Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease



Abstract
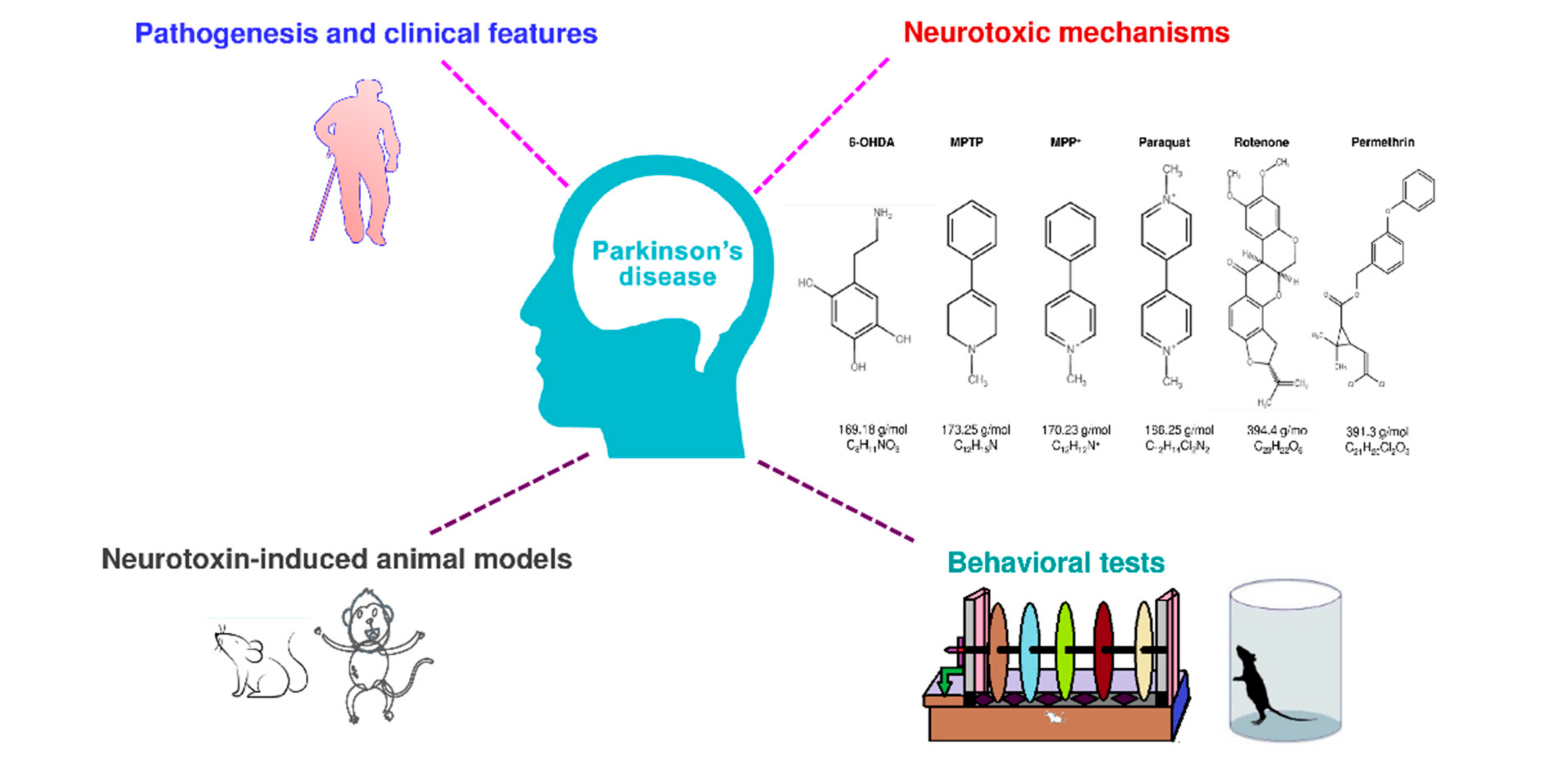
:1. Background
2. Pathogenesis and Symptoms of PD
2.1. α-Synuclein Misfolding and Aggregation
2.2. Impaired Functioning of Mitochondria
2.3. Dysregulation of Protein Clearance Control
2.4. Neuroinflammation
3. Neurotoxins Used to Induce PD In Vivo Models
3.1. 6-OHDA
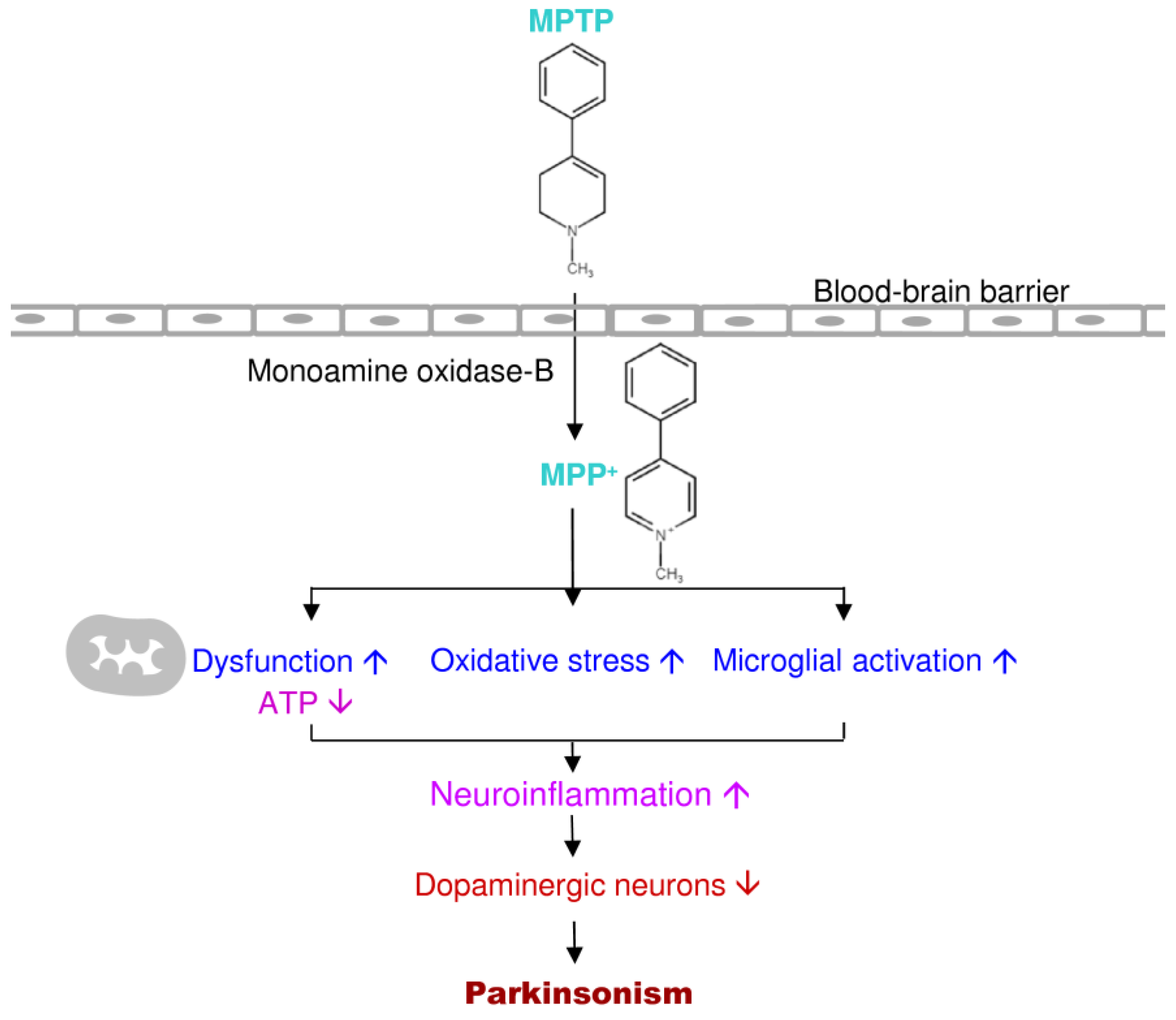
3.2. MPTP
3.3. MPP+
3.4. Paraquat
3.5. Rotenone
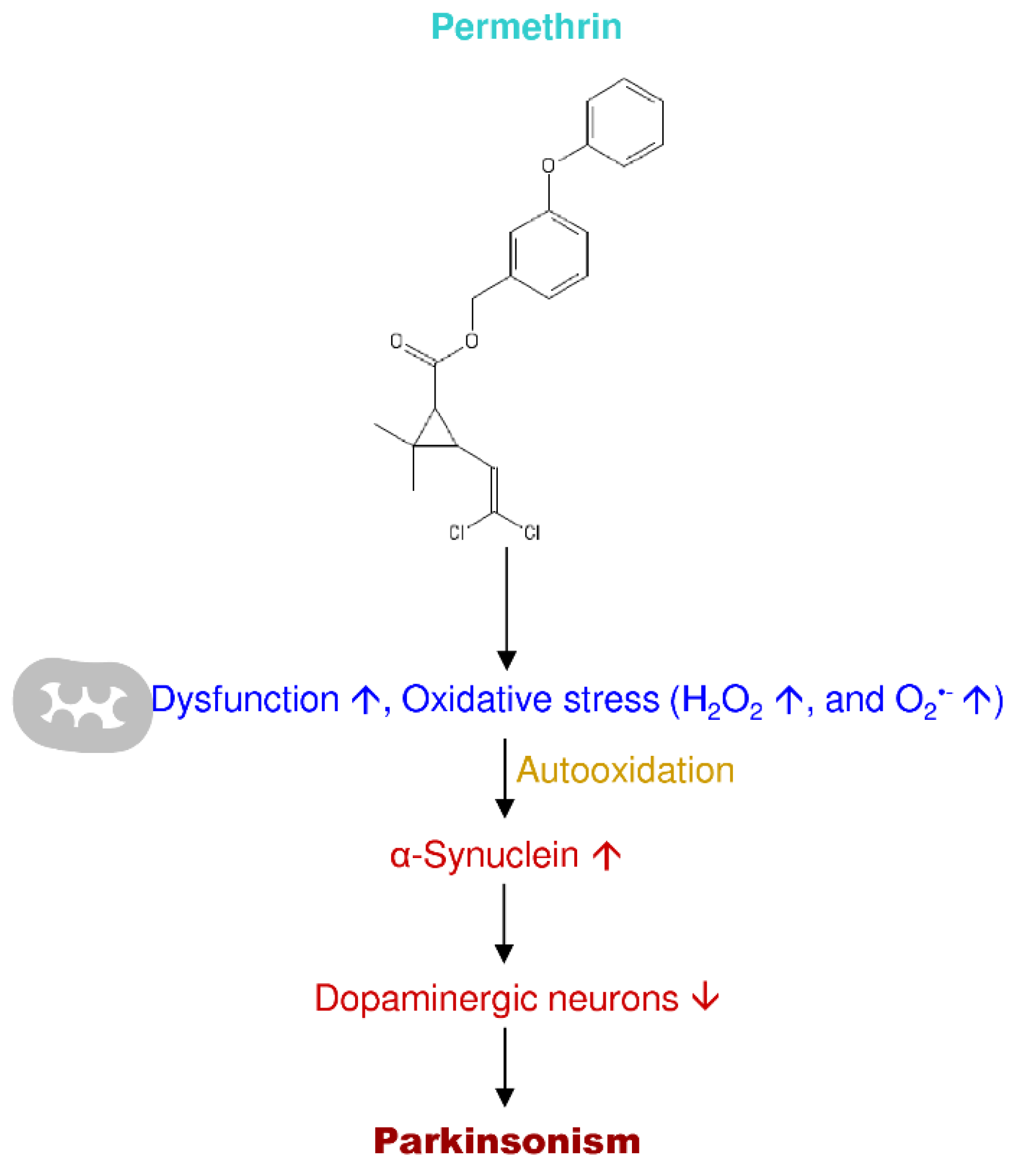
3.6. Permethrin
4. Neurotoxin-Induced Experimental In Vivo Models of PD
5. Discussion
6. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chiueh, C.C. Dopamine in the extrapyramidal motor function. A study based upon the MPTP-induced primate model of Parkinsonism. Ann. N. Y. Acad. Sci. 1988, 515, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Pankratz, N.; Foroud, T. Genetics of Parkinson Disease. Genet. Med. 2007, 9, 801–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Kowal, S.L.; Dall, T.M.; Chakrabarti, R.; Storm, M.V.; Jain, A. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord. 2013, 28, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Beck, J.C.; Bower, J.H.; Roberts, E.; Ritz, B.; Ross, G.W.; Abbott, R.D.; Savica, R.; Van Den Eeden, S.K.; Willis, A.W.; et al. Prevalence of Parkinson’s disease across North America. NPJ Parkinson’s Dis. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyers, K.R.; Dorencamp, D.G.; Suzuki, K. Amyotrophic lateral sclerosis with diffuse neurofibrillary changes: Report of a case. Arch. Neurol. 1974, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Le, W.-D.; Xie, W.-J.; Pan, T.-H.; Zhang, X.; Jankovic, J. Genetic analysis of parkin co-regulated gene (PACRG) in patients with early-onset parkinsonism. Neurosci. Lett. 2005, 382, 297–299. [Google Scholar] [CrossRef]
- Kilarski, L.L.; Pearson, J.P.; Newsway, V.; Majounie, E.; Knipe, M.D.; Misbahuddin, A.; Chinnery, P.F.; Burn, D.J.; Clarke, C.E.; Marion, M.H.; et al. Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov. Disord. 2012, 27, 1522–1529. [Google Scholar] [CrossRef]
- Haber, S.N. The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 2014, 282, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Uneven pattern of dopamine loss in the striatum of patients with idiopathic Parkinson’s disease. N. Engl. J. Med. 1988, 318, 876–880. [Google Scholar] [CrossRef]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The role of the nitric oxide pathway in brain injury and its treatment—From bench to bedside. Exp. Neurol. 2015, 263, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Boje, K.M. Nitric oxide neurotoxicity in neurodegenerative diseases. Front. Biosci. 2004, 9, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-W.; Lin, C.C.; Chen, Y.-H.; Yang, H.-B.; Hung, S.-Y. Celastrol inhibits dopaminergic neuronal death of Parkinson’s disease through activating mitophagy. Antioxidants 2020, 9, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.L. Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr. Genom. 2013, 14, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarffe, L.A.; Stevens, D.A.; Dawson, V.L.; Dawson, T.M. Parkin and PINK1: Much more than mitophagy. Trends Neurosci. 2014, 37, 315–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkanli, N.; Ay, A. The relationship between alpha-synuclein (SNCA) gene polymorphisms and development risk of Parkinson’s disease. Synucleins Biochem. Role Dis. 2019. [Google Scholar] [CrossRef] [Green Version]
- Sonne, J.; Reddy, V.; Beato, M.R. Neuroanatomy, substantia nigra. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Stoker, T.B.; Torsney, K.M.; Barker, R.A. Emerging treatment approaches for Parkinson’s disease. Front. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W.; Antonini, A.; Zijlmans, J.C.; Burkhard, P.R.; Vingerhoets, F. Levodopa in the treatment of Parkinson’s disease: An old drug still going strong. Clin. Interv. Aging 2010, 5, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.-J.; Liu, L.-Y.; Peng, W.-B.; Rao, J.; Liu, Z.; Cui, L.-L. The effectiveness of creatine treatment for Parkinson’s disease: An updated meta-analysis of randomized controlled trials. BMC Neurol. 2017, 17, 105. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.-S.; Geng, W.-S.; Jia, J.-J. Neurotoxin-induced animal models of parkinson disease: Pathogenic mechanism and assessment. ASN Neuro 2018, 10. [Google Scholar] [CrossRef]
- Dean, D.C.; Sojkova, J.; Hurley, S.; Kecskemeti, S.; Okonkwo, O.; Bendlin, B.B.; Theisen, F.; Johnson, S.C.; Alexander, A.L.; Gallagher, C.L. Alterations of myelin content in Parkinson’s disease: A cross-sectional neuroimaging study. PLoS ONE 2016, 11, e0163774. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Tampellini, D.; Gerlach, M.; Riederer, P.; Fariello, R.G.; Sulzer, D. Substantia nigra neuromelanin: Structure, synthesis, and molecular behaviour. Mol. Pathol. 2001, 54, 414–418. [Google Scholar] [PubMed]
- Elstner, M.; Müller, S.K.; Leidolt, L.; Laub, C.; Krieg, L.; Schlaudraff, F.; Liss, B.; Morris, C.; Turnbull, D.M.; Masliah, E.; et al. Neuromelanin, neurotransmitter status and brainstem location determine the differential vulnerability of catecholaminergic neurons to mitochondrial DNA deletions. Mol. Brain 2011, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson’s disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Sauer, H.; Bjorklund, A. Dopaminergic neuronal degeneration and motor impairments following axon terminal lesion by instrastriatal 6-hydroxydopamine in the rat. Neuroscience 1996, 72, 641–653. [Google Scholar] [CrossRef]
- Amano, S.; Kegelmeyer, D.; Hong, S.L. Rethinking energy in parkinsonian motor symptoms: A potential role for neural metabolic deficits. Front. Syst. Neurosci. 2015, 8. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Mazzoni, P.; Shabbott, B.; Cortés, J.C. Motor control abnormalities in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Navntoft, C.A.; Dreyer, J.K. How compensation breaks down in Parkinson’s disease: Insights from modeling of denervated striatum. Mov. Disord. 2016, 31, 280–289. [Google Scholar] [CrossRef]
- Winner, B.; Vogt-Weisenhorn, D.M.; Lie, C.D.; Blümcke, I.; Winkler, J. Cellular repair strategies in Parkinson’s disease. Ther. Adv. Neurol. Disord. 2009, 2, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, F.; Flavin, W.P.; Iqbal, S.; Pacelli, C.; Sri Renganathan, S.D.; Trudeau, L.-E.; Campbell, E.M.; Fraser, P.E.; Tandon, A. Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J. Biol. Chem. 2016, 291, 4374–4385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Xia, Y.; Wan, F.; Ma, K.; Guo, X.; Kou, L.; Yin, S.; Han, C.; Liu, L.; Huang, J.; et al. New perspectives on roles of alpha-synuclein in Parkinson’s disease. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Periquet, M.; Fulga, T.; Myllykangas, L.; Schlossmacher, M.G.; Feany, M.B. Aggregated-synuclein mediates dopaminergic neurotoxicity in vivo. J. Neurosci. 2007, 27, 3338–3346. [Google Scholar] [CrossRef]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Vasili, E.; Dominguez-Meijide, A.; Outeiro, T.F. Spreading of α-synuclein and tau: A systematic comparison of the mechanisms involved. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Kopin, I.J. Features of the dopaminergic neurotoxin MPTP. Ann. N. Y. Acad. Sci. 1992, 648, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.R.; Chesselet, M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial dysfunction in Parkinson’s disease—Cause or consequence? Biology (Basel) 2019, 8, 38. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial dysfunction in Parkinson’s disease: New mechanistic insights and therapeutic perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18. [Google Scholar] [CrossRef] [Green Version]
