Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes


Abstract
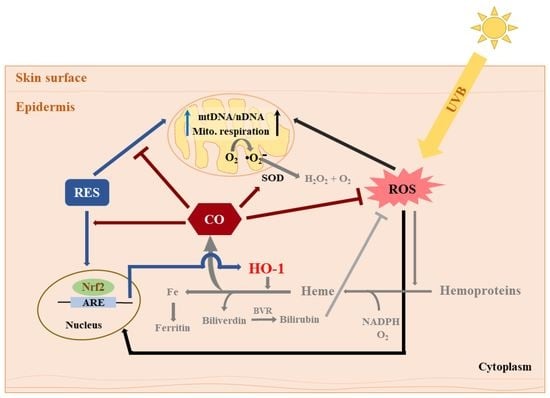
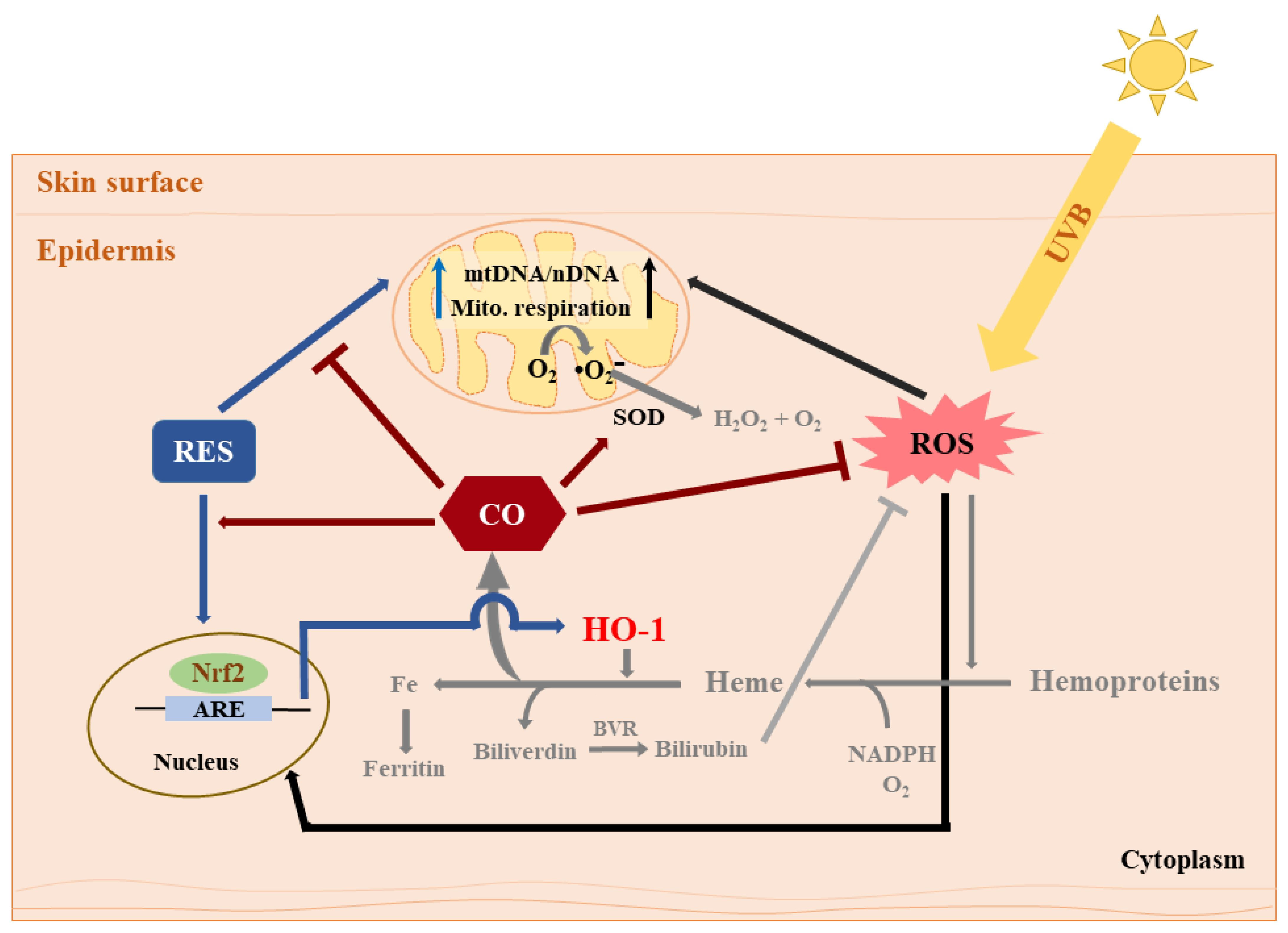
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture
2.3. Cell Viability
2.4. Measurement of UVB-Induced Intracellular ROS Production
2.5. Time-Course Analysis of H2O2-Induced Intracellular ROS Generation
2.6. Measurement of Mitochondrial DNA Quantity by Quantitative Real-Time PCR
2.7. Measurement of Mitochondrial Oxygen Consumption Rate (OCR)
2.8. Preparation of Cytosolic Extract
2.9. Protein Electrophoresis and Western Blotting
2.10. Statistical Analysis
3. Results
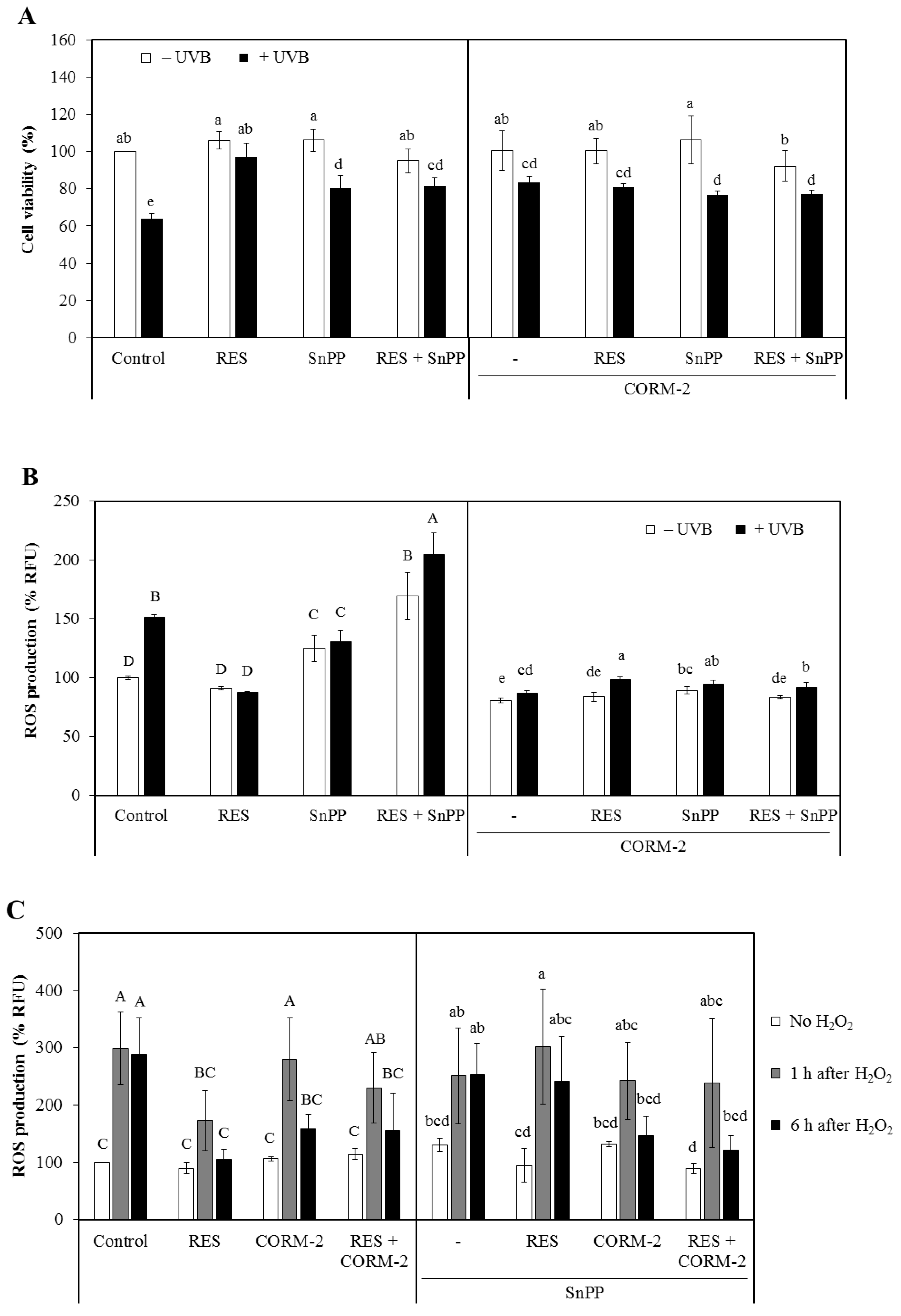
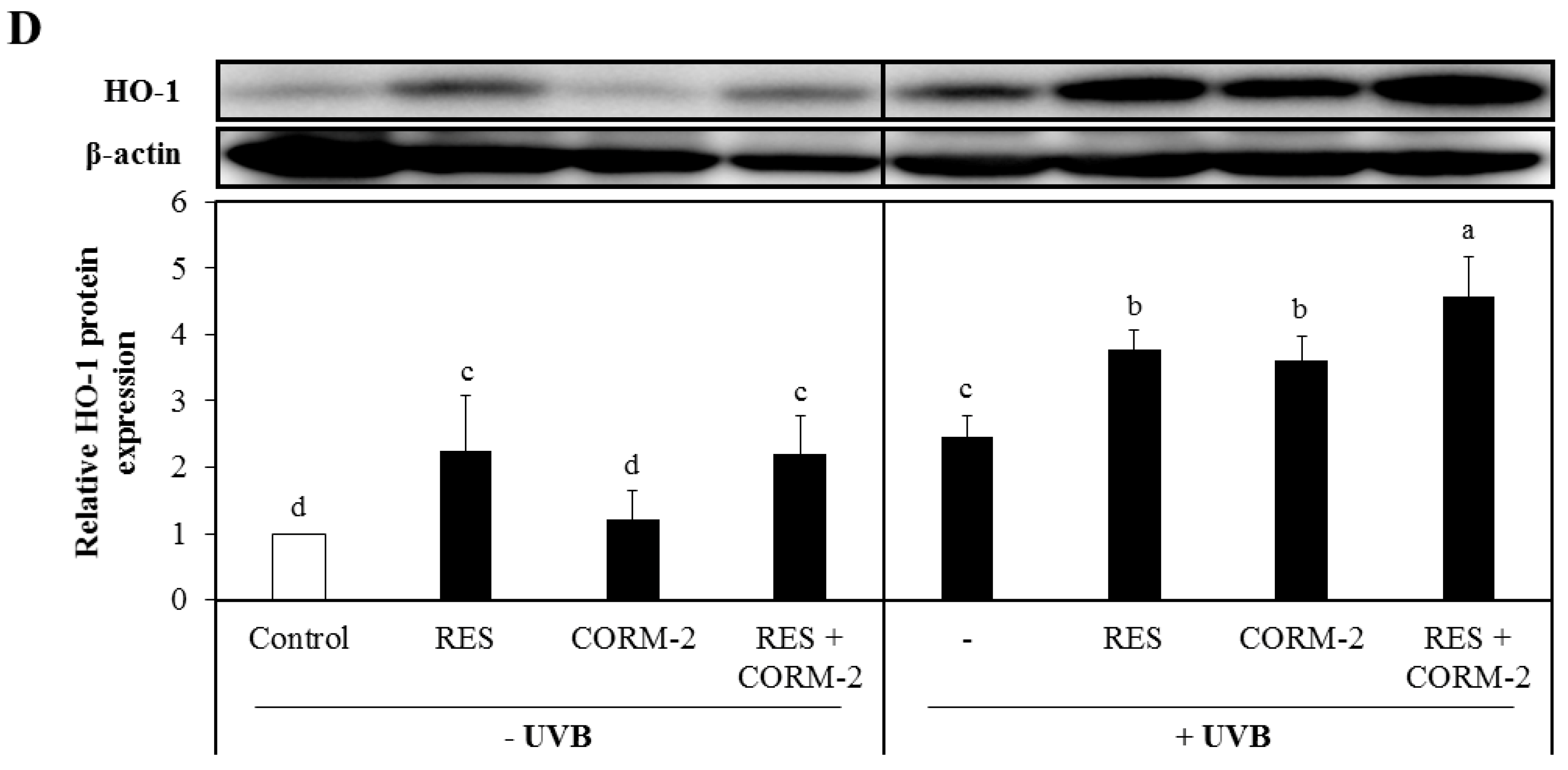
3.1. Intracellular CO Protects HaCaT Cells from UVB- and H2O2-Induced Oxidative Damage Possibly via HO-1 Regulation
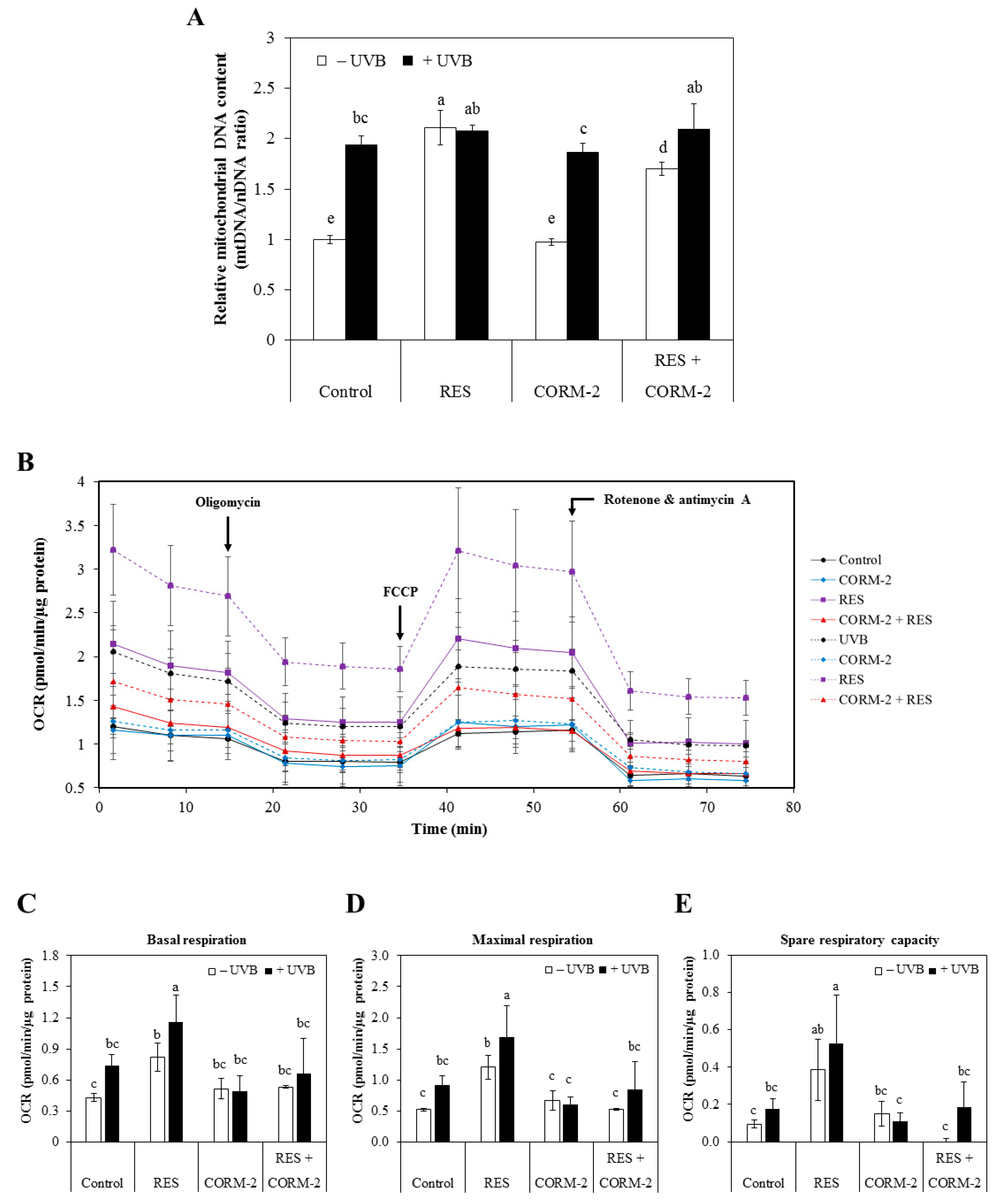
3.2. Intracellular CO Alleviates UVB-Induced Oxidative Damage by Modulating Mitochondrial Functionality
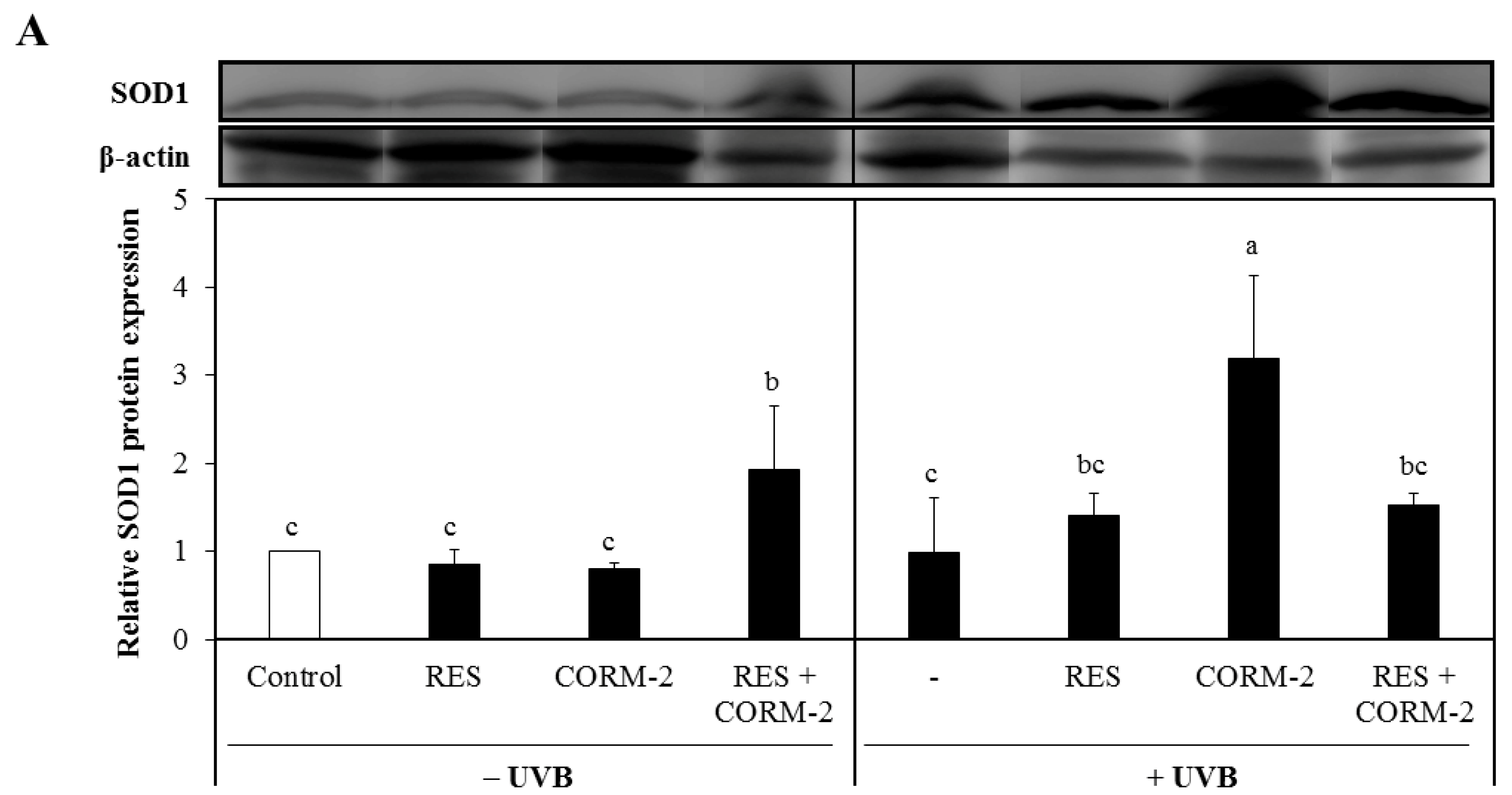
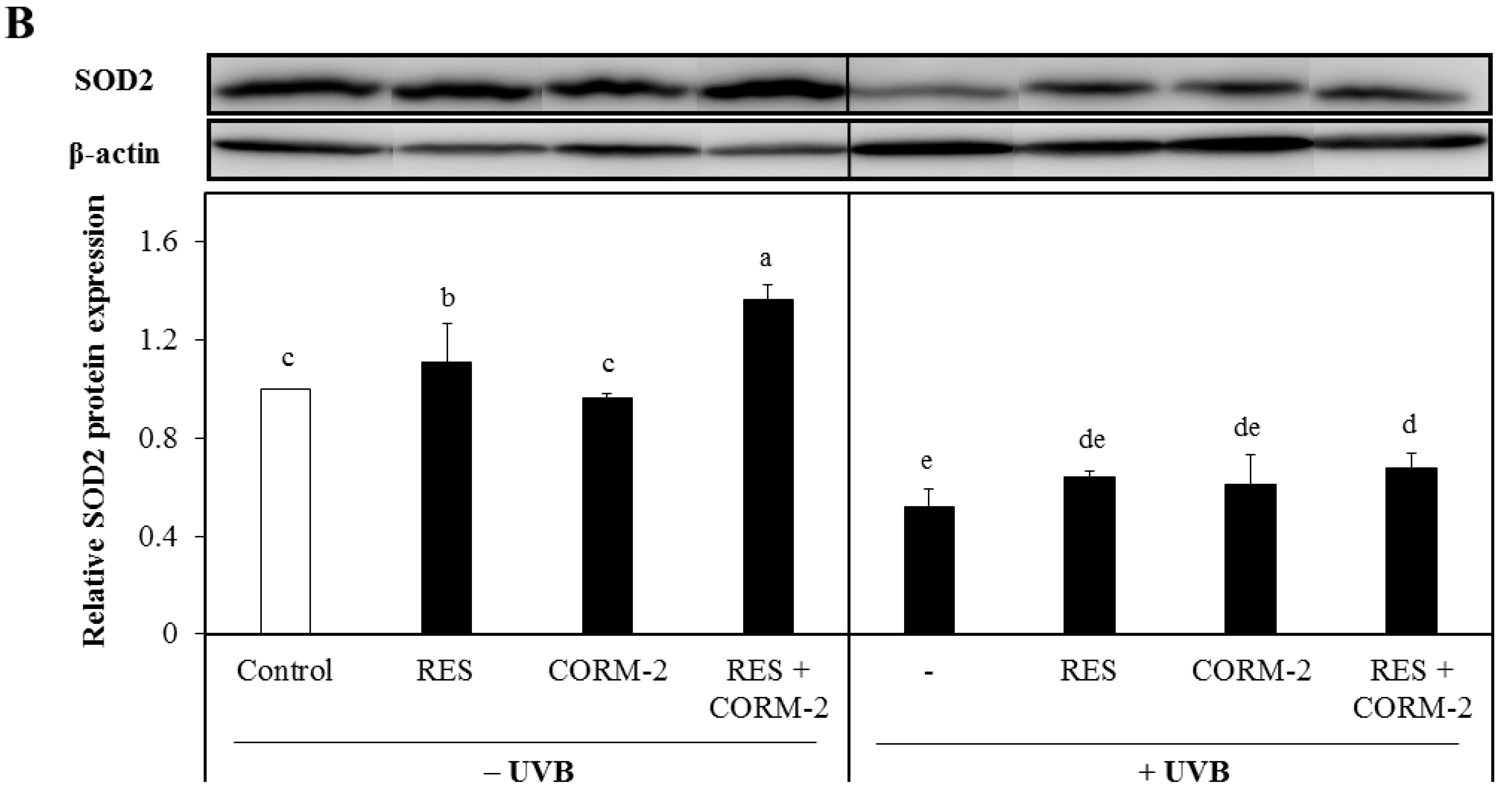
3.3. Intracellular CO Increases Protein Expression of the Antioxidant Enzymes SOD1 and SOD2
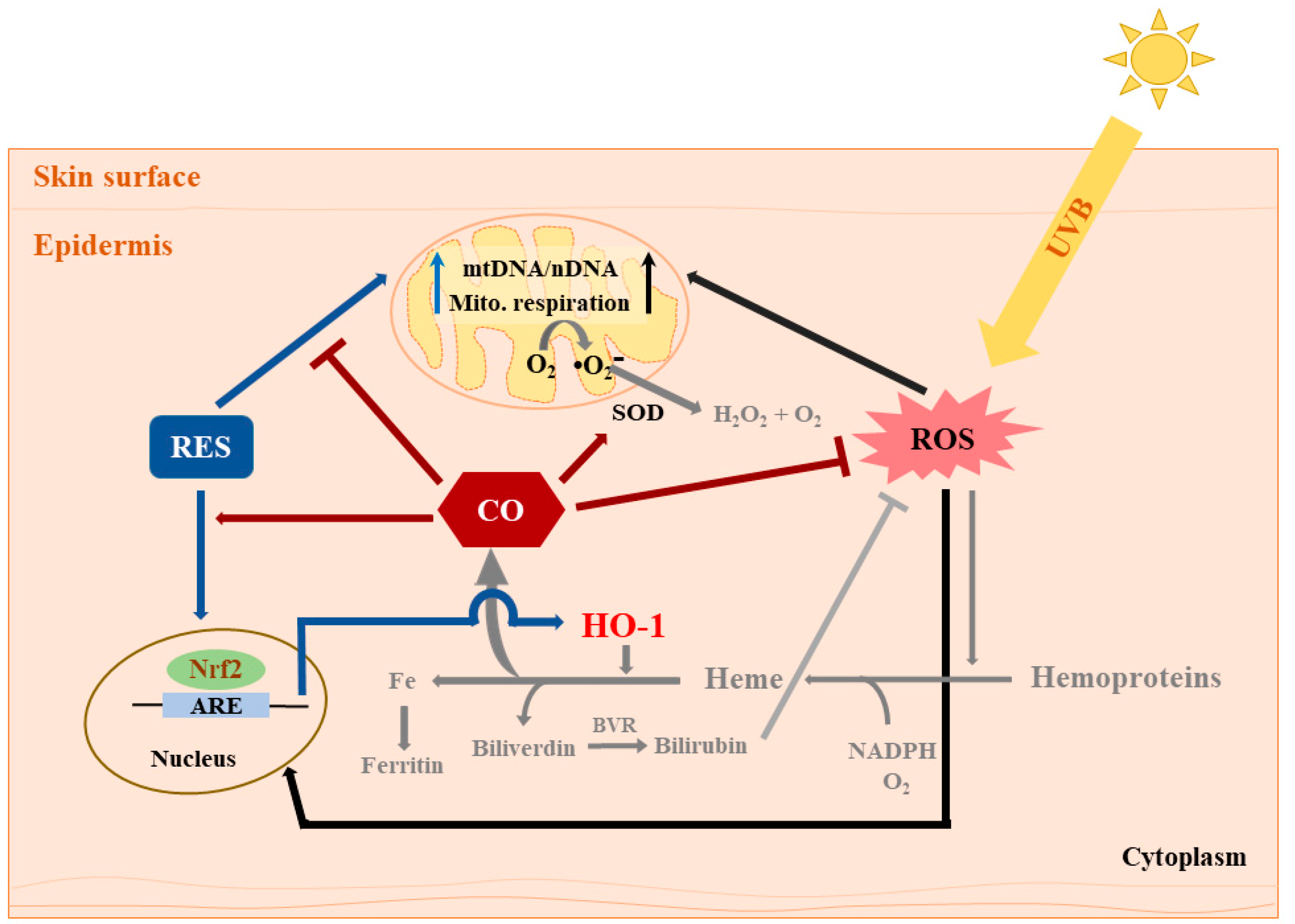
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Amaro-Ortiz, A.; Yan, B.; D’Orazio, J.A. Ultraviolet radiation, aging and the skin: Prevention of damage by topical cAMP manipulation. Molecules 2014, 19, 6202–6219. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Epidermis and Its Renewal by Stem Cells. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Okuyama, R.; Tagami, H.; Aiba, S. Notch signaling: Its role in epidermal homeostasis and in the pathogenesis of skin diseases. J. Dermatol. Sci. 2008, 49, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Rittie, L. Cellular mechanisms of skin repair in humans and other mammals. J. Cell Commun. Signal. 