The Antioxidant Role of One-Carbon Metabolism on Stroke

 ,
, 
Abstract
:1. Introduction
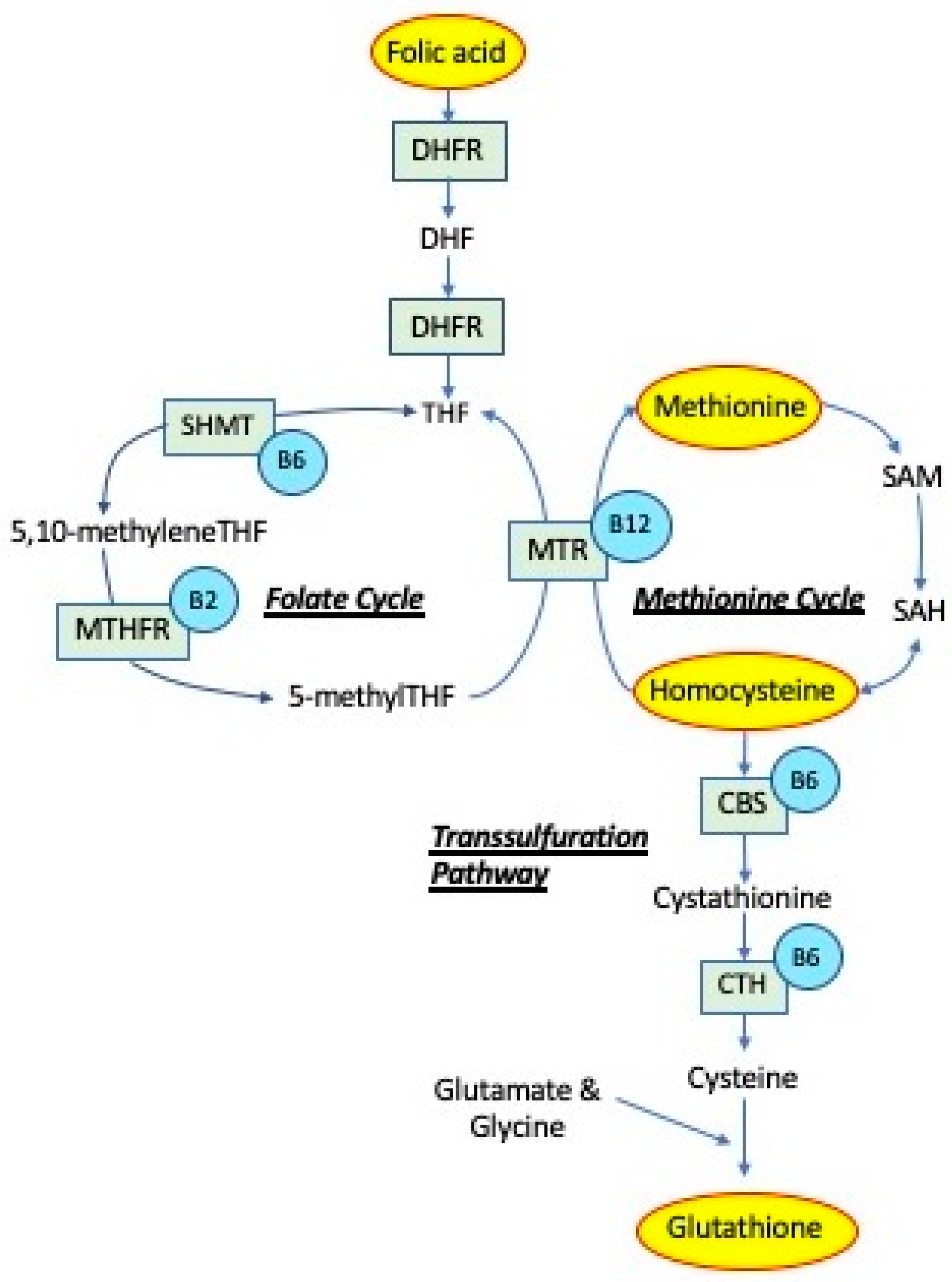
2. Folates
3. Dietary Supplementation of 1C
4. Methionine
5. Transsulfuration
5.1. Cystathionine β-synthase (CBS)
5.2. Glutathione
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Murray, L.; Emmerson, J.; Jadavji, N.M. Roles of Folate in Neurological Function. In Folic Acid: Sources, Health Effects and Role in Disease; Nova Publishers Science Inc.: Hauppauge, NY, USA, 2017. [Google Scholar]
- Ashjazadeh, N.; Fathi, M.; Shariat, A. Evaluation of homocysteine level as a risk factor among patients with ischemic stroke and its subtypes. Iran. J. Med. Sci. 2013, 38, 233–239. [Google Scholar] [PubMed]
- Han, L.; Wu, Q.; Wang, C.; Hao, Y.; Zhao, J.; Zhang, L.; Fan, R.; Liu, Y.; Li, R.; Chen, Z.; et al. Homocysteine, Ischemic Stroke, and Coronary Heart Disease in Hypertensive Patients: A Population-Based, Prospective Cohort Study. Stroke 2015, 46, 17777–17786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, R.; Rivera, I.; Blom, H.J.; Jakobs, C.; Tavares de Almeida, I. Homocysteine metabolism, hyperhomocysteinaemia and vascular disease: An overview. J. Inherit. Metab. Dis. 2006, 29, 3–20. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar]
- Yoo, J.H.; Chung, C.S.; Kang, S.S. Relation of plasma homocyst(e)ine to cerebral infarction and cerebral atherosclerosis. Stroke 1998, 29, 2478–2483. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Watanabe, M.; Nagai, Y.; Hoshi, T.; Takasawa, M.; Nukata, M.; Taguchi, A.; Kitagawa, K.; Kinoshita, N.; Matsumoto, M. Association of plasma homocysteine concentration with atherosclerotic carotid plaques and lacunar infarction. Stroke 2002, 33, 1493–1496. [Google Scholar] [CrossRef] [Green Version]
- Perry, I.J.; Morris, R.W.; Ebrahim, S.B.; Shaper, A.G.; Refsum, H.; Ueland, P.M. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 1995, 346, 1395–1398. [Google Scholar] [CrossRef]
- Kittner, S.J.; Giles, W.H.; Macko, R.F.; Hebel, J.R.; Wozniak, M.A.; Wityk, R.J.; Stolley, P.D.; Stern, B.J.; Sloan, M.A.; Sherwin, R.; et al. Homocyst(e)ine and Risk of Cerebral Infarction in a Biracial Population: The Stroke Prevention in Young Women Study. Stroke 1999, 30, 1554–1560. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Han, L.; Wu, Q.; Zhuo, R.; Liu, K.; Zhao, J.; Zhang, L.; Hao, Y.; Fan, R.; Liu, Y.; et al. Association between homocysteine and incidence of ischemic stroke in subjects with essential hypertension: A matched case-control study. Clin. Exp. Hypertens. 2015, 37, 557–562. [Google Scholar] [CrossRef]
- The Homocysteine Studies Collaboration Homocysteine and Risk of Ischemic Heart Disease and Stroke. Am. Med. Assoc. 2002, 288, 2015–2023. [CrossRef]
- Pan, S.; Liu, H.; Gao, F.; Luo, H.; Lin, H.; Meng, L.; Jiang, C.; Guo, Y.; Chi, J.; Guo, H. Folic acid delays development of atherosclerosis in low-density lipoprotein receptor-deficient mice. J. Cell. Mol. Med. 2018, 22, 3183–3191. [Google Scholar] [CrossRef]
- Loscalzo, J. The oxidant stress of hyperhomocyst(e)inemia. J. Clin. Investig. 1996, 98, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stühlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.C.; Lominadze, D.; Roberts, A.M. Homocysteine in microvascular endothelial cell barrier permeability. Cell Biochem. Biophys. 2005, 43, 37–44. [Google Scholar] [CrossRef]
- Jin, L.; Caldwell, R.B.; Li-Masters, T.; Caldwell, R.W. Homocysteine induces endothelial dysfunction via inhibition of arginine transport. J. Physiol. Pharmacol. 2007, 58, 191–206. [Google Scholar] [PubMed]
- Laursen, J.B.; Somers, M.; Kurz, S.; Mccann, L.; Warnholtz, A.; Freeman, B.A.; Tarpey, M.; Fukai, T.; Harrison, D.G. Implications for Interactions Between Peroxynitrite. Circulation 2001, 103, 1282–1289. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Münzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [Green Version]
- Milstien, S.; Katusic, Z. Oxidation of tetrahydrobiopterin by peroxynitrite: Implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 1999, 263, 681–684. [Google Scholar] [CrossRef]
- Zhang, F.; Slungaard, A.; Gregory, M.; Vercellotti; Iadecola, C. Superoxide-dependent cerebrovascular effects of homocysteine. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1998, 274, R1704–R1711. [Google Scholar] [CrossRef]
- Outinen, P.A.; Sood, S.K.; Liaw, P.C.Y.; Sarge, K.D.; Maeda, N.; Hirsh, J.; Ribau, J.; Podor, T.J.; Weitz, J.I.; Austin, R.C. Characterization of the stress-inducing effects of homocysteine. Biochem. J. 1998, 332, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, G.N.; Upchurch, G.R.; Loscalzo, J. Homocysteine, oxidative stress, and vascular disease. Hosp. Pract. 1997, 32, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Upchurch, G.R.; Welche, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayal, S.; Wilson, K.M.; Leo, L.; Arning, E.; Bottiglieri, T.; Lentz, S.R. Enhanced susceptibility to arterial thrombosis in a murine model of hyperhomocysteinemia. Blood 2006, 108, 2237–2243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starkebaum, G.; Harlan, J.M. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J. Clin. Investig. 1986, 77, 1370–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ardlie, N.G.; Selley, M.L.; Simons, L.A. Platelet activation by oxidatively modified low density lipoproteins. Atherosclerosis 1989, 76, 117–124. [Google Scholar] [CrossRef]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.K.C.; Kan, M.Y. Homocysteine-induced endothelial dysfunction. Ann. Nutr. Metab. 2015, 67, 1–12. [Google Scholar] [CrossRef]
- Jakubowski, H.; Zhang, L.; Bardeguez, A.; Aviv, A. Homocysteine Thiolactone and Protein Homocysteinylation in Human Endothelial Cells. Circ. Res. 2000, 87, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacCoss, M.J.; Fukagawa, N.K.; Matthews, D.E. Measurement of homocysteine concentrations and stable isotope tracer enrichments in human plasma. Anal. Chem. 1999, 71, 4527–4533. [Google Scholar] [CrossRef] [PubMed]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- James, S.J.; Melnyk, S.; Pogribna, M.; Pogribny, I.P.; Caudill, M.A. Elevation in S-Adenosylhomocysteine and DNA Hypomethylation: Potential Epigenetic Mechanism for Homocysteine-Related Pathology. J. Nutr. 2002, 2361–2366. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yoon, W.; Goldschmidt-Clermont, P.J. DNA methylation and athersclerosis. J. Nutr. 2002, 131, 2406S–2409S. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yoshizumi, M.; Lai, K.; Tsai, J.-C.C.; Perrella, M.A.; Haber, E.; Lee, M.-E.E. Inhibition of growth and p21ras methylation in vascular endothelial cells by homocysteine but not cysteine. J. Biol. Chem. 1997, 272, 25380–25385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, B.H.; Broadhurst, D.I.; Mandal, R.; Wishart, D.S.; Boylan, G.B.; Kenny, L.C.; Murray, D.M. The Metabolomic Profile of Umbilical Cord Blood in Neonatal Hypoxic Ischaemic Encephalopathy. PLoS ONE 2012, 7, e50520. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373. [Google Scholar] [CrossRef] [Green Version]
- Hwang, I.K.; Yoo, K.-Y.; Suh, H.-W.; Kim, Y.S.; Kwon, D.Y.; Kwon, Y.-G.; Yoo, J.-H.; Won, M.-H. Folic acid deficiency increases delayed neuronal death, DNA damage, platelet endothelial cell adhesion molecule-1 immunoreactivity, and gliosis in the hippocampus after transient cerebral ischemia. J. Neurosci. Res. 2008, 86, 2003–2015. [Google Scholar] [CrossRef]
- Van Guelpen, B.; Hultdin, J.; Johansson, I.; Stegmayr, B.; Hallmans, G.; Nilsson, T.K.; Weinehall, L.; Witthöft, C.; Palmqvist, R.; Winkvist, A. Folate, vitamin B12, and risk of ischemic and hemorrhagic stroke: A prospective, nested case-referent study of plasma concentrations and dietary intake. Stroke 2005, 36, 1426–1431. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Huang, G.; Chen, S.; Gou, Y.; Dong, Z.; Zhang, X. Folic acid deficiency increases brain cell injury via autophagy enhancement after focal cerebral ischemia. J. Nutr. Biochem. 2016, 38, 41–49. [Google Scholar] [CrossRef] [PubMed]
- McNulty, H.; Pentieva, K. Folate bioavailability. Folate Health Dis. Second Ed. 2004, 63, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A. Folate bioavailability: Implications for establishing dietary recommendations and optimizing status. Am. J. Clin. Nutr. 2010, 91, 1455S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, S.C.; Gupta, E. Das Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.A.; Renna, S.A.; Khitun, E.; Ortiz, M.; Moriarty, D.J.; Caudill, M.A. Ethnicity and race influence the folate status response to controlled folate intakes in young women. J. Nutr. 2004, 134, 1786–1792. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Emmerson, J.; Willmore, W.G.; MacFarlane, A.J.; Smith, P. B-vitamin and choline supplementation increases neuroplasticity and recovery after stroke. Neurobiol. Dis. 2017, 103, 89–100. [Google Scholar] [CrossRef]
- Zhang, X.; Huang, G.; Liu, H.; Chang, H.; Wilson, J.X. Folic acid enhances Notch signaling, hippocampal neurogenesis, and cognitive function in a rat model of cerebral ischemia. Nutr. Neurosci. 2012, 15, 55–61. [Google Scholar] [CrossRef]
- Davis, C.K.; Nampoothiri, S.S.; Rajanikant, G.K. Folic Acid Exerts Post-Ischemic Neuroprotection In Vitro Through HIF-1α Stabilization. Mol. Neurobiol. 2018, 55, 8328–8345. [Google Scholar] [CrossRef]
- Hankey, G.J.; Ford, A.H.; Yi, Q.; Eikelboom, J.W.; Lees, K.R.; Chen, C.; Xavier, D.; Navarro, J.C.; Ranawaka, U.K.; Uddin, W.; et al. Effect of B vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: A prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke 2013, 44, 2232–2239. [Google Scholar] [CrossRef] [Green Version]
- Galan, P.; Kesse-Guyot, E.; Czernichow, S.; Briancon, S.; Blacher, J.; Hercberg, S. Effects of B vitamins and omega 3 fatty acids on cardiovascular diseases: A randomised placebo controlled trial. BMJ 2011, 342, 36. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, Y.; Li, P.; Li, J.; Bao, H.; Zhang, Y.; Wang, B.; Sun, N.; Wang, J.; He, M.; et al. Association between percent decline in serum total homocysteine and risk of first stroke. Neurology 2017, 89, 2101–2107. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Li, J.; Qin, X.; Huang, Y.; Wang, X.; Gottesman, R.F.; Tang, G.; Wang, B.; Chen, D.; He, M.; et al. Efficacy of folic acid therapy in primary prevention of stroke among adults with hypertension in China: The CSPPT randomized clinical trial. JAMA J. Am. Med. Assoc. 2015, 313, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Qin, X.; Wang, J.; Sun, N.; Zeng, Q.; Xu, X.; Liu, L.; Xu, X.; Wang, X. Efficacy of folic acid supplementation in stroke prevention: New insight from a meta-analysis. Int. J. Clin. Pract. 2012, 66, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chi, F.L.; Xie, T.H.; Zhou, Y.H. Effect of B-vitamin supplementation on stroke: A meta-analysis of randomized controlled trials. PLoS ONE 2013, 8, e81577. [Google Scholar] [CrossRef] [Green Version]
- Tian, T.; Yang, K.-Q.; Cui, J.-G.; Zhou, L.-L.; Zhou, X.-L. Folic Acid Supplementation for Stroke Prevention in Patients with Cardiovascular Disease. Am. J. Med. Sci. 2017, 354, 379–387. [Google Scholar] [CrossRef]
- Zeng, R.; Xu, C.H.; Xu, Y.N.; Wang, Y.L.; Wang, M. The effect of folate fortification on folic acid-based homocysteine-lowering intervention and stroke risk: A meta-analysis. Public Health Nutr. 2015, 18, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.; Li, J.; Spence, J.D.; Zhang, Y.; Li, Y.; Wang, X.; Wang, B.; Sun, N.; Chen, F.; Guo, J.; et al. Folic Acid Therapy Reduces the First Stroke Risk Associated with Hypercholesterolemia Among Hypertensive Patients. Stroke 2016, 47, 2805–2812. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Huang, X.; Zhao, M.; Xu, B.; Xu, R.; Song, Y.; Yu, Y.; Yang, W.; Zhang, J.; Liu, L.; et al. Platelet Count Affects Efficacy of Folic Acid in Preventing First Stroke. J. Am. Coll. Cardiol. 2018, 71, 2136–2146. [Google Scholar] [CrossRef]
- Hankey, G.J.; Eikelboom, J.W.; Yi, Q.; Lees, K.R.; Chen, C.; Xavier, D.; Navarro, J.C.; Ranawaka, U.K.; Uddin, W.; Ricci, S.; et al. Antiplatelet therapy and the effects of B vitamins in patients with previous stroke or transient ischaemic attack: A post-hoc subanalysis of VITATOPS, a randomised, placebo-controlled trial. Lancet Neurol. 2012, 11, 512–520. [Google Scholar] [CrossRef] [Green Version]
- Assanelli, D.; Bonanome, A.; Pezzini, A.; Albertini, F.; Maccalli, P.; Grassi, M.; Archetti, S.; Negrini, R.; Visioli, F. Folic acid and Vitamin E supplementation effects on homocysteinemia, endothelial function and plasma antioxidant capacity in young myocardial-infarction patients. Pharmacol. Res. 2004, 49, 79–84. [Google Scholar] [CrossRef]
- Aghamohammadi, V.; Gargari, B.P.; Aliasgharzadeh, A. Effect of Folic Acid Supplementation on Homocysteine, Serum Total Antioxidant Capacity, and Malondialdehyde in Patients with Type 2 Diabetes Mellitus. J. Am. Coll. Nutr. 2011, 30, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.M.; Pérez-Cejas, A.; Abreu-González, P.; Ramos, L.; Argueso, M.; Cáceres, J.J.; Solé-Violán, J.; Jiménez, A. Association between total antioxidant capacity and mortality in ischemic stroke patients. Ann. Intensive Care 2016, 6, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dam, K.; Füchtemeier, M.; Farr, T.; Boehm-Strum, P.; Foddis, M.; Dirnagal, U.; Jadavji, N.M. Deficiencies in methylenetetrahydrofolate reductase and dietary folic acid alter choline metabolism during chronic hypoperfusion. Behav. Brain Res. 2017, 321, 201–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, N.; Arboleya, S.; Allison, J.; Kaliszewska, A.; Higarza, S.G.; Gueimonde, M.; Arias, J.L. The relationship between choline bioavailability from diet, intestinal microbiota composition, and its modulation of human diseases. Nutrients 2020, 12, 2340. [Google Scholar] [CrossRef] [PubMed]
- Mödinger, Y.; Schön, C.; Wilhelm, M.; Hals, P.A. Plasma kinetics of choline and choline metabolites after a single dose of SuperbaBoostTM krill oil or choline bitartrate in healthy volunteers. Nutrients 2019, 11, 2548. [Google Scholar] [CrossRef] [Green Version]
- Borges, A.A.; Ei-Batah, P.N.; Yamashita, L.F.; dos Santos Santana, A.; Lopes, A.C.; Freymuller-Haapalainen, E.; Coimbra, C.G.; Sinigaglia-Coimbra, R. Neuroprotective effect of oral choline administration after global brain ischemia in rats. Nutr. Neurosci. 2015, 18, 265–274. [Google Scholar] [CrossRef]
- Guseva, M.V.; Hopkins, D.M.; Scheff, S.W.; Pauly, J.R. Dietary Choline Supplementation Improves Behavioral, Histological, and Neurochemical Outcomes in a Rat Model of Traumatic Brain Injury. J. Neurotrauma 2008, 25, 975–983. [Google Scholar] [CrossRef] [Green Version]
- Adibhatla, R.M.; Hatcher, J.F.; Dempsey, R.J. Effects of Citicoline on Phospholipid and Glutathione Levels in Transient Cerebral Ischemia. Stroke. 2001, 32, 2376–2381. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, O.; Cardenas, A.; Pradillo, J.M.; Morales, J.R.; Ortego, F.; Sobrino, T.; Castillo, J.; Moro, M.A.; Lizasoain, I.; Cardenas, A.; et al. A chronic treatment with CDP-choline improves functional recovery and increases neuronal plasticity after experimental stroke. Neurobiol. Dis. 2007, 26, 105–111. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Jiménez, A.; Peres, H.; Rubio, V.C.; Oliva-Teles, A. The effect of hypoxia on intermediary metabolism and oxidative status in gilthead sea bream (Sparus aurata) fed on diets supplemented with methionine and white tea. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2012, 155, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Elango, R. Methionine Nutrition and Metabolism: Insights from Animal Studies to Inform Human Nutrition—PubMed. J. Nutr. 2020, 150, 2518S–2523S. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Blokhin, I.O.; Wilson, K.M.; Dhanesha, N.; Doddapattar, P.; Grumbach, I.M.; Chauhan, A.K.; Lentz, S.R. Protein methionine oxidation augments reperfusion injury in acute ischemic stroke. JCI Insight 2016, 1, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Swanson, R.A.; Shiraishi, K.; Morton, M.T.; Sharp, F.R. Methionine sulfoximine reduces cortical infarct size in rats after middle cerebral artery occlusion. Stroke 1990, 21, 322–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yermolaieva, O.; Xu, R.; Schinstock, C.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Hoshi, T. Methionine sulfoxide reductase A protects neuronal cells against brief hypoxia/reoxygenation. Proc. Natl. Acad. Sci. USA 2004, 101, 1159–1164. [Google Scholar] [CrossRef] [Green Version]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the transsulfuration pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Takano, N.; Peng, Y.J.; Kumar, G.K.; Luo, W.; Hu, H.; Shimoda, L.A.; Suematsu, M.; Prabhakar, N.R.; Semenza, G.L. Hypoxia-inducible factors regulate human and rat cystathionine â-synthase gene expression. Biochem. J. 2014, 458, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Xu, G.; Wen, Q.; Peng, X.; Chen, H.; Zhang, J.; Xu, S.; Zhang, C.; Zhang, M.; Ma, J.; et al. CBS promoter hypermethylation increases the risk of hypertension and stroke. Clinics 2019, 74, e630. [Google Scholar] [CrossRef] [PubMed]
- Sikora, M.; Lewandowska, I.; Kupc, M.; Kubalska, J.; Graban, A.; Marczak, Ł.; Kaźmierski, R.; Jakubowski, H. Serum proteome alterations in human cystathionine β-synthase deficiency and ischemic stroke subtypes. Int. J. Mol. Sci. 2019, 20, 3096. [Google Scholar] [CrossRef] [Green Version]
- Boers Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. N. Engl. J. Med. 1974, 306, 802–805.
- Dhar, I.; Svingen, G.F.T.; Ueland, P.M.; Lysne, V.; Svenningsson, M.M.; Tell, G.S.; Nygård, O.K. Plasma cystathionine and risk of incident stroke in patients with suspected stable angina pectoris. J. Am. Heart Assoc. 2018, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wong, P.T.H.; Qu, K.; Chimon, G.N.; Seah, A.B.H.; Hui, M.C.; Meng, C.W.; Yee, K.N.; Rumpel, H.; Halliwell, B.; Chen, C.P.L.H. High plasma cyst(e)ine level may indicate poor clinical outcome in patients with acute stroke: Possible involvement of hydrogen sulfide. J. Neuropathol. Exp. Neurol. 2006, 65, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, T.M.; Wierzbicka, G.T.; Sillau, A.H.; Bowman, B.B.; Jones, D.P. Bioavailability of dietary glutathione: Effect on plasma concentration. Am. J. Physiol. Gastrointest. Liver Physiol. 1990, 259, G524–G529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richie, J.P.; Nichenametla, S.; Neidig, W.; Calcagnotto, A.; Haley, J.S.; Schell, T.D.; Muscat, J.E. Randomized controlled trial of oral glutathione supplementation on body stores of glutathione. Eur. J. Nutr. 2015, 54, 251–263. [Google Scholar] [CrossRef]
- Coimbra-Costa, D.; Alva, N.; Duran, M.; Carbonell, T.; Rama, R. Oxidative stress and apoptosis after acute respiratory hypoxia and reoxygenation in rat brain. Redox Biol. 2017, 12, 216–225. [Google Scholar] [CrossRef]
- Dayal, S.; Brown, K.L.; Weydert, C.J.; Oberley, L.W.; Arning, E.; Bottiglieri, T.; Faraci, F.M.; Lentz, S.R. Deficiency of glutathione peroxidase-1 sensitizes hyperhomocysteinemic mice to endothelial dysfunction. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Park, J.; Oh, Y.; Lee, J.E. Glutathione suppresses cerebral infarct volume and cell death after ischemic injury: Involvement of FOXO3 inactivation and Bcl2 expression. Oxid. Med. Cell. Longev. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Garcia, M.; Mulvagh, S.L.; Merz, C.N.B.; Buring, J.E.; Manson, J.A.E. Cardiovascular disease in women: Clinical perspectives. Circ. Res. 2016, 118, 1273–1293. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Q.; Wang, J.; Zhuang, Q.K.; Zhang, Y.Y. Combination of thrombolytic therapy and neuroprotective therapy in acute ischemic stroke: Is it important? Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 416–422. [Google Scholar]


{kind=link}
| Reference | Model System and Sex Used | Design | Major Findings |
|---|---|---|---|
| [42] | Sprague-Dawley rats (male) | Using focal cerebral ischemia model and folic acid deficiency to investigate neuronal autophagy in neuronal cells. | Autophagy was induced in rats subjected to ischemic insults. Further autophagy activation in cortex neurons caused by folic acid deficiency was confirmed by the increase in the LC3II/LC3I ratio and beclin 1 expression. The increased formation of autophagosomes. results suggest that cerebral cortex cell injury by folic acid deficiency in ischemic brains is partially mediated by the activation of autophagy. Oxidative injury seems to be involved in excessive activation of autophagy caused by folic acid deficiency. |
| [40] | Mongolian gerbils (male) | Examined whether folic acid deficiency and transient forebrain ischemia enhances neuronal damage and gliosis via oxidative stress in the gerbil hippocampus. | After transient cerebral ischemia Folic acid deficiency increases delayed neuronal death, DNA damage, platelet endothelial cell adhesion molecule 1 immunoreactivity, and gliosis in the hippocampus. |
| [47] | C57BL/6J mice (male) | Investigated the impact of dietary deficiency of folic acid and supplementation after with folic acid, vitmain B12, riboflavin and choline promoted recovery after photothrombosis ischemic damage. | Combination of B-vitamins, including folic acid, riboflavin and vitamin B12 with choline supplementation promotes some degree of functional improvement following ischemic damage. Additionally, study shows increased neuroplasticity markers deltaFosB and brain derived neurotrophic factor (BDNF), as well as increased levels of nuclear factor erythroid 2–related factor 2 (Nrf2) and superoxide dismutase 2 (SOD2), indicators of anti-oxidant activity |
| [67] | Wistar Hannover rats (male) | The impact of choline supplementation on the survival of hippocampal neurons following transient forebrain ischemia | There was no difference between choline treated rats up to 200 mg/kg/day and controls (vehicle-treated animals). Choline administered at 400 mg/kg/day provided a significant neuroprotection to ischemic animals at the dose. |
| [68] | Sprague-Dawley rats (male) | The aim of this study was to determine whether the low-potency and elective alpha-7 neuronal nicotinic cholinergic receptor (nAChR) agonist choline could be a useful treatment for improvement of neurological outcome in a rat model of traumatic brain injury (TBI). | Choline supplemented animals show improved memory retention tests; dietary choline supplementation was associated with cortical tissue preservation; choline supplementation attenuates TBI-induced decreases in cortical levels of alpha-7 nAChR. |
| [69] | Gerbils (male) | Examined changes and effects of citocoline on phospholipids and glutathione synthesis after transient ischemia and reperfusion. Citcoline is a precursor to choline, which can be metabolized to methionine (1C metabolism), which may be further converted to glutathione, which is one of the primary endogenous antioxidant defense systems in the brain. | The study demonstrated that citicoline supplementation improved phospholipid membrane short-term, while the neuroprotective factors (demonstrated by glutathione) were more significant 3+ days post infarct. |
| [70] | Sprague-Dawley rats (male) | Investigated whether a chronic treatment with CDP-choline starting 24 h after is middle cerebral artery occlusion in the rat. | Increased neuronal plasticity and contribution to sensorimotor function recovery when chronically treated with CDP-choline. |
| [74] | C57BL/6J mice (male) and human umbical vein endothelial cells | Study tried to determine if NF-kB (nuclear factor kappa-light-chain-enhancer of activated B) activation is increased by methionine oxidation after ischemic stroke. | The main findings are that using the oxidation of methionine and of CaMMKII Met281/282, reactive oxygen species (ROS) promote the NF-kB pathway in endothelial cells in vitro and in vivo. Furthermore, NF-kB pathway activation and cerebral ischemia/reperfusion injury can be prevented by MsrA, through the expression of MsrA in nonhematopoietic cells. Finally, mice deficient in methionine sulfoxide reductase A (MsrA-/-) have decreased outcome after stroke, but outcome can be protected against by inhibition of the NF-kB pathway or CaMKII. |
| [75] | Sprague-Dawley rats (male) | The focus of this study was to look at the protective effects of methionine sulfoximine (MSO) in middle cerebral artery occlusion, which was used as an animal model for stroke. | The main findings of this study are that intraperitoneal MSO injection prevented significant infarct volume in the rat cerebral cortex but not the basal ganglia after middle cerebral artery occlusion. MSO administration was found to increase cortical glycogen by 81% 24hours after administration, but did not change glucose levels significantly. Methionine sulfoximine might be changing the presynaptic cells by interrupting the astrocyte-neuron glutamate shuttle and impairing neuronal glutamine synthetase. |
| [76] | PC12 Cells | To determine whether overexpression of methionine sulfoxide reductase A (MSRA) impacts protection in cells after hypoxia and reoxygenation injury. | The main findings of this study are that brief hypoxia and reoxygenation cause significant and quick changes in reactive oxygen species (ROS) levels in the cells. This increase in ROS causes a depolarization of the mitochondrial membrane of the cells, thereby facilitating cell death, as well as an increase in apoptosis of cells due to the ROS. Methionine sulfoxide reductase type A (MSRA) is an antioxidant that promotes the reduction of met-O in proteins to methionine and, when overexpressed, was found to protect against these cellular injuries caused by hypoxia/reoxygenation. MSRA was found to lower the ROS levels in cells caused by hypoxia/reoxygenation therefore playing a protective role against oxidative stress causing cell injury. |
| [78] | Cells: U87-MG, PC12, human lung microvascular and aortic endothelial cells, and primary vascular smooth muscle Sprague-Dawley Wistar rats cell cultures | Study examined the relationship of hypoxia and expression of cystathionine beta synthase (CBS). | mRNA and protein expression of CBS were increased after hypoxia. The increase may be mediated by hypoxia-inducible factors (HIFs) in the cell models. |
| [86] | Sprague-Dawley rats (male) | The study evaluate the effects of exposition to acute severe repiratory hypoxia followed by reoxygenation in brain injury. | After hypoxia and reoxygenation, oxidative stress and apoptosis were increased. |
| [88] | Sprague-Dawley rats (male) and endothelial cells | The study examined whether glutathione reduces cerebral infarct size after middle cerebral artery occulsion | In vivo GSH-deficiency resulted in increased cerebral infarction volume. GSH reduced brain infarct volume. the expression of claudin-5 associated with brain infarct formation.We also examined activation of the PI3K/Akt pathway, inactivation of FOXO3, and expression of Bcl2 to assess the role of GSH in promoting cell survival in response to ischemic injury. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burgess, K.; Bennett, C.; Mosnier, H.; Kwatra, N.; Bethel, F.; Jadavji, N.M. The Antioxidant Role of One-Carbon Metabolism on Stroke. Antioxidants 2020, 9, 1141. https://doi.org/10.3390/antiox9111141
Burgess K, Bennett C, Mosnier H, Kwatra N, Bethel F, Jadavji NM. The Antioxidant Role of One-Carbon Metabolism on Stroke. Antioxidants. 2020; 9(11):1141. https://doi.org/10.3390/antiox9111141
Chicago/Turabian StyleBurgess, Kassidy, Calli Bennett, Hannah Mosnier, Neha Kwatra, Forrest Bethel, and Nafisa M. Jadavji. 2020. "The Antioxidant Role of One-Carbon Metabolism on Stroke" Antioxidants 9, no. 11: 1141. https://doi.org/10.3390/antiox9111141
APA StyleBurgess, K., Bennett, C., Mosnier, H., Kwatra, N., Bethel, F., & Jadavji, N. M. (2020). The Antioxidant Role of One-Carbon Metabolism on Stroke. Antioxidants, 9(11), 1141. https://doi.org/10.3390/antiox9111141