioxidants 7, no. 11: 168. https://doi.org/10.3390/antiox7110168
APA StyleNordzieke, D. E., & Medraño-Fernandez, I. (2018). The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants, 7(11), 168. https://doi.org/10.3390/antiox7110168