2016, 10, 103–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villeneuve, N.F.; Sun, Z.; Chen, W.M.; Zhang, D.D. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle 2009, 8, 3255–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch-Machin, M.A. The role of mitochondria in ageing and carcinogenesis. Clin. Exp. Dermatol. 2006, 31, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef] [PubMed]
- Hansford, R.G.; Hogue, B.A.; Mildaziene, V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J. Bioenerg. Biomembr. 1997, 29, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.M.; Wipf, P.; Durham, A.; Epperly, M.W.; Greenberger, J.S.; Falo, L.D., Jr. Targeting mitochondrial oxidative stress to mitigate UV-induced skin damage. Front. Pharmacol. 2018, 9, 920. [Google Scholar] [CrossRef]
- Amaral, S.; Redmann, K.; Sanchez, V.; Mallidis, C.; Ramalho-Santos, J.; Schlatt, S. UVB irradiation as a tool to assess ROS-induced damage in human spermatozoa. Andrology 2013, 1, 707–714. [Google Scholar] [CrossRef]
- Heck, D.E.; Vetrano, A.M.; Mariano, T.M.; Laskin, J.D. UVB light stimulates production of reactive oxygen species: Unexpected role for catalase. J. Biol. Chem. 2003, 278, 22432–22436. [Google Scholar] [CrossRef]
- Pillai, S.; Oresajo, C.; Hayward, J. Ultraviolet radiation and skin aging: Roles of reactive oxygen species, inflammation and protease activation, and strategies for prevention of inflammation-induced matrix degradation—A review. Int. J. Cosmet. Sci. 2005, 27, 17–34. [Google Scholar] [CrossRef]
- Subedi, L.; Lee, T.H.; Wahedi, H.M.; Baek, S.H.; Kim, S.Y. Resveratrol-enriched rice attenuates UVB-ROS-induced skin aging via downregulation of inflammatory cascades. Oxidative Med. Cell. Longev. 2017, 2017, 8379539. [Google Scholar] [CrossRef]
- Kim, J.; Oh, J.; Averilla, J.N.; Kim, H.J.; Kim, J.S.; Kim, J.S. Grape peel extract and resveratrol inhibit wrinkle formation in mice model through activation of Nrf2/HO-1 signaling pathway. J. Food Sci. 2019, 84, 1600–1608. [Google Scholar] [CrossRef]
- Chen, C.Y.; Jang, J.H.; Li, M.H.; Surh, Y.J. Resveratrol upregulates heme oxygenase-1 expression via activation of NF-E2-related factor 2 in PC12 cells. Biochem. Biophys. Res. Commun. 2005, 331, 993–1000. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, X.; Hu, Y.; Zhang, Z. The protective role of resveratrol in the sodium arsenite-induced oxidative damage via modulation of intracellular GSH homeostasis. Biol. Trace Elem. Res. 2013, 155, 119–131. [Google Scholar] [CrossRef]
- Yu, M.; Xu, M.J.; Liu, Y.; Yang, W.; Rong, Y.; Yao, P.; Yan, H.; Wang, D.; Liu, L.G. Nrf2/ARE is the potential pathway to protect Sprague-Dawley rats against oxidative stress induced by quinocetone. Regul. Toxicol. Pharmacol. 2013, 66, 279–285. [Google Scholar] [CrossRef]
- Zheng, X.; Wang, G.; Bin, P.; Meng, T.; Niu, Y.; Yang, M.; Zhang, L.; Duan, H.; Yu, T.; Dai, Y.; et al. Time-course effects of antioxidants and phase II enzymes on diesel exhaust particles-induced oxidative damage in the mouse lung. Toxicol. Appl. Pharmacol. 2019, 366, 25–34. [Google Scholar] [CrossRef]
- Saunier, E.; Antonio, S.; Regazzetti, A.; Auzeil, N.; Laprevote, O.; Shay, J.W.; Coumoul, X.; Barouki, R.; Benelli, C.; Huc, L.; et al. Resveratrol reverses the Warburg effect by targeting the pyruvate dehydrogenase complex in colon cancer cells. Sci. Rep. 2017, 7, 6945. [Google Scholar] [CrossRef]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 234–235, 249–263. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar]
- Alonso, J.R.; Cardellach, F.; Lopez, S.; Casademont, J.; Miro, O. Carbon monoxide specifically inhibits cytochrome c oxidase of human mitochondrial respiratory chain. Pharmacol. Toxicol. 2003, 93, 142–146. [Google Scholar] [CrossRef]
- Adach, W.; Olas, B. Carbon monoxide and its donors—Their implications for medicine. Future Med. Chem. 2019, 11, 61–73. [Google Scholar] [CrossRef]
- Vreman, H.J.; Wong, R.J.; Stevenson, D.K. Carbon monoxide in breath, blood, and other tissues. In Carbon Monoxide Toxicity; CRC Press: Boca Raton, FL, USA, 2000; pp. 19–60. [Google Scholar]
- Ryter, S.W.; Otterbein, L.E. Carbon monoxide in biology and medicine. Bioessays 2004, 26, 270–280. [Google Scholar] [CrossRef]
- Rodkey, F.L.; O’Neal, J.D.; Collison, H.A.; Uddin, D.E. Relative affinity of hemoglobin S and hemoglobin A for carbon monoxide and oxygen. Clin. Chem. 1974, 20, 83–84. [Google Scholar]
- Cheng, Y.; Mitchell-Flack, M.J.; Wang, A.; Levy, R.J. Carbon monoxide modulates cytochrome oxidase activity and oxidative stress in the developing murine brain during isoflurane exposure. Free Radic. Biol. Med. 2015, 86, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Zuckerbraun, B.S.; Chin, B.Y.; Bilban, M.; d’Avila, J.C.; Rao, J.; Billiar, T.R.; Otterbein, L.E. Carbon monoxide signals via inhibition of cytochrome c oxidase and generation of mitochondrial reactive oxygen species. FASEB J. 2007, 21, 1099–1106. [Google Scholar] [CrossRef]
- Allanson, M.; Reeve, V.E. Carbon monoxide signalling reduces photocarcinogenesis in the hairless mouse. Cancer Immunol. Immunother. 2007, 56, 1807–1815. [Google Scholar] [CrossRef]
- Allanson, M.; Reeve, V.E. Ultraviolet A (320–400 nm) modulation of ultraviolet B (290–320 nm)-induced immune suppression is mediated by carbon monoxide. J. Investig. Dermatol. 2005, 124, 644–650. [Google Scholar] [CrossRef]
- Araujo, J.A.; Zhang, M.; Yin, F. Heme oxygenase-1, oxidation, inflammation, and atherosclerosis. Front. Pharmacol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef]
- Foresti, R.; Shurey, C.; Ansari, T.; Sibbons, P.; Mann, B.E.; Johnson, T.R.; Green, C.J.; Motterlini, R. Reviewing the use of carbon monoxide-releasing molecules (CO-RMs) in biology: Implications in endotoxin-mediated vascular dysfunction. Cell. Mol. Biol. 2005, 51, 409–423. [Google Scholar]
- Poljak-Blazi, M.; Jaganjac, M.; Sabol, I.; Mihaljevic, B.; Matovina, M.; Grce, M. Effect of ferric ions on reactive oxygen species formation, cervical cancer cell lines growth and E6/E7 oncogene expression. Toxicol. In Vitro 2011, 25, 160–166. [Google Scholar] [CrossRef]
- Cha, J.W.; Piao, M.J.; Kim, K.C.; Yao, C.W.; Zheng, J.; Kim, S.M.; Hyun, C.L.; Ahn, Y.S.; Hyun, J.W. The polyphenol chlorogenic acid attenuates UVB-mediated oxidative stress in human HaCaT keratinocytes. Biomol. Ther. (Seoul) 2014, 22, 136–142. [Google Scholar] [CrossRef]
- Masaki, H.; Atsumi, T.; Sakurai, H. Detection of hydrogen-peroxide and hydroxyl radicals in murine skin fibroblasts under Uvb irradiation. Biochem. Biophys. Res. Commun. 1995, 206, 474–479. [Google Scholar] [CrossRef]
- He, Y.Y.; Hader, D.P. UV-B-induced formation of reactive oxygen species and oxidative damage of the cyanobacterium Anabaena sp.: Protective effects of ascorbic acid and N-acetyl-L-cysteine. J. Photochem. Photobiol. B 2002, 66, 115–124. [Google Scholar] [CrossRef]
- Kim, D.H.; Byamba, D.; Wu, W.H.; Kim, T.G.; Lee, M.G. Different characteristics of reactive oxygen species production by human keratinocyte cell line cells in response to allergens and irritants. Exp. Dermatol. 2012, 21, 99–103. [Google Scholar] [CrossRef]
- Kulms, D.; Zeise, E.; Poppelmann, B.; Schwarz, T. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene 2002, 21, 5844–5851. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Oehrl, W.; Elsner, P.; Thiele, J.J. The role of H2O2 as a mediator of UVB-induced apoptosis in keratinocytes. Free Radic. Res. 2003, 37, 655–663. [Google Scholar] [CrossRef]
- Paz, M.L.; Gonzalez Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet B irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed. 2008, 24, 115–122. [Google Scholar] [CrossRef]
- Ungvari, Z.; Sonntag, W.E.; de Cabo, R.; Baur, J.A.; Csiszar, A. Mitochondrial protection by resveratrol. Exerc. Sport Sci. Rev. 2011, 39, 128–132. [Google Scholar] [CrossRef]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Plitzko, B.; Loesgen, S. Measurement of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in culture cells for assessment of the energy metabolism. Bio-Protocol 2018, 8, e2850. [Google Scholar] [CrossRef]
- Porter, C.; Hurren, N.M.; Cotter, M.V.; Bhattarai, N.; Reidy, P.T.; Dillon, E.L.; Durham, W.J.; Tuvdendorj, D.; Sheffield-Moore, M.; Volpi, E.; et al. Mitochondrial respiratory capacity and coupling control decline with age in human skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2015, 309, E224–E232. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef]
