The Writers, Readers, and Erasers in Redox Regulation of GAPDH


Abstract
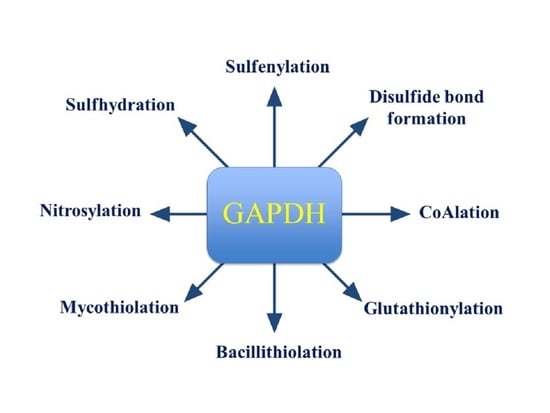
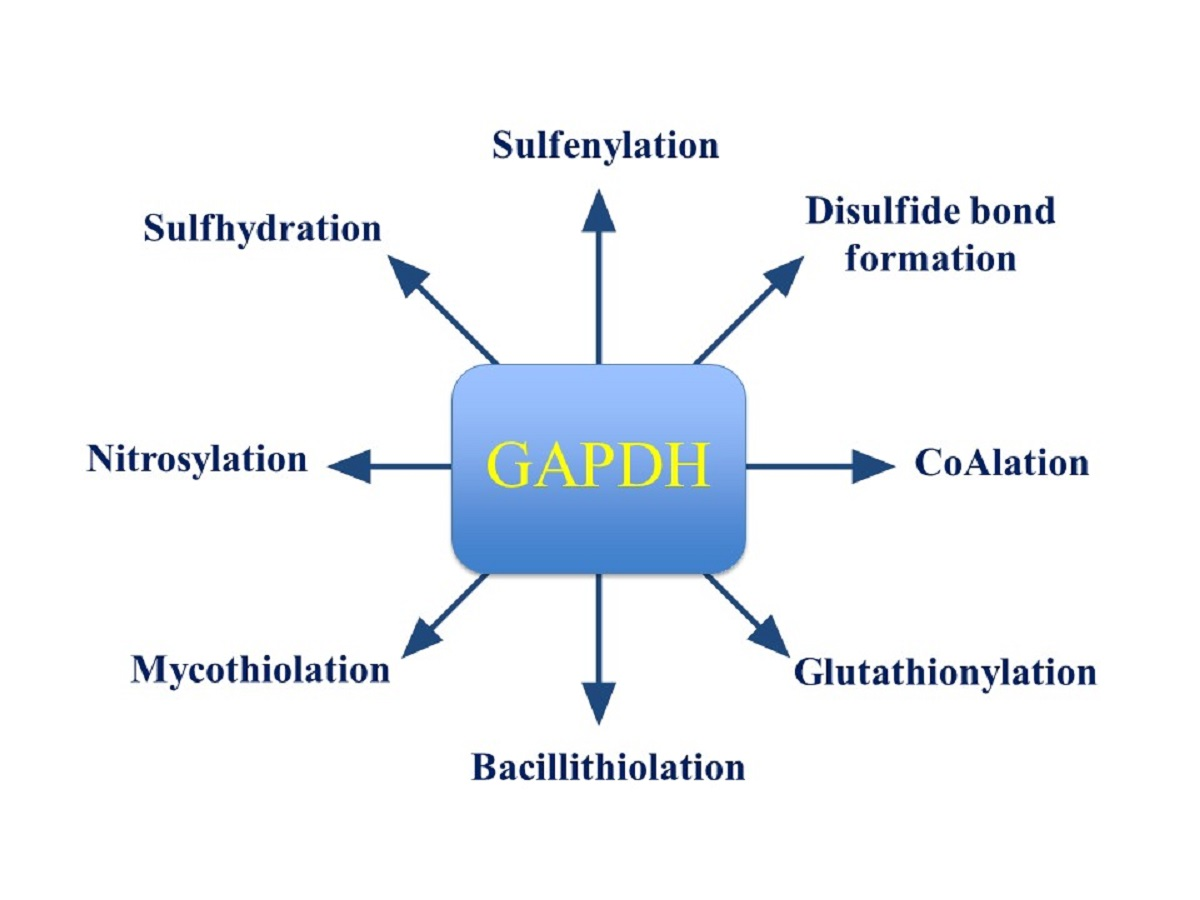
:
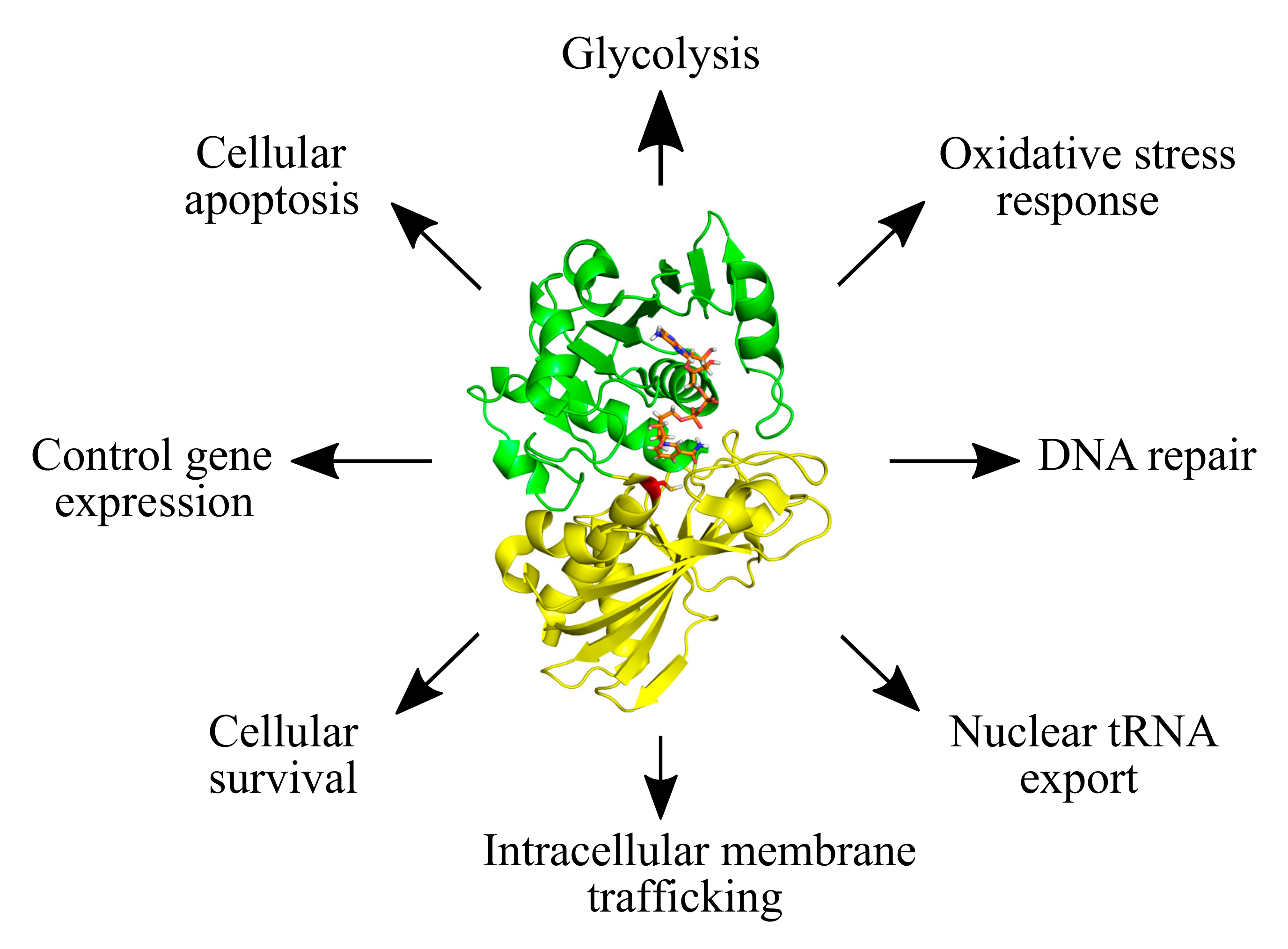
1. Introduction
2. GAPDH Modifications by Redox Writers
2.1. Key Features of Redox Writers
2.2. Redox Sensing by GAPDH and Its Regulatory Consequences
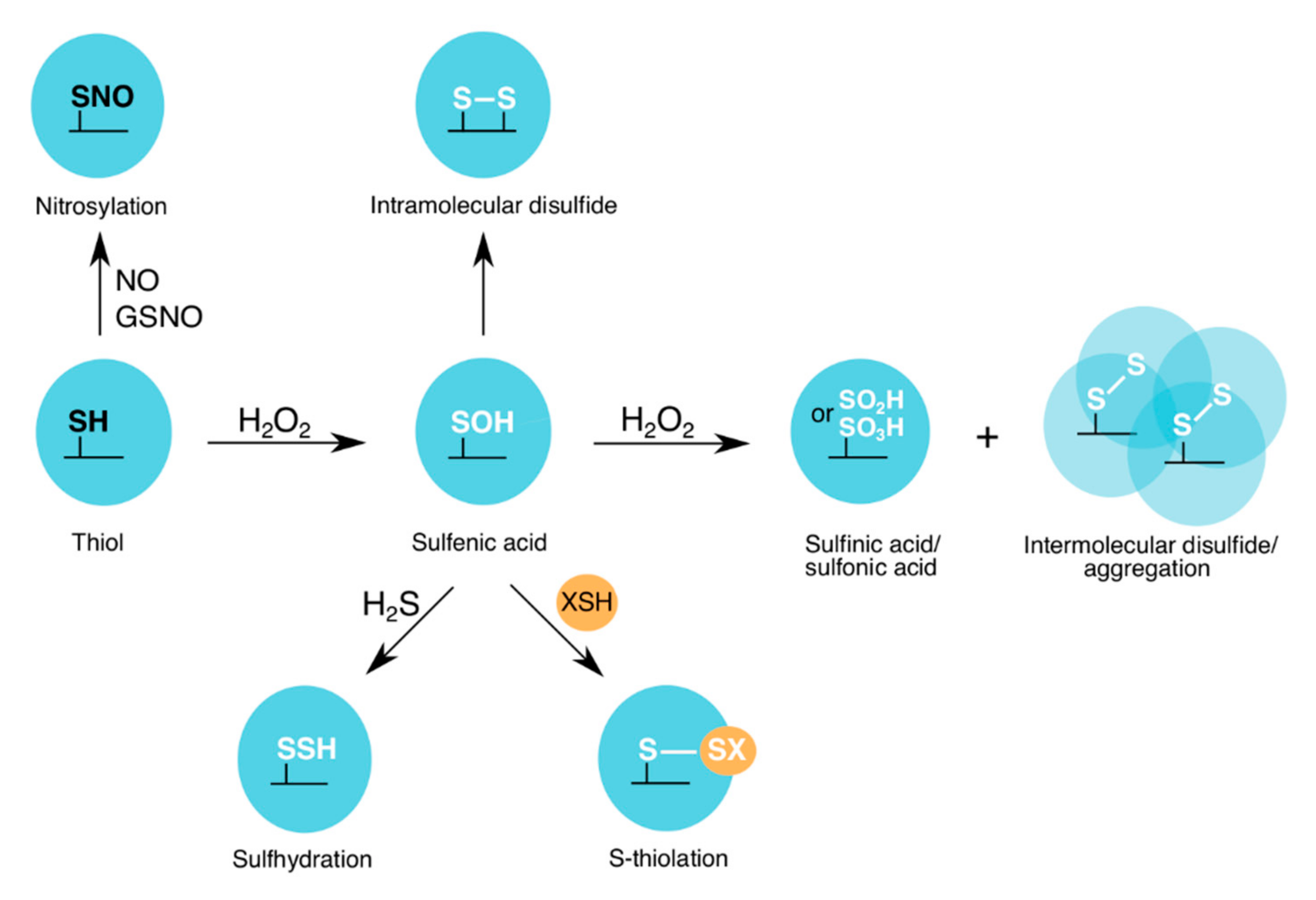
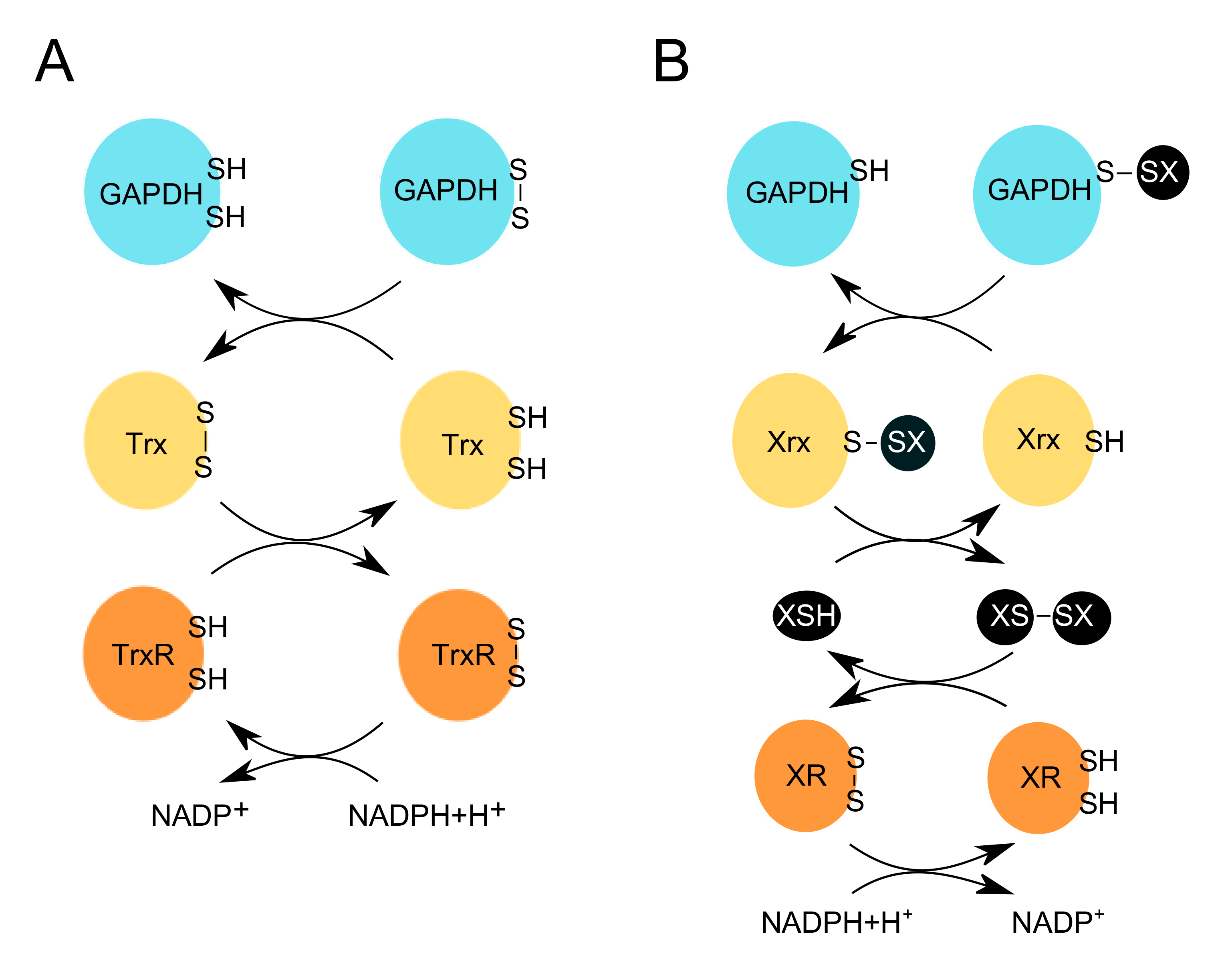
2.2.1. Sulfenylation and S-Thiolation of GAPDH

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cysteine Oxidation to Sulfenic, Sulfinic and Sulfonic Acids or Disulfide Bond Formation | |||||