- Mouton-Liger, F.; Jacoupy, M.; Corvol, J.-C.; Corti, O. PINK1/Parkin-dependent mitochondrial surveillance: From pleiotropy to Parkinson’s disease. Front. Mol. Neurosci. 2017, 10. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, Parkin and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Cookson, M.R. Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Helley, M.P.; Pinnell, J.; Sportelli, C.; Tieu, K. Mitochondria: A common target for genetic mutations and environmental toxicants in Parkinson’s disease. Front. Genet. 2017, 8. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-synuclein physiology and pathology: A perspective on cellular structures and organelles. Front. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Kausar, S.; Wang, F.; Cui, H. The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cells 2018, 7, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mhyre, T.R.; Boyd, J.T.; Hamill, R.W.; Maguire-Zeiss, K.A. Parkinson’s disease. Subcell. Biochem. 2012, 65, 389–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, Y.; Morishima-Kawashima, M.; Nixon, R. The ubiquitin–proteasome system and the autophagic–lysosomal system in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [Green Version]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Petrucelli, L. A critical evaluation of the ubiquitin–proteasome system in Parkinson’s disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Sala, G.; Marinig, D.; Arosio, A.; Ferrarese, C. Role of chaperone-mediated autophagy dysfunctions in the pathogenesis of Parkinson’s disease. Front. Mol. Neurosci. 2016, 9. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Autophagic pathways in Parkinson disease and related disorders. Expert. Rev. Mol. Med. 2011, 13, e8. [Google Scholar] [CrossRef]
- Monaco, A.; Fraldi, A. Protein aggregation and dysfunction of autophagy-lysosomal pathway: A vicious cycle in lysosomal storage diseases. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Lopes da Fonseca, T.; Villar-Piqué, A.; Outeiro, T.F. The interplay between alpha-synuclein clearance and spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef] [Green Version]
- Plotegher, N.; Civiero, L. Neuronal autophagy, α-synuclein clearance, and LRRK2 regulation: A lost equilibrium in parkinsonian brain. J. Neurosci. 2012, 32, 14851–14853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsden, C.D.; Obeso, J.A.; Zarranz, J.J.; Lang, A.E. The anatomical basis of symptomatic hemidystonia. Brain 1985, 108, 463–483. [Google Scholar] [CrossRef] [PubMed]
- Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Coleman, S.; O’Brien, J.T.; Brooks, D.J.; Barker, R.A.; Burn, D.J. The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 2013, 80, 276–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marconi, R.; Lefebvre-Caparros, D.; Bonnet, A.-M.; Vidailhet, M.; Dubois, B.; Agid, Y. Levodopa-induced dyskinesias in Parkinson’s disease phenomenology and pathophysiology. Mov. Disord. 1994, 9, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Ko, H.W.; Bok, E.; Park, E.S.; Huh, S.H.; Nam, J.H.; Jin, B.W. The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep. 2010, 43, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.C.; Vyas, S.; Hunot, S. Neuroinflammation in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18 (Suppl. S1), S210–S212. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Liou, H.-C.; Kang, K.-H.; Wu, R.-M.; Wen, C.-C.; Fu, W.-M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef] [Green Version]
- Troncoso-Escudero, P.; Parra, A.; Nassif, M.; Vidal, R.L. Outside in: Unraveling the role of neuroinflammation in the progression of Parkinson’s disease. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking neuroinflammation and neurodegeneration in Parkinson’s disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef] [Green Version]
- Ambani, L.M.; Woert, M.H.V.; Murphy, S. Brain peroxidase and catalase in parkinson disease. Arch. Neurol. 1975, 32, 114–118. [Google Scholar] [CrossRef]
- Kin, K.; Yasuhara, T.; Kameda, M.; Date, I. Animal models for Parkinson’s disease research: Trends in the 2000s. Int. J. Mol. Sci. 2019, 20, 5402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlbarg, V.; Lambert, J.; Butler, B.; Felfli, M.; Valabrègue, R.; Privat, A.-L.; Lehéricy, S.; Petiet, A. Alterations of the nigrostriatal pathway in a 6-OHDA rat model of Parkinson’s disease evaluated with multimodal MRI. PLoS ONE 2018, 13, e0202597. [Google Scholar] [CrossRef] [PubMed]
- Bové, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of Parkinson’s disease. NeuroRx 2005, 2, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Chotibut, T.; Apple, D.M.; Jefferis, R.; Salvatore, M.F. Dopamine transporter loss in 6-OHDA Parkinson’s model is unmet by parallel reduction in dopamine uptake. PLoS ONE 2012, 7, e52322. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Lim, H.-W.; More, S.V.; Kim, B.-W.; Koppula, S.; Kim, I.S.; Choi, D.-K. The role of free radicals in the aging brain and Parkinson’s disease: Convergence and parallelism. Int. J. Mol. Sci. 2012, 13, 10478–10504. [Google Scholar] [CrossRef] [Green Version]
- Glinka, Y.; Gassen, M.; Youdim, M.B.H. Mechanism of 6-hydroxydopamine neurotoxicity. In Advances in Research on Neurodegeneration; Riederer, P., Calne, D.B., Horowski, R., Mizuno, Y., Poewe, W., Youdim, M.B.H., Eds.; Springer Vienna: Vienna, Austria, 1997; Volume 50, pp. 55–66. [Google Scholar]
- Varešlija, D.; Tipton, K.F.; Davey, G.P.; McDonald, A.G. 6-Hydroxydopamine: A far from simple neurotoxin. J. Neural. Transm. 2020, 127, 213–230. [Google Scholar] [CrossRef]
- Rodriguez-Pallares, J.; Parga, J.A.; Muñoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef]
- Liu, Q.; Qin, Q.; Sun, H.; Zhong, D.; An, R.; Tian, Y.; Chen, H.; Jin, J.; Wang, H.; Li, G. Neuroprotective effect of olfactory ensheathing cells co-transfected with Nurr1 and Ngn2 in both in vitro and in vivo models of Parkinson’s disease. Life Sci. 2018, 194, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Boix, J.; Padel, T.; Paul, G. A partial lesion model of Parkinson’s disease in mice—Characterization of a 6-OHDA-induced medial forebrain bundle lesion. Behav. Brain Res. 2015, 284, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Jalewa, J.; Sharma, M.K.; Gengler, S.; Hölscher, C. A novel GLP-1/GIP dual receptor agonist protects from 6-OHDA lesion in a rat model of Parkinson’s disease. Neuropharmacology 2017, 117, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; Parr-Brownlie, L.C.; Duncan, E.J.; Black, M.A.; Gemmell, N.J.; Dearden, P.K.; Reynolds, J.N.J. Striatal mRNA expression patterns underlying peak dose l-DOPA-induced dyskinesia in the 6-OHDA hemiparkinsonian rat. Neuroscience 2016, 324, 238–251. [Google Scholar] [CrossRef]
- Ribeiro, R.P.; Santos, D.B.; Colle, D.; Naime, A.A.; Gonçalves, C.L.; Ghizoni, H.; Hort, M.A.; Godoi, M.; Dias, P.F.; Braga, A.L.; et al. Decreased forelimb ability in mice intracerebroventricularly injected with low dose 6-hydroxidopamine: A model on the dissociation of bradykinesia from hypokinesia. Behav. Brain Res. 2016, 305, 30–36. [Google Scholar] [CrossRef]
- Lai, C.-L.; Lu, C.-C.; Lin, H.-C.; Sung, Y.-F.; Wu, Y.-P.; Hong, J.-S.; Peng, G.-S. Valproate is protective against 6-OHDA-induced dopaminergic neurodegeneration in rodent midbrain: A potential role of BDNF up-regulation. J. Formos. Med Assoc. 2019, 118, 420–428. [Google Scholar] [CrossRef]
- Vieira, J.C.F.; Bassani, T.B.; Santiago, R.M.; Guaita, G.d.O.; Zanoveli, J.M.; da Cunha, C.; Vital, M.A.B.F. Anxiety-like behavior induced by 6-OHDA animal model of Parkinson’s disease may be related to a dysregulation of neurotransmitter systems in brain areas related to anxiety. Behav. Brain Res. 2019, 371, 111981. [Google Scholar] [CrossRef]
- Penttinen, A.-M.; Suleymanova, I.; Albert, K.; Anttila, J.; Voutilainen, M.H.; Airavaara, M. Characterization of a new low-dose 6-hydroxydopamine model of Parkinson’s disease in rat. J. Neurosci. Res. 2016, 94, 318–328. [Google Scholar] [CrossRef]
- Quiroga-Varela, A.; Aguilar, E.; Iglesias, E.; Obeso, J.A.; Marin, C. Short- and long-term effects induced by repeated 6-OHDA intraventricular administration: A new progressive and bilateral rodent model of Parkinson’s disease. Neuroscience 2017, 361, 144–156. [Google Scholar] [CrossRef]
- Oliveira-Giacomelli, Á.; Carolina, M.A.; de Souza, H.D.N.; Corrêa-Velloso, J.; de Jesus Santos, A.P.; Baranova, J.; Ulrich, H. P2Y6 and P2X7 receptor antagonism exerts neuroprotective/neuroregenerative effects in an animal model of Parkinson’s disease. Front. Cell. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Park, S.E.; Song, K.-I.; Kim, H.; Chung, S.; Youn, I. Graded 6-OHDA-induced dopamine depletion in the nigrostriatal pathway evokes progressive pathological neuronal activities in the subthalamic nucleus of a hemi-parkinsonian mouse. Behav. Brain Res. 2018, 344, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Guimarães Marques, M.J.; Real, C.C.; Victorino, D.B.; Britto, L.R.; Cavalheiro, E.A.; Scorza, F.A.; Ferraz, H.B.; Scorza, C.A. Endogenous protection against the 6-OHDA model of Parkinson’s disease in the Amazonian rodent Proechimys. Neurosci. Lett. 2019, 709, 134381. [Google Scholar] [CrossRef]
- Sarookhani, M.R.; Haghdoost-Yazdi, H.; Sarbazi-Golezari, A.; Babayan-Tazehkand, A.; Rastgoo, N. Involvement of adenosine triphosphate-sensitive potassium channels in the neuroprotective activity of hydrogen sulfide in the 6-hydroxydopamine-induced animal model of Parkinson’s disease. Behav. Pharmacol. 2018, 29, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Romero-Sánchez, H.A.; Mendieta, L.; Austrich-Olivares, A.M.; Garza-Mouriño, G.; Benitez-Diaz Mirón, M.; Coen, A.; Godínez-Chaparro, B. Unilateral lesion of the nigroestriatal pathway with 6-OHDA induced allodynia and hyperalgesia reverted by pramipexol in rats. Eur. J. Pharmacol. 2020, 869, 172814. [Google Scholar] [CrossRef]
- Huang, N.; Zhang, Y.; Chen, M.; Jin, H.; Nie, J.; Luo, Y.; Zhou, S.; Shi, J.; Jin, F. Resveratrol delays 6-hydroxydopamine-induced apoptosis by activating the PI3K/Akt signaling pathway. Exp. Gerontol. 2019, 124, 110653. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Krishnamurthy, S. Rebamipide mitigates impairments in mitochondrial function and bioenergetics with α-synuclein pathology in 6-OHDA-induced Hemiparkinson’s model in rats. Neurotox. Res. 2019, 35, 542–562. [Google Scholar] [CrossRef]
- Lima, F.A.V.; Joventino, I.P.; Joventino, F.P.; de Almeida, A.C.; Neves, K.R.T.; do Carmo, M.R.; Leal, L.K.A.M.; de Andrade, G.M.; de Barros Viana, G.S. Neuroprotective activities of spirulina platensis in the 6-OHDA model of Parkinson’s disease are related to its anti-inflammatory effects. Neurochem. Res. 2017, 42, 3390–3400. [Google Scholar] [CrossRef]
- Szot, P.; Franklin, A.; Miguelez, C.; Wang, Y.; Vidaurrazaga, I.; Ugedo, L.; Sikkema, C.; Wilkinson, C.W.; Raskind, M.A. Depressive-like behavior observed with a minimal loss of locus coeruleus (LC) neurons following administration of 6-hydroxydopamine is associated with electrophysiological changes and reversed with precursors of norepinephrine. Neuropharmacology 2016, 101, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Kamińska, K.; Lenda, T.; Konieczny, J.; Czarnecka, A.; Lorenc-Koci, E. Depressive-like neurochemical and behavioral markers of Parkinson’s disease after 6-OHDA administered unilaterally to the rat medial forebrain bundle. Pharmacol. Rep. 2017, 69, 985–994. [Google Scholar] [CrossRef]
- Haddadi, R.; Eyvari Brooshghalan, S.; Farajniya, S.; Mohajjel Nayebi, A.; Sharifi, H. Short-term treatment with silymarin improved 6-OHDA-induced catalepsy and motor imbalance in hemi-parkisonian rats. Adv. Pharm. Bull. 2015, 5, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Haddadi, R.; Poursina, M.; Zeraati, F.; Nadi, F. Gastrodin microinjection suppresses 6-OHDA-induced motor impairments in parkinsonian rats: Insights into oxidative balance and microglial activation in SNc. Inflammopharmacology 2018, 26, 1305–1316. [Google Scholar] [CrossRef]
- Kwan, C.; Frouni, I.; Bédard, D.; Hamadjida, A.; Huot, P. Ondansetron, a highly selective 5-HT3 receptor antagonist, reduces L-DOPA-induced dyskinesia in the 6-OHDA-lesioned rat model of Parkinson’s disease. Eur. J. Pharmacol. 2020, 871, 172914. [Google Scholar] [CrossRef]
- Gomes, F.A.; Flores, R.A.; Bruxel, M.A.; da Silva, F.N.; Moreira, E.L.G.; Zoccal, D.B.; Prediger, R.D.; Rafacho, A. Glucose Homeostasis Is Not Affected in a Murine Model of Parkinson’s Disease Induced by 6-OHDA. Front. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Voronin, M.V.; Kadnikov, I.A.; Voronkov, D.N.; Seredenin, S.B. Chaperone Sigma1R mediates the neuroprotective action of afobazole in the 6-OHDA model of Parkinson’s disease. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Konieczny, J.; Czarnecka, A.; Lenda, T.; Kamińska, K.; Antkiewicz-Michaluk, L. The significance of rotational behavior and sensitivity of striatal dopamine receptors in hemiparkinsonian rats: A comparative study of lactacystin and 6-OHDA. Neuroscience 2017, 340, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-W.; Hsueh, S.-C.; Lai, J.-H.; Chen, Y.-H.; Kang, S.-J.; Chen, K.-Y.; Hsieh, T.-H.; Hoffer, B.J.; Li, Y.; Greig, N.H.; et al. Glucose-dependent insulinotropic polypeptide mitigates 6-OHDA-induced behavioral impairments in parkinsonian rats. Int. J. Mol. Sci. 2018, 19, 1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-T.; Kao, L.-T.; Shih, J.-H.; Chien, W.-C.; Chiu, C.-H.; Ma, K.-H.; Huang, Y.-S.; Cheng, C.-Y.; Shiue, C.-Y.; Li, I.H. The effect of dextromethorphan use in Parkinson’s disease: A 6-hydroxydopamine rat model and population-based study. Eur. J. Pharmacol. 2019, 862, 172639. [Google Scholar] [CrossRef] [PubMed]
- Su, R.; Sun, M.; Wang, W.; Zhang, J.; Zhang, L.; Zhen, J.; Qian, Y.; Zheng, Y.; Wang, X. A novel immunosuppressor, (5R)-5-hydroxytriptolide, alleviates movement disorder and neuroinflammation in a 6-OHDA hemiparkinsonian rat model. Aging Dis. 2017, 8, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.-F.; Xie, C.-L.; Lin, J.-Y.; Wang, M.-H.; Wang, X.-J.; Liu, Z.-G. Lipoic acid alleviates L-DOPA-induced dyskinesia in 6-OHDA parkinsonian rats via anti-oxidative stress. Mol. Med. Rep. 2018, 17, 1118–1124. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Tuazon, J.P.; Corey, S.; Bonsack, B.; Acosta, S.; Ehrhart, J.; Sanberg, P.R.; Borlongan, C.V. A gutsy move for cell-based regenerative medicine in Parkinson’s disease: Targeting the gut microbiome to sequester inflammation and neurotoxicity. Stem Cell Rev. Rep. 2019, 15, 690–702. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Shu, H.; Li, L.; Zhen, T.; Zhao, J.; Zhou, X.; Luo, W. L-DOPA-induced motor impairment and overexpression of corticostriatal synaptic components are improved by the mGluR5 antagonist MPEP in 6-OHDA-lesioned rats. ASN Neuro 2018, 10, 1759091418811021. [Google Scholar] [CrossRef] [PubMed]
- Crabbé, M.; Van der Perren, A.; Weerasekera, A.; Himmelreich, U.; Baekelandt, V.; Van Laere, K.; Casteels, C. Altered mGluR5 binding potential and glutamine concentration in the 6-OHDA rat model of acute Parkinson’s disease and levodopa-induced dyskinesia. Neurobiol. Aging 2018, 61, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Soares de Souza, B.; Roversi, K.; Schuh, T.; Poli, A.; Takahashi, R.N.; Prediger, R.D. Temporal development of behavioral impairments in rats following locus coeruleus lesion induced by 6-hydroxydopamine: Involvement of β3-adrenergic receptors. Neuropharmacology 2019, 151, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Long, H.; Zhou, F.; Zhu, W.; Ruan, J.; Zhao, Y.; Lu, Y. Echinacoside’s nigrostriatal dopaminergic protection against 6-OHDA-Induced endoplasmic reticulum stress through reducing the accumulation of Seipin. J. Cell. Mol. Med. 2017, 21, 3761–3775. [Google Scholar] [CrossRef] [Green Version]
- Vaz, R.L.; Chapela, D.; Coelho, J.E.; Lopes, L.V.; Ferreira, J.J.; Afonso, N.D.; Sousa, S.; Outeiro, T.F. Tapentadol prevents motor impairments in a mouse model of dyskinesia. Neuroscience 2020, 424, 58–71. [Google Scholar] [CrossRef]
- Manouchehrabadi, M.; Farhadi, M.; Azizi, Z.; Torkaman-Boutorabi, A. Carvacrol protects against 6-Hydroxydopamine-induced neurotoxicity in in vivo and in vitro models of Parkinson’s disease. Neurotox. Res. 2020, 37, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Shi, C.; Cao, J.; Sun, Y.; Zhao, X.; Guo, Y.; Wang, C.; Lei, H.; Jiang, H.; Ablat, N.; et al. Neuroprotective effects of a standardized flavonoid extract of safflower against neurotoxin-induced cellular and animal models of Parkinson’s disease. Sci. Rep. 2016, 6, 22135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuliano, C.; Siani, F.; Mus, L.; Ghezzi, C.; Cerri, S.; Pacchetti, B.; Bigogno, C.; Blandini, F. Neuroprotective effects of lignan 7-hydroxymatairesinol (HMR/lignan) in a rodent model of Parkinson’s disease. Nutrition 2020, 69, 110494. [Google Scholar] [CrossRef]
- Wattanathorn, J.; Sutalangka, C. Novel food supplement “CP1” improves motor deficit, cognitive function, and neurodegeneration in animal model of Parkinson’s disease. Rejuvenation Res. 2015, 19, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cheng, L.; Wei, X.; Yuan, Z.; Wu, Y.; Wang, S.; Ren, Z.; Liu, X.; Liu, H. Tetramethylpyrazine analogue CXC195 protects against dopaminergic neuronal apoptosis via activation of PI3K/Akt/GSK3β signaling pathway in 6-OHDA-induced Parkinson’s disease mice. Neurochem. Res. 2017, 42, 1141–1150. [Google Scholar] [CrossRef]
- Ren, M.; Han, M.; Wei, X.; Guo, Y.; Shi, H.; Zhang, X.; Perez, R.G.; Lou, H. FTY720 attenuates 6-OHDA-associated dopaminergic degeneration in cellular and mouse parkinsonian models. Neurochem. Res. 2017, 42, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W. The MPTP story. J. Parkinsons Dis. 2017, 7, S11–S19. [Google Scholar] [CrossRef] [Green Version]
- Ballard, P.A.; Tetrud, J.W.; Langston, J. Permanent human parkinsonism due to 1-methy 1–4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurology 1985. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, G.C.; Williams, A.C.; Markey, S.P.; Ebert, M.H.; Caine, E.D.; Reichert, C.M.; Kopin, I.J. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979, 1, 249–254. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V.; Djaldetti, R.; Liberatore, G.; Vila, M.; Vukosavic, S.; Almer, G. The parkinsonian toxin MPTP: Action and mechanism. Restor. Neurol. Neurosci. 2000, 16, 135–142. [Google Scholar]
- Ransom, B.R.; Kunis, D.M.; Irwin, I.; Langston, J.W. Astrocytes convert the Parkinsonism inducing neurotoxin, MPTP, to its active metabolite, MPP+. Neurosci. Lett. 1987, 75, 323–328. [Google Scholar] [CrossRef]
- Uchida, S.-I.; Soshiroda, K.; Okita, E.; Kawai-Uchida, M.; Mori, A.; Jenner, P.; Kanda, T. The adenosine A2A receptor antagonist, istradefylline enhances anti-parkinsonian activity induced by combined treatment with low doses of L-DOPA and dopamine agonists in MPTP-treated common marmosets. Eur. J. Pharmacol. 2015, 766, 25–30. [Google Scholar] [CrossRef]
- Lin, J.G.; Chen, C.J.; Yang, H.B.; Chen, Y.H.; Hung, S.Y. Electroacupuncture promotes recovery of motor function and reduces dopaminergic neuron degeneration in rodent models of Parkinson’s disease. Int. J. Mol. Sci. 2017, 18, 1846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Lee, S.Y.; Shin, J.; Hwang, J.-T.; Jeon, H.N.; Bae, H. Dose-dependent neuroprotective effect of standardized bee venom phospholipase A2 against MPTP-induced Parkinson’s disease in mice. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, S.; Zhu, D.; Tang, X.; Che, Y.; Feng, X. The parthenolide derivative ACT001 synergizes with low doses of L-DOPA to improve MPTP-induced Parkinson’s disease in mice. Behav. Brain Res. 2020, 379, 112337. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, D.; Jiang, P.; Tang, X.; Lang, Q.; Yu, Q.; Zhang, S.; Che, Y.; Feng, X. Resveratrol synergizes with low doses of L-DOPA to improve MPTP-induced Parkinson disease in mice. Behav. Brain Res. 2019, 367, 10–18. [Google Scholar] [CrossRef]
- Biju, K.C.; Evans, R.C.; Shrestha, K.; Carlisle, D.C.B.; Gelfond, J.; Clark, R.A. Methylene blue ameliorates olfactory dysfunction and motor deficits in a chronic MPTP/probenecid mouse model of Parkinson’s disease. Neuroscience 2018, 380, 111–122. [Google Scholar] [CrossRef]
- Datta, I.; Mekha, S.R.; Kaushal, A.; Ganapathy, K.; Razdan, R. Influence of intranasal exposure of MPTP in multiple doses on liver functions and transition from non-motor to motor symptoms in a rat PD model. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2020, 393, 147–165. [Google Scholar] [CrossRef]
- Roostalu, U.; Salinas, C.B.G.; Thorbek, D.D.; Skytte, J.L.; Fabricius, K.; Barkholt, P.; John, L.M.; Jurtz, V.I.; Knudsen, L.B.; Jelsing, J.; et al. Quantitative whole-brain 3D imaging of tyrosine hydroxylase-labeled neuron architecture in the mouse MPTP model of Parkinson’s disease. Dis. Models Mech. 2019, 12, dmm042200. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.-F.; Wu, L.; Du, Z.-R.; Chen, L.; Xu, A.-L.; Chen, X.-H.; Teng, J.-J.; Wong, M.-S. Neuroprotective properties of icariin in MPTP-induced mouse model of Parkinson’s disease: Involvement of PI3K/Akt and MEK/ERK signaling pathways. Phytomedicine 2017, 25, 93–99. [Google Scholar] [CrossRef]
- Zheng, M.; Liu, C.; Fan, Y.; Yan, P.; Shi, D.; Zhang, Y. Neuroprotection by Paeoniflorin in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 2017, 116, 412–420. [Google Scholar] [CrossRef]
- Ko, W.K.D.; Camus, S.M.; Li, Q.; Yang, J.; McGuire, S.; Pioli, E.Y.; Bezard, E. An evaluation of istradefylline treatment on Parkinsonian motor and cognitive deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated macaque models. Neuropharmacology 2016, 110, 48–58. [Google Scholar] [CrossRef]
- Zhou, T.; Zhu, M.; Liang, Z. (-)-Epigallocatechin-3-gallate modulates peripheral immunity in the MPTP-induced mouse model of Parkinson’s disease. Mol. Med. Rep. 2018, 17, 4883–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.V.; Cheung, C.Y.; Lyu, H.; Chan, H.Y.; Li, Y.; Bian, Z.X.; Wang, K.K.W.; Poon, W.S. Baicalein enhances the effect of low dose Levodopa on the gait deficits and protects dopaminergic neurons in experimental Parkinsonism. J. Clin. Neurosci. 2019, 64, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Zeng, S.; Tang, R.; Tao, G.; Chan, P. MPTP induces systemic Parkinsonism in middle-aged cynomolgus monkeys: Clinical evolution and outcomes. Neurosci. Bull. 2016, 33, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Lee, Y.; Kim, B.S.; Park, J.; Yang, S.; Yoon, H.-J.; Yoo, J.; Park, H.S.; Hong, J.-J.; Koo, B.-S.; et al. A non-human primate model for stable chronic Parkinson’s disease induced by MPTP administration based on individual behavioral quantification. J. Neurosci. Methods 2019, 311, 277–287. [Google Scholar] [CrossRef]
- Franke, S.K.; van Kesteren, R.E.; Wubben, J.A.M.; Hofman, S.; Paliukhovich, I.; van der Schors, R.C.; van Nierop, P.; Smit, A.B.; Philippens, I.H.C.H.M. Progression and recovery of Parkinsonism in a chronic progressive MPTP-induction model in the marmoset without persistent molecular and cellular damage. Neuroscience 2016, 312, 247–259. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Glud, A.N.; Møller, A.; Mogensen, P.; Bender, D.; Sørensen, J.C.; Doudet, D.; Bjarkam, C.R. Continuous MPTP intoxication in the Göttingen minipig results in chronic parkinsonian deficits. Acta Neurobiol. Exp. (Wars) 2016, 76, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.-J.; Kwon, K.-C.; Song, H.-K.; Kim, K.-S.; Jung, Y.-S.; Hwang, D.-Y.; Cho, J.-Y. Comparative analysis of dose-dependent neurotoxic response to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57BL/6 N mice derived from three different sources. Lab. Anim. Res. 2019, 35, 10. [Google Scholar] [CrossRef]
- Hu, A.-L.; Song, S.; Li, Y.; Xu, S.-F.; Zhang, F.; Li, C.; Liu, J. Mercury sulfide-containing Hua-Feng-Dan and 70W (Rannasangpei) protect against LPS plus MPTP-induced neurotoxicity and disturbance of gut microbiota in mice. J. Ethnopharmacol. 2020, 254, 112674. [Google Scholar] [CrossRef]
- Yun, J.-W.; Ahn, J.-B.; Kwon, E.; Ahn, J.H.; Park, H.W.; Heo, H.; Park, J.-S.; Kim, H.; Paek, S.H.; Kang, B.-C. Behavior, PET and histology in novel regimen of MPTP marmoset model of Parkinson’s disease for long-term stem cell therapy. Tissue Eng. Regen. Med. 2015, 13, 100–109. [Google Scholar] [CrossRef]
- Arbez, N.; He, X.; Huang, Y.; Ren, M.; Liang, Y.; Nucifora, F.C.; Wang, X.; Pei, Z.; Tessarolo, L.; Smith, W.W.; et al. G2019S-LRRK2 mutation enhances MPTP-linked Parkinsonism in mice. Hum. Mol. Genet. 2020, 29, 580–590. [Google Scholar] [CrossRef]
- Guo, Z.; Xu, S.; Du, N.; Liu, J.; Huang, Y.; Han, M. Neuroprotective effects of stemazole in the MPTP-induced acute model of Parkinson’s disease: Involvement of the dopamine system. Neurosci. Lett. 2016, 616, 152–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, F.; Seguella, L.; Gigli, S.; Hanieh, P.N.; Del Favero, E.; Cantù, L.; Pesce, M.; Sarnelli, G.; Marianecci, C.; Esposito, G.; et al. inPentasomes: An innovative nose-to-brain pentamidine delivery blunts MPTP parkinsonism in mice. J. Control. Release 2019, 294, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Nataraj, J.; Manivasagam, T.; Thenmozhi, A.J.; Essa, M.M. Lutein protects dopaminergic neurons against MPTP-induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutr. Neurosci. 2016, 19, 237–246. [Google Scholar] [CrossRef]
- Zhou, H.; Niu, L.; Meng, L.; Lin, Z.; Zou, J.; Xia, X.; Huang, X.; Zhou, W.; Bian, T.; Zheng, H. Noninvasive ultrasound deep brain stimulation for the treatment of Parkinson’s disease model mouse. Research (Wash. DC) 2019, 2019, 1748489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.J.; Panhelainen, A.; Schmitz, Y.; Larsen, K.E.; Kanter, E.; Wu, M.; Sulzer, D.; Mosharov, E.V. Changes in neuronal dopamine homeostasis following 1-Methyl-4-phenylpyridinium (MPP+) exposure. J. Biol. Chem. 2015, 290, 6799–6809. [Google Scholar] [CrossRef] [Green Version]
- Miller, G.; Kirby, M.L.; Levey, A.; Bloomquist, J. Heptachlor alters expression and function of dopamine transporters. NeuroToxicology 1999, 20, 631–637. [Google Scholar]
- Hsieh, Y.-C.; Mounsey, R.B.; Teismann, P. MPP+-induced toxicity in the presence of dopamine is mediated by COX-2 through oxidative stress. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Lotharius, J.; O’Malley, K.L. The parkinsonism-inducing drug 1-Methyl-4-phenylpyridinium triggers intracellular dopamine oxidation: A novel mechanism of toxicity. J. Biol. Chem. 2000, 275, 38581–38588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, M.; Neis, V.B.; Matheus, F.C.; Cunha, M.P.; Rosa, P.B.; Ribeiro, C.M.; Rodrigues, A.L.S.; Prediger, R.D. Effects of agmatine on depressive-like behavior induced by intracerebroventricular administration of 1-Methyl-4-phenylpyridinium (MPP+). Neurotox. Res. 2015, 28, 222–231. [Google Scholar] [CrossRef]
- Cunha, M.P.; Pazini, F.L.; Lieberknecht, V.; Budni, J.; Oliveira, Á.; Rosa, J.M.; Mancini, G.; Mazzardo, L.; Colla, A.R.; Leite, M.C.; et al. MPP+-lesioned mice: An experimental model of motor, emotional, memory/learning, and striatal neurochemical dysfunctions. Mol. Neurobiol. 2017, 54, 6356–6377. [Google Scholar] [CrossRef]
- Pérez-Barrón, G.; Ávila-Acevedo, J.G.; García-Bores, A.M.; Montes, S.; García-Jiménez, S.; León-Rivera, I.; Rubio-Osornio, M.; Monroy-Noyola, A. Neuroprotective effect of Buddleja cordata methanolic extract in the 1-methyl-4-phenylpyridinium Parkinson’s disease rat model. J. Nat. Med. 2015, 69, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Osornio, M.; Gorostieta-Salas, E.; Montes, S.; Pérez-Severiano, F.; Rubio, C.; Gómez, C.; Ríos, C.; Guevara, J. Epicatechin reduces striatal MPP+-induced damage in rats through slight increases in SOD-Cu,Zn activity. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Vidal, Y.; Monroy-Noyola, A.; Anaya-Ramos, L.; Arteaga-Silva, M.; Mendez-Armenta, M.; Ostoa-Saloma, P.; Díaz-Zaragoza, M.; Morales-Montor, J.; Ríos, C.; Montes, S. β-Estradiol-3-benzoate confers neuroprotection in Parkinson MPP+ rat model through inhibition of lipid peroxidation. Steroids 2017, 126, 7–14. [Google Scholar] [CrossRef]
- Aguirre-Vidal, Y.; Montes, S.; Tristan-López, L.; Anaya-Ramos, L.; Teiber, J.; Ríos, C.; Baron-Flores, V.; Monroy-Noyola, A. The neuroprotective effect of lovastatin on MPP+-induced neurotoxicity is not mediated by PON2. NeuroToxicology 2015, 48, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, Z.; Cao, B.-B.; Qiu, Y.-H.; Peng, Y.-P. TGF-β1 neuroprotection via inhibition of microglial activation in a rat model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2017, 12, 433–446. [Google Scholar] [CrossRef]
- Boon, W.R. The Chemistry and Mode of Action of the Bipyridylium Herbicides Diquat and Paraquat. Outlook Agric. 1964, 4, 163–170. [Google Scholar] [CrossRef]
- Smith, L.L. Mechanism of paraquat toxicity in lung and its relevance to treatment. Hum. Toxicol. 1987, 6, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Li, S.; Rodriguez-Rocha, H.; Burns, M.; Panayiotidis, M.I. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2010, 188, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Dinis-Oliveira, R.J.; Duarte, J.A.; Sánchez-Navarro, A.; Remião, F.; Bastos, M.L.; Carvalho, F. Paraquat poisonings: Mechanisms of lung toxicity, clinical features, and treatment. Crit. Rev. Toxicol. 2008, 38, 13–71. [Google Scholar] [CrossRef]
- Wunnapuk, K.; Mohammed, F.; Gawarammana, I.; Liu, X.; Verbeeck, R.K.; Buckley, N.A.; Roberts, M.S.; Musuamba, F.T. Prediction of paraquat exposure and toxicity in clinically ill poisoned patients: A model based approach. Br. J. Clin. Pharmacol. 2014, 78, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Carr, R.J.; Bilton, R.F.; Atkinson, T. Mechanism of biodegradation of paraquat by Lipomyces starkeyi. Appl. Environ. Microbiol. 1985, 49, 1290–1294. [Google Scholar] [CrossRef] [Green Version]
- Murray, R.E.; Gibson, J.E. Paraquat disposition in rats, guinea pigs and monkeys. Toxicol. Appl. Pharmacol. 1974, 27, 283–291. [Google Scholar] [CrossRef]
- Shimada, H.; Furuno, H.; Hirai, K.-I.; Koyama, J.; Ariyama, J.; Simamura, E. Paraquat detoxicative system in the mouse liver postmitochondrial fraction. Arch. Biochem. Biophys. 2002, 402, 149–157. [Google Scholar] [CrossRef]
- Philippot, G.; Stenerlöw, B.; Fredriksson, A.; Sundell-Bergman, S.; Eriksson, P.; Buratovic, S. Developmental effects of neonatal fractionated co-exposure to low-dose gamma radiation and paraquat on behaviour in adult mice. J. Appl. Toxicol. 2019, 39, 582–589. [Google Scholar] [CrossRef] [PubMed]
- Rudyk, C.A.; McNeill, J.; Prowse, N.; Dwyer, Z.; Farmer, K.; Litteljohn, D.; Caldwell, W.; Hayley, S. Age and chronicity of administration dramatically influenced the impact of low dose paraquat exposure on behavior and hypothalamic-pituitary-adrenal activity. Front. Aging Neurosci. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, L.; Bove, C.; Coleman, F.H.; Le, K.; Subramanian, M.P.; Venkiteswaran, K.; Subramanian, T.; Travagli, R.A. Ingestion of subthreshold doses of environmental toxins induces ascending Parkinsonism in the rat. NPJ Parkinson’s Dis. 2018, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Rudyk, C.; Dwyer, Z.; McNeill, J.; Salmaso, N.; Farmer, K.; Prowse, N.; Hayley, S. Chronic unpredictable stress influenced the behavioral but not the neurodegenerative impact of paraquat. Neurobiol. Stress 2019, 11, 100179. [Google Scholar] [CrossRef] [PubMed]
- Cristóvão, A.C.; Campos, F.L.; Je, G.; Esteves, M.; Guhathakurta, S.; Yang, L.; Beal, M.F.; Fonseca, B.M.; Salgado, A.J.; Queiroz, J.; et al. Characterization of a Parkinson’s disease rat model using an upgraded paraquat exposure paradigm. Eur. J. Neurosci. 2020. [Google Scholar] [CrossRef]
- Ait-Bali, Y.; Ba-M’hamed, S.; Bennis, M. Prenatal Paraquat exposure induces neurobehavioral and cognitive changes in mice offspring. Environ. Toxicol. Pharmacol. 2016, 48, 53–62. [Google Scholar] [CrossRef]
- Heydari, M.; Mokhtari-Zaer, A.; Amin, F.; Memarzia, A.; Saadat, S.; Hosseini, M.; Boskabady, M.H. The effect of Zataria multiflora hydroalcoholic extract on memory and lung changes induced by rats that inhaled paraquat. Nutr. Neurosci. 2019, 1–14. [Google Scholar] [CrossRef]
- Gonçalves, C.; dos Santos, D.B.; Portilho, S.S.; Lopes, M.W.; Ghizoni, H.; de Souza, V.; Mack, J.M.; Naime, A.A.; Dafre, A.L.; de Souza Brocardo, P.; et al. Lipopolysaccharide-induced striatal nitrosative stress and impaired social recognition memory are not magnified by Paraquat coexposure. Neurochem. Res. 2018, 43, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular senescence is induced by the environmental neurotoxin paraquat and contributes to neuropathology linked to Parkinson’s disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caito, S.W.; Aschner, M. Mitochondrial redox dysfunction and environmental exposures. Antioxid. Redox Signal. 2015, 23, 578–595. [Google Scholar] [CrossRef]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.L.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef] [Green Version]
- Xiong, N.; Huang, J.; Zhang, Z.; Zhang, Z.; Xiong, J.; Liu, X.; Jia, M.; Wang, F.; Chen, C.; Cao, X.; et al. Stereotaxical infusion of rotenone: A reliable rodent model for Parkinson’s disease. PLoS ONE 2009, 4, e7878. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Beiram, R.; Azimullah, S.; Meeran, M.F.N.; Ojha, S.K.; Adem, A.; Jalal, F.Y. Lycopodium attenuates loss of dopaminergic neurons by suppressing oxidative stress and neuroinflammation in a rat model of Parkinson’s disease. Molecules 2019, 24, 2182. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Wang, X.; Chen, H.; Yan, Z.; Wang, M.; Li, Y. Neurochemical and behavior deficits in rats with iron and rotenone Co-treatment: Role of redox imbalance and neuroprotection by Biochanin A. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Zhang, Z.-N.; Zhang, J.-S.; Xiang, J.; Yu, Z.-H.; Zhang, W.; Cai, M.; Li, X.-T.; Wu, T.; Li, W.-W.; Cai, D.-F. Subcutaneous rotenone rat model of Parkinson’s disease: Dose exploration study. Brain Res. 2017, 1655, 104–113. [Google Scholar] [CrossRef]
- Bandookwala, M.; Sahu, A.K.; Thakkar, D.; Sharma, M.; Khairnar, A.; Sengupta, P. Edaravone-caffeine combination for the effective management of rotenone induced Parkinson’s disease in rats: An evidence based affirmative from a comparative analysis of behavior and biomarker expression. Neurosci. Lett. 2019, 711, 134438. [Google Scholar] [CrossRef]
- Palle, S.; Neerati, P. Improved neuroprotective effect of resveratrol nanoparticles as evinced by abrogation of rotenone-induced behavioral deficits and oxidative and mitochondrial dysfunctions in rat model of Parkinson’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Alikatte, K.; Palle, S.; Kumar, J.R.; Pathakala, N. Fisetin improved rotenone-induced behavioral deficits, oxidative changes, and mitochondrial dysfunctions in rat model of Parkinson’s disease. J. Diet. Suppl. 2020, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Darbinyan, L.V.; Hambardzumyan, L.E.; Simonyan, K.V.; Chavushyan, V.A.; Manukyan, L.P.; Badalyan, S.A.; Khalaji, N.; Sarkisian, V.H. Protective effects of curcumin against rotenone-induced rat model of Parkinson’s disease: In vivo electrophysiological and behavioral study. Metab. Brain Dis. 2017, 32, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Dhanalakshmi, C.; Janakiraman, U.; Manivasagam, T.; Justin Thenmozhi, A.; Essa, M.M.; Kalandar, A.; Khan, M.A.S.; Guillemin, G.J. Vanillin attenuated behavioural impairments, neurochemical deficts, oxidative stress and apoptosis against rotenone induced rat model of Parkinson’s disease. Neurochem. Res. 2016, 41, 1899–1910. [Google Scholar] [CrossRef]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef]
- Carriere, C.H.; Kang, N.H.; Niles, L.P. Chronic low-dose melatonin treatment maintains nigrostriatal integrity in an intrastriatal rotenone model of Parkinson’s disease. Brain Res. 2016, 1633, 115–125. [Google Scholar] [CrossRef]
- Ross, M.K. Pyrethroids. In Encyclopedia of Environmental Health; Nriagu, J.O., Ed.; Elsevier: Burlington, IA, USA, 2011; pp. 702–708. [Google Scholar] [CrossRef]
- Gaughan, L.C.; Unai, T.; Casida, J.E. Permethrin metabolism in rats. J. Agric. Food Chem. 1977, 25, 9–17. [Google Scholar] [CrossRef]
- Bordoni, L.; Nasuti, C.; Di Stefano, A.; Marinelli, L.; Gabbianelli, R. Epigenetic memory of early-life parental perturbation: Dopamine decrease and DNA methylation changes in offspring. Oxid. Med. Cell. Longev. 2019, 2019, 1472623. [Google Scholar] [CrossRef]
- Gassner, B.; Wüthrich, A.; Scholtysik, G.; Solioz, M. The pyrethroids permethrin and cyhalothrin are potent inhibitors of the mitochondrial complex I. J. Pharmacol. Exp. Ther. 1997, 281, 855–860. [Google Scholar]
- Drago, B.; Shah, N.S.; Shah, S.H. Acute permethrin neurotoxicity: Variable presentations, high index of suspicion. Toxicol. Rep. 2014, 1, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, L.; Gabbianelli, R. Mitochondrial DNA and neurodegeneration: Any role for dietary antioxidants? Antioxidants (Basel) 2020, 9, 764. [Google Scholar] [CrossRef]
- Saito, H.; Hara, K.; Tominaga, T.; Nakashima, K.; Tanemura, K. Early-life exposure to low levels of permethrin exerts impairments in learning and memory with the effects on neuronal and glial population in adult male mice. J. Appl. Toxicol. 2019, 39, 1651–1662. [Google Scholar] [CrossRef]
- Nasuti, C.; Brunori, G.; Eusepi, P.; Marinelli, L.; Ciccocioppo, R.; Gabbianelli, R. Early life exposure to permethrin: A progressive animal model of Parkinson’s disease. J. Pharmacol. Toxicol. Methods 2017, 83, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Golde, T.E.; Tourenne, C.L. Animal models of neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1370–1379. [Google Scholar] [CrossRef] [PubMed]
- Jagmag, S.A.; Tripathi, N.; Shukla, S.D.; Maiti, S.; Khurana, S. Evaluation of models of Parkinson’s disease. Front. Neurosci. 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Alexander, G.E. Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin. Neurosci. 2004, 6, 259–280. [Google Scholar]
- Magrinelli, F.; Picelli, A.; Tocco, P.; Federico, A.; Roncari, L.; Smania, N.; Zanette, G.; Tamburin, S. Pathophysiology of motor dysfunction in Parkinson’s disease as the rationale for drug treatment and rehabilitation. Parkinsons Dis. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Potashkin, J.A.; Blume, S.R.; Runkle, N.K. Limitations of animal models of Parkinson’s disease. Parkinson’s Dis. 2011, 2011, 658083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.E.; Winslow, J.T. Non-human primates: Model animals for developmental psychopathology. Neuropsychopharmacology 2009, 34, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical perspective: Models of Parkinson’s disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [Green Version]
- Masato, A.; Plotegher, N.; Boassa, D.; Bubacco, L. Impaired dopamine metabolism in Parkinson’s disease pathogenesis. Mol. Neurodegener. 2019, 14. [Google Scholar] [CrossRef] [Green Version]
- Kouli, A.; Torsney, K.M.; Kuan, W.-L. Parkinson’s disease: Etiology, neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Doherty, K.M.; Hardy, J. Parkin disease and the lewy body conundrum. Mov. Disord. 2013, 28, 702–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulopoulos, M.; Levy, O.A.; Alcalay, R.N. The neuropathology of genetic Parkinson’s disease. Mov. Disord. 2012, 27, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Skogar, O.; Nilsson, M. Distribution of non-motor symptoms in idiopathic Parkinson’s disease and secondary Parkinsonism. J. Multidiscip. Healthc. 2018, 11, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Tibar, H.; El Bayad, K.; Bouhouche, A.; Ait Ben Haddou, E.H.; Benomar, A.; Yahyaoui, M.; Benazzouz, A.; Regragui, W. Non-motor symptoms of Parkinson’s disease and their impact on quality of life in a cohort of Moroccan patients. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.M.; Jaumotte, J.D.; Signore, A.P.; Zigmond, M.J. Effects of 6-hydroxydopamine on primary cultures of substantia nigra: Specific damage to dopamine neurons and the impact of glial cell line-derived neurotrophic factor. J. Neurochem. 2004, 89, 776–787. [Google Scholar] [CrossRef]
- Nishijima, H.; Tomiyama, M. What mechanisms are responsible for the reuptake of levodopa-derived dopamine in parkinsonian striatum? Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Konnova, E.A.; Swanberg, M. Animal models of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects; Stoker, T.B., Greenland, J.C., Eds.; Codon Publications: Brisbane, Australia, 2018. [Google Scholar]
- Zeiss, C.J.; Allore, H.G.; Beck, A.P. Established patterns of animal study design undermine translation of disease-modifying therapies for Parkinson’s disease. PLoS ONE 2017, 12, e0171790. [Google Scholar] [CrossRef]
- Michel, P.P.; Hefti, F. Toxicity of 6-hydroxydopamine and dopamine for dopaminergic neurons in culture. J. Neurosci. Res. 1990, 26, 428–435. [Google Scholar] [CrossRef]
- Streeter, A.J.; Harvison, P.J.; Nelson, S.D.; Baillie, T.A. Cross-linking of protein molecules by the reactive metabolite of acetaminophen, N-acetyl-p-benzoquinone imine, and related quinoid compounds. Adv. Exp. Med. Biol. 1986, 197, 727–737. [Google Scholar] [CrossRef]
- Graham, D.G. Oxidative pathways for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 1978, 14, 633–643. [Google Scholar] [PubMed]
- Campbell, J.C.; Seiden, L.S. Performance influence on the development of tolerance to amphetamine. Pharmacol. Biochem. Behav. 1973, 1, 703–708. [Google Scholar] [CrossRef]
- Miyanishi, K.; Choudhury, M.E.; Watanabe, M.; Kubo, M.; Nomoto, M.; Yano, H.; Tanaka, J. Behavioral tests predicting striatal dopamine level in a rat hemi-Parkinson’s disease model. Neurochem. Int. 2019, 122, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.S.; Larson, G.A.; Swant, J.; Sen, N.; Javitch, J.A.; Zahniser, N.R.; De Felice, L.J.; Khoshbouei, H. Amphetamine and methamphetamine differentially affect dopamine transporters in vitro and in vivo. J. Biol. Chem. 2009, 284, 2978–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Björklund, A.; Dunnett, S.B. The amphetamine induced rotation test: A re-assessment of its use as a tool to monitor motor impairment and functional recovery in rodent models of Parkinson’s disease. J. Parkinsons Dis. 2019, 9, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Ungerstedt, U.; Ljungberg, T.; Steg, G. Behavioral, physiological, and neurochemical changes after 6-hydroxydopamine-induced degeneration of the nigro-striatal dopamine neurons. Adv. Neurol. 1974, 5, 421–426. [Google Scholar]
- Perese, D.A.; Ulman, J.; Viola, J.; Ewing, S.E.; Bankiewicz, K.S. A 6-hydroxydopamine-induced selective parkinsonian rat model. Brain Res. 1989, 494, 285–293. [Google Scholar] [CrossRef]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Kopin, I.J. MPTP: An industrial chemical and contaminant of illicit narcotics stimulates a new era in research on Parkinson’s disease. Environ. Health Perspect. 1987, 75, 45–51. [Google Scholar] [CrossRef]
- Meredith, G.E.; Rademacher, D.J. MPTP mouse models of Parkinson’s disease: An update. J. Parkinsons Dis. 2011, 1, 19–33. [Google Scholar] [CrossRef] [Green Version]
- Shimoji, M.; Zhang, L.; Mandir, A.S.; Dawson, V.L.; Dawson, T.M. Absence of inclusion body formation in the MPTP mouse model of Parkinson’s disease. Brain Res. Mol. Brain Res. 2005, 134, 103–108. [Google Scholar] [CrossRef]
- Nisticò, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322. [Google Scholar] [CrossRef] [Green Version]
- Barbeau, A.; Dallaire, L.; Buu, N.T.; Poirier, J.; Rucinska, E. Comparative behavioral, biochemical and pigmentary effects of MPTP, MPP+ and paraquat in rana pipiens. Life Sci. 1985, 37, 1529–1538. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar] [CrossRef] [Green Version]
- Nasuti, C.; Falcioni, M.L.; Nwankwo, I.E.; Cantalamessa, F.; Gabbianelli, R. Effect of permethrin plus antioxidants on locomotor activity and striatum in adolescent rats. Toxicology 2008, 251, 45–50. [Google Scholar] [CrossRef]
- Gillette, J.S.; Bloomquist, J.R. Differential up-regulation of striatal dopamine transporter and alpha-synuclein by the pyrethroid insecticide permethrin. Toxicol. Appl. Pharmacol. 2003, 192, 287–293. [Google Scholar] [CrossRef]
- Mazurski, E.J.; Beninger, R.J. Stimulant effects of apomorphine and (+)-amphetamine in rats with varied habituation to test environment. Pharmacol. Biochem. Behav. 1988, 29, 249–255. [Google Scholar] [CrossRef]
- Cooper, J.F.; Van Raamsdonk, J.M. Modeling Parkinson’s disease in C. elegans. J. Parkinsons Dis. 2018, 8, 17–32. [Google Scholar] [CrossRef] [Green Version]
- Maulik, M.; Mitra, S.; Bult-Ito, A.; Taylor, B.E.; Vayndorf, E.M. Behavioral phenotyping and pathological indicators of Parkinson’s disease in C. elegans models. Front. Genet. 2017, 8, 77. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Neurotoxin | IUPAC Name | Lesion Types | Pathogenesis | PD Symptoms |
|---|---|---|---|---|
| 6-OHDA | 5-(2-aminoethyl) benzene-1,2,4-triol |
|
| Rotational motor behavior ↑ after injections(Postural asymmetry) (Bilateral injections: akinesia). |
| MPTP | 1-Methyl-4-phenyl-1,2,3,6-tetra hydropyridine |
|
| Motor imbalance ↑ in primates (Hypokinesia). Motor disturbance ↑ in acute rodents. |
| MPP+ | 1-Methyl-4-phenyl pyridinium |
| ||
| Paraquat | 1,1′-dimethyl-4,4′-bipyridinium dichloride |
|
| No clear motor defects, but slightly ↓ ambulatory behaviors, stereotypy and rotational activities. |
| Rotenone | (2R,6aS,12aS)-1,2,6,6a,12,12 a-hexahydro-2-isopropenyl-8,9-dimethoxychromeno [3,4-b] furo (2,3-h)chromen-6-one |
|
| Motor disturbances ↑ in rodents. |
| Permethrin | 3-phenoxybenzyl-(1R,S)-cis,trans-3-(2,2-dichlorovinyl)-2,2-dimethylcyclopropane carboxylate | Oral administration |
| Motor disturbances ↑. Learning/ memory impairments ↑. |
| 6-OHDA Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 4 µg/µL/min/rat (in saline-containing 0.02% ascorbic acid) | Sprague Dawley rats | Unilateral injection (right striatum) |
| [85] |
| 0.3, 0.7, 1, or 3.6 µg/µL. (in 0.02% ice-cold ascorbate/saline solution and used within 3 h) | Male Rgs5gfp/+ reporter mice | Unilateral injection (midforebrain) |
| [86] |
| 5 µg in 5 µL at 1 µL/min (2 mg/mL 6-OHDA) injection rate (in 0.01% ascorbic acid prepared in saline and protected from light and stored at 20 °C) | Male Sprague Dawley rats | Unilateral injection (right midforebrain) |
| [87] |
| 6 µg in 3 µL at 0.3 µL/min injection rate (in 3 µL of 0.9% saline solution containing 0.008% L-ascorbic acid) | Male Wistar rats | Unilateral injection (left midforebrain) |
| [88] |
| 40 µg/site (in saline containing 0.1% ascorbic acid) | Male Swiss mice | Intracerebroventricular |
| [89] |
| 5/10/15 µg in 4 µL (in saline) | Male Sprague Dawley rats | Intracranial injection (right midforebrain) |
| [90] |
| 6 µg in 1 µL at 0.33 µL/min injection rate for 3 min (6 µg in 1 µL of artificial cerebrospinal fluid, supplemented with 0.2% ascorbic acid) | Male Wistar rats | Bilateral intranigral injection |
| [91] |
| 6 µg in three sites at 0.5 µL/min injection rate (in saline with 0.02% ascorbic acid) | Male Wistar rats | Intrastriatal injection (left midforebrain) |
| [92] |
| 100, 300, 500, 700, or 1000 µg in 4 µL (100 µg/day 1st–10th day) at 1 µL/min injection rate (in 0.02% ascorbate solution) | Male Sprague Dawley rats | Intracerebroventricular |
| [93] |
| 7 µg/µL (in 0.9% saline solution containing 0.02% ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (right midforebrain) |
| [94] |
| 0.5, 1, 2, or 4 µg/µL in 1 µL at 0.5 µL/min injection rate (in saline with 0.1% ascorbic acid) | C57BL/6 mice | Unilateral injection (right midforebrain) |
| [95] |
| 6, 12, or 24 µg in 0.5 µL (in 0.3% ascorbic acid in saline) | Proechimys (Spiny rats) | Unilateral injection (right striatum) |
| [96] |
| 4 µL (4 µg/µL) into two Sites (in isotonic saline containing 0.2 mg/mL of ascorbic acid) | Male Wistar rats | Unilateral injection (midforebrain) |
| [97] |
| 6, 10, or 16 µg in 2 µL at 0.1 µL/min (in 0.02% ascorbic acid) | Male Wistar rats | Unilateral injection (left substantia nigra) |
| [98] |
| 8 µg in 4 µL (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (substantia nigra) |
| [99] |
| 20 µg 6-OHDA in 4 µl at 1 µL/min injection rate (in saline and ascorbic acid) | Charles Foster strain of male albino rats | Unilateral injection (striatum) |
| [100] |
| 6 µg in 1 µL (in saline and ascorbic acid) | Male Wistar rats | Unilateral injection (Intracerebral infusion) |
| [101] |
| 5, 10, or 14 µg/µL (in saline and ascorbic acid) | Male C57BL/6 mice | Bilateral administration (into locus coeruleus) |
| [102] |
| 8, 12, or 16 µg/4 µL at 0.5 mL/min injection rate (in saline and ascorbic acid) | Male Wistar Han rats | Unilateral injection (midforebrain) |
| [103] |
| 8 µg/2 µL/rat at 0.2 µL/min injection rate (in saline and ascorbic acid) | Male Wistar rats | Intranigral injection |
| [104] |
| 8 µg/2 µL/rat at 0.2 µL/min injection rate (in saline and ascorbic acid) | Male Wistar rats | Unilateral injection |
| [105] |
| 17.5 µg in 2.5 µL at 0.5 µL/min injection rate (saline and ascorbic acid) | Female Sprague Dawley rats | Unilateral injection (right midforebrain) |
| [106] |
| 20 µg in 3 µL at 1 µL/min injection rate (in saline and ascorbic acid) | Male Wistar rats | Bilaterally into dorsolateral striatum |
| [107] |
| 5 µg in 1 µL at 0.5 µL/min injection rate (in saline and ascorbic acid) | Male mice (CD-1) | Intrastriatal (right striatum) |
| [108] |
| 8 µg/2 µL (in saline and ascorbic acid) | Male Wistar rats | Unilateral injection |
| [109] |
| 8 µg/ rat (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (left midforebrain) |
| [110] |
| 20 µg/4 µL at 0.5 µL/min injection rate (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (midforebrain) |
| [111] |
| 20 µg in 4 µL (2 µL/site) at 0.2 µL/min injection rate (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (left striatum) |
| [112] |
| 16 µg/4 µL/rat (in 0.2% ascorbic acid) | Female Sprague Dawley rats | Unilateral injection (right midforebrain) |
| [113] |
| 8 µg/3 µL (in saline) | Male Sprague Dawley rats | Intracranial stereotaxic administration (left substantia nigra) |
| [114] |
| 16 µg (8 µL/site) at 0.5 µL/min injection rate (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (right midforebrain) |
| [115] |
| 24 µg/4 µL (in 0.05% ascorbate saline) | Female Wistar rats | Unilateral injection |
| [116] |
| 5, 10, or 20 µg/2 µL/hemisphere at 1 µL/min injection rate (in saline and ascorbic acid) | Male Wistar rats | Bilateral injection |
| [117] |
| 8 µg/4 µL at 0.5 µL/min injection rate (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (right substantia nigra) |
| [118] |
| 10 µg/2 µL at 0.5 µL/min injection rate (in saline and ascorbic acid) | Male CD1 wild-type mice | Intrastriatal injection |
| [119] |
| 6 µg/2.5 µL (in saline and ascorbic acid) | Male albino Wistar rats | Intrastriatal injection |
| [120] |
| 12 µg/6 µL at 1 µL/min injection rate (in saline and ascorbic acid) | Male Sprague Dawley rats | Unilateral intracerebral injection |
| [121] |
| 20 µg/3 µL at 1 µL/min injection rate (in 0.02% ascorbic acid) | Male Sprague Dawley rats | Unilateral injection (right striatum) |
| [122] |
| 6 µg/2 µL (in saline and ascorbic acid) | Male Wistar rats | Intranigral injection (right substantia nigra) |
| [123] |
| 4 µg/2 µL (2 µg/µL for 2 sites) (in saline and ascorbic acid) | Male C57BL/6 mice | Intrastriatal injection (right striatum) |
| [124] |
| 6 µg/2 µL (in saline and ascorbic acid) | Male C57BL/6 mice | Intrastriatal injection (right striatum) |
| [125] |
| MPTP Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 2 mg/kg/injection for five days, a total of 10 mg/kg(in saline) | Common marmosets (Callithrix jacchus) | Subcutaneously |
| [132] |
| 10 mg/kg/injection for three days, a total of 30 mg/kg (in saline) | Male C57BL6 mice | Intraperitoneally |
| [13] |
| 10 mg/kg/injection for three days, a total of 30 mg/kg (in saline) | Male C57BL6 mice | Intraperitoneally |
| [133] |
| 20 mg/kg/injection four times/day at 2-h intervals, a total of 80 mg/kg (in saline) | Male C57BL/6 J mice | Intraperitoneally |
| [134] |
| 15 mg/kg/injection for seven days, a total of 105 mg/kg (in saline) | Female BALB/c mice | Intraperitoneally |
| [135] |
| 15 mg/kg/injection for seven days, a total of 105 mg/kg (in saline) | Female BALB/c mice | Intraperitoneally |
| [136] |
| 25 mg/kg/injection at 3.5-day intervals for five weeks, total 125 mg/kg (in saline) | Male C57BL/6 J mice | Subcutaneously |
| [137] |
| 0.1 mg/nostril (2–three doses, baseline and 7th day) (in 10% w/v ethanol and 0.9% saline) | Male albino Wistar rats | Intranasal administration |
| [138] |
| 20 mg/kg/injection at 2-h intervals; a total 40 mg/kg (in saline) | Male C57Bl/6 mice | Intraperitoneally |
| [139] |
| 15 mg/kg/injection four times/day at 2-h intervals; total 60 mg (0.1% ethanol in saline) | Female C57BL/6 mice | Intraperitoneally |
| [140] |
| 30 mg/kg/day for eight days (in saline) | Male C57BL/6 mice | Intraperitoneally |
| [141] |
| 0.2 mg/kg/day, until Parkinsonian motor signs appeared (in saline) | Captive-bred monkeys (M. fascicularis) | Intravenous |
| [142] |
| 30 mg/kg/day for five days, a total of 150 mg/kg (in saline) | Male C57BL/6 J mice | Intraperitoneally |
| [143] |
| 30 mg/kg/day for five days, a total of 150 mg/kg (in saline) | Male C57BL/6 mice | Intraperitoneally |
| [144] |
| 0.2 mg/kg/day at 1 mL/min injection rate until the appearance of typical PD (in saline) | Cynomolgus monkeys (M. fascicularis) | Intraperitoneally |
| [145] |
| 0.2 mg/kg (in saline) | Adult female cynomolgus monkeys (M. fascicularis) | Intramuscularly |
| [146] |
| 0.5 mg/kg/week for five weeks, a total of 2.5 mg/kg (in saline) | Marmoset monkeys (C. jacchus) | Subcutaneously |
| [147] |
| 10 mg/mL, a subcutaneously implanted device on the right side of the back | Female Gottingen minipigs | Subcutaneously |
| [148] |
| 10, 20, or 30 mg/kg/injection four times/day at 2-h intervals (in saline) | Male C57BL/6 N mouse | Intraperitoneally |
| [149] |
| 15 mg/kg/day for four days, a total of 60 mg/kg (in saline) | Male C57BL/6 mice | Intraperitoneally |
| [150] |
| 2 mg/kg for two days and then 1 mg/kg for next three days, total 7 mg/kg | Male common marmoset (C. jacchus) | Subcutaneously |
| [151] |
| 2.5 mg/kg/injection twice per day, a total of 5 mg (in saline) | Male mice (G2019S-LRRK2 mutation) | Subcutaneously |
| [152] |
| 20 mg/kg/injection four times/day at 2-h intervals, a total of 80 mg/kg (in saline) | Male C57BL/6 mice | Intraperitoneally |
| [153] |
| 25 mg/kg/day for four days, a total of 100 mg/kg (in H2O) | Male C57Bl/6 J mice | Intraperitoneally |
| [154] |
| 30 mg/kg/injection for four days, a total of 120 mg/kg (in saline) | Male C57BL/6 mice | Intraperitoneally |
| [155] |
| 30 mg/kg/day for five days, a total of 150 mg/kg (in saline) | Male C57BL/6 J mice | Intraperitoneally |
| [156] |
| MPP+ Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 1.8 µg/site (in saline) | Male C57BL6 mice | Unilateral (intracerebroventricular) injection |
| [161] |
| 1.8–18 µg (in saline) | Male C57BL6 mice | Unilateral (intracerebroventricular) injection |
| [162] |
| 8 µg/site (in saline) | Sprague Dawley rats | Unilateral injection | Apomorphine-induced rotation (5 mg/kg, intraperitoneally) and locomotor activity: 8 days after MPP+ administration. | [133] |
| 10 µg/site (in 8 µL saline) | Male Wistar rats (strain NIH) | Unilateral (intrastriatal) injection | Apomorphine-induced rotational behavior (1 mg/kg apomorphine, subcutaneously): 6 days after MPP+ administration. | [163] |
| 10 µg/site in (in 8 µL saline) | Male Wistar rats | Unilateral (intrastriatal) injection | Apomorphine-induced rotational behavior (1 mg/kg apomorphine, subcutaneously): 6 days after MPP+ administration. | [164] |
| 15 µg/site (in 8 µL saline) | Male Wistar rats | Unilateral (intrastriatal) injection | Apomorphine-induced rotational behavior (1 mg/kg apomorphine, subcutaneously): 6 days after MPP+ administration. | [165] |
| 15 µg/site (in 8 µL saline) | Male Wistar rats (NIH strain) | Unilateral (intrastriatal) injection | Apomorphine-induced rotational behavior (1 mg/kg apomorphine, subcutaneously): 6 days after MPP+ administration. | [166] |
| 30 µg/site (in 4 µL saline) | Sprague Dawley rats | Unilateral (intrastriatal) injection |
| [167] |
| Paraquat Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 0.02 or 0.2 mg/kg postnatal day 10 and 11 (in H2O and sonicated with a 20% fat emulsion vehicle) | Male C57Bl/6 mice | Oral administration |
| [176] |
| 1 or 10 mg/kg, two times/week for three weeks (on the 1st, 5th, 8th, 12th, 15th, and 19th days) (in saline) | Male C57/BL6 mice | Intraperitoneally |
| [177] |
| 1 mg/kg/day for seven days, a total of 7 mg/kg (in saline) | Male Sprague Dawley rats | Oral administration |
| [178] |
| 10 mg/kg twice per week for three weeks, a total of 60 mg/kg (in saline) | Male C57BL6/J mice | Intraperitoneally |
| [179] |
| 2.5 mg/kg/day for four weeks; 0.25 µL/h injection rate (in saline) | Male Wistar rats | Subcutaneously (chronic exposure using osmotic minipumps implanted slightly posterior back to the shoulder blades) |
| [180] |
| 10–20 mg/kg (200 g/L gramoxone, from 1st day of pregnancy (G0) to G21) | Male and female Swiss mice | Oral administration |
| [181] |
| 27 or 54 mg/m3 eight times (on the 1st, 3rd, 5th, 7th, 9th, 13th, and 15th day), each time 30 min, for 16 days. | Sprague Dawley rats | Aerosol administration |
| [182] |
| 5 mg/kg four times (on the 2nd, 4th, 6th, and 7th day; a total of 20 mg/kg) (in saline) | Male Swiss mice | Intraperitoneally |
| [183] |
| 7 mg/kg with two-day intervals for a total of six doses; a total of 42 mg/kg (in saline) | C57Bl6/J (p16-3MR) mice | Intraperitoneally | Cylinder test: before the sacrifice. | [184] |
| Rotenone Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 0.5 mg/mL at 1 mL/kg/day for 35 days (in sunflower oil) | Sprague Dawley rat pups | Intraperitoneally |
| [190] |
| 1.5, 2, or 2.5 mg/kg day for five weeks (in sunflower oil) | Male Wistar rats | Subcutaneously |
| [191] |
| 2 mg/kg (in chloroform and 0.5% carboxymethyl cellulose solution) | Male Sprague Dawley rats | Subcutaneously |
| [192] |
| 2 mg/kg/day for 35 days (in sunflower oil at 2 mg/mL) | Male albino Wistar rats | Subcutaneously |
| [193] |
| 2 mg/kg/day for 35 days (2 mg/mL of rotenone in 98% sunflower oil and 2% dimethyl sulfoxide) | Male albino Wistar rats | Subcutaneously | Cylinder test: on the 36th day. | [194] |
| 2.5 mg/kg/day for 21 days (in sunflower oil) | Adult male Wistar albino rats | Intraperitoneally | Cylinder test: baseline, 1st, 2nd, 3rd, 4th, 5th, and 6th week. | [195] |
| 2.5 mg/kg/day for 10 or 45 days (in sunflower oil) | Male albino Wistar rats | Intraperitoneally |
| [196] |
| 3 mg/kg/day for 10 days (in 2% dimethyl sulfoxide and 98% polyethylene glycol 400) | Male albino rats | Subcutaneously |
| [197] |
| 4 µg /site; a total of 12 µg for three sites (in 2 μL of dimethyl sulfoxide and saline) | Male Sprague Dawley rats | Unilateral injection (into right striatum) | Postural instability test: two weeks after rotenone injection. | [198] |
| Permethrin Dose | Animal | Lesion Type | Description of Behavioral Tests Performed | Reference |
|---|---|---|---|---|
| 0.05 mg/kg/day (in drinking water) | Male and female C57BL/6 mice | Oral administration |
| [205] |
| 34 mg/kg/daily from postnatal day 6th–21st (in corn oil) | Male and female Wistar rats | Oral administration |
| [206] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prasad, E.M.; Hung, S.-Y. Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease. Antioxidants 2020, 9, 1007. https://doi.org/10.3390/antiox9101007
Prasad EM, Hung S-Y. Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease. Antioxidants. 2020; 9(10):1007. https://doi.org/10.3390/antiox9101007
Chicago/Turabian StylePrasad, E. Maruthi, and Shih-Ya Hung. 2020. "Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease" Antioxidants 9, no. 10: 1007. https://doi.org/10.3390/antiox9101007
APA StylePrasad, E. M., & Hung, S. -Y. (2020). Behavioral Tests in Neurotoxin-Induced Animal Models of Parkinson’s Disease. Antioxidants, 9(10), 1007. https://doi.org/10.3390/antiox9101007