- Sheu, S.J.; Liu, N.C.; Ou, C.C.; Bee, Y.S.; Chen, S.C.; Lin, H.C.; Chan, J.Y. Resveratrol stimulates mitochondrial bioenergetics to protect retinal pigment epithelial cells from oxidative damage. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6426–6438. [Google Scholar] [CrossRef]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef]
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum. Mol. Genet. 2008, 17, 3303–3317. [Google Scholar] [CrossRef] [Green Version]
- Zemanovic, S.; Ivanov, M.V.; Ivanova, L.V.; Bhatnagar, A.; Michalkiewicz, T.; Teng, R.J.; Kumar, S.; Rathore, R.; Pritchard, K.A., Jr.; Konduri, G.G.; et al. Dynamic phosphorylation of the C terminus of Hsp70 regulates the mitochondrial import of SOD2 and redox balance. Cell Rep. 2018, 25, 2605–2616. [Google Scholar] [CrossRef]
- Milani, P.; Amadio, M.; Laforenza, U.; Dell’Orco, M.; Diamanti, L.; Sardone, V.; Gagliardi, S.; Govoni, S.; Ceroni, M.; Pascale, A.; et al. Posttranscriptional regulation of SOD1 gene expression under oxidative stress: Potential role of ELAV proteins in sporadic ALS. Neurobiol. Dis. 2013, 60, 51–60. [Google Scholar] [CrossRef]
- Farris, P.; Krutmann, J.; Li, Y.H.; McDaniel, D.; Krol, Y. Resveratrol: A unique antioxidant offering a multi-mechanistic approach for treating aging skin. J. Drugs Dermatol. 2013, 12, 1389–1394. [Google Scholar]
- Averilla, J.N.; Oh, J.; Wu, Z.; Liu, K.H.; Jang, C.H.; Kim, H.J.; Kim, J.S.; Kim, J.S. Improved extraction of resveratrol and antioxidants from grape peel using heat and enzymatic treatments. J. Sci. Food Agric. 2019, 99, 4043–4053. [Google Scholar] [CrossRef]
- Jang, C.H.; Moon, N.; Oh, J.; Kim, J.S. Luteolin Shifts Oxaliplatin-Induced Cell Cycle Arrest at G(0)/G(1) to Apoptosis in HCT116 Human Colorectal Carcinoma Cells. Nutrients 2019, 11, 770. [Google Scholar] [CrossRef]
- Jansen, T.; Daiber, A. Direct antioxidant properties of bilirubin and biliverdin. is there a role for biliverdin reductase? Front. Pharmacol. 2012, 3, 30. [Google Scholar] [CrossRef]
- Queiroga, C.S.; Almeida, A.S.; Vieira, H.L. Carbon monoxide targeting mitochondria. Biochem. Res. Int. 2012, 2012, 749845. [Google Scholar] [CrossRef]
- Rayamajhi, N.; Kim, S.K.; Go, H.; Joe, Y.; Callaway, Z.; Kang, J.G.; Ryter, S.W.; Chung, H.T. Quercetin induces mitochondrial biogenesis through activation of HO-1 in HepG2 cells. Oxidative Med. Cell. Longev. 2013, 2013, 154279. [Google Scholar] [CrossRef]
- Gruber, H.E.; Watts, J.A.; Riley, F.E.; Fulkerson, M.B.; Norton, H.J.; Hanley, E.N., Jr. Mitochondrial bioenergetics, mass, and morphology are altered in cells of the degenerating human annulus. J. Orthop. Res. 2013, 31, 1270–1275. [Google Scholar] [CrossRef]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of respiration extends C. elegans life span via reactive oxygen species that increase HIF-1 activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Averilla, J.N.; Oh, J.; Kim, J.-S. Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes. Antioxidants 2019, 8, 432. https://doi.org/10.3390/antiox8100432
Averilla JN, Oh J, Kim J-S. Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes. Antioxidants. 2019; 8(10):432. https://doi.org/10.3390/antiox8100432
Chicago/Turabian StyleAverilla, Janice N., Jisun Oh, and Jong-Sang Kim. 2019. "Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes" Antioxidants 8, no. 10: 432. https://doi.org/10.3390/antiox8100432
APA StyleAverilla, J. N., Oh, J., & Kim, J. -S. (2019). Carbon Monoxide Partially Mediates Protective Effect of Resveratrol Against UVB-Induced Oxidative Stress in Human Keratinocytes. Antioxidants, 8(10), 432. https://doi.org/10.3390/antiox8100432