|---|---|---|---|---|---|
| Target Protein | Oxidant | Cys Residue | oxPTM | Influence on Enzymatic Activity | Ref. |
| H. sapien GAPDH | H2O2 | Cys152 | Sulfenic and sulfonic acids | Inhibition | [23] |
| Rabbit muscle GAPDH | H2O2 | Cys150–Cys154 | Intramolecular disulfide bonds | Inhibition | [58] |
| A. thaliana GAPC1 | H2O2 | Cys155 | Sulfenic, sulfinic and sulfonic acids | Inhibition | [59] |
| C.diphtheriae GAPDH | H2O2, NaOCl | Cys153, Cys153–Cys157 | Sulfonic acid, and intramolecular disulfide bond | Inhibition | [47] |
2.2.2. Nitrosylation and Sulfhydration of GAPDH
2.2.3. Methionine Oxidation, Cysteine Disulfide Formation and GAPDH Aggregation
| Regulatory S-Thiolations on GAPDH | ||||||
| Target Protein | Oxidant or Molecule | Residue | oxPTM | Influence on Enzymatic Activity | Recycling | Ref. |
| P. falciparum GAPDH | GSSG | Cys153 | Glutathion ylation | Reversible inhibition | Grx1, Trx, and plasmo- redoxin | [70] |
| Rabbit muscle GAPDH | H2O2 (+GSH) | Cys150 | Glutathionylation | Reversible inhibition | Excess GSH, and Trx | [58] |
| A. thaliana GAPC1 | GSSG, and H2O2 (>+GSH) | Cys155 * | Glutathionylation | Reversible inhibition | GrxC1, and Trx | [59] |
| A. thaliana A4-GAPDH | GSSG, and H2O2 (+GSH) | Cys149 | Glutathionylation | Reversible inhibition | Grx1, and Grx3 | [94,95] |
| H. sapiens GAPDH | H2O2 (+GSH) | Cys152 | Glutathionylation | Reversible inhibition | DTT | [23] |
| C. diphtheriae GAPDH | H2O2 (+MSH), and NaOCl (+MSH) | Cys153 | Mycothiolation | Reversible inhibition | Mrx1, and Trx | [47] |
| S. aureus GAPDH | H2O2 (+BSH), and NaOCl (+BSH) | Cys151 | Bacillithiolation | Reversible inhibition | Brx | [46] |
| S. aureus GAPDH1 | CoASSCoA | Cys151 | CoAlation | Reversible inhibition | DTT | [20] |
| Citobacter sp. 5-77 GAPDH | CoASSCoA, NaOCl (+CoA), and H2O2 (+CoA) | Cys149 | CoAlation | Reversible inhibition | Excess DTT, GSH, and CoA | [62] |
| Nitrosylation and Sulfhydration of GAPDH | ||||||
| Target Protein | Oxidant or Molecule | Residue | oxPTM | Influence on Enzymatic Activity | Recycling | Ref. |
| A. thaliana GAPC1 | GSNO | Cys155 * | Nitrosylation | Reversible inhibition | GSH | [72] |
| GAPDH (SH-Sy5Y cell extract) | SNO-Trx1 | Cys247 | Nitrosylation | Reversible inhibition | Reduced Trx1 | [96,97] |
| GAPDH (HEK293 extract) | H2S | Cys152 | Sulfhydration | Activity increase | DTT | [17] |
3. Decoding of the GAPDH Redox Communication
3.1. Readers of Sulfenylated and S-Thiolated GAPDH
3.2. Readers of Nitrosylated GAPDH
3.2.1. Nuclear Transportation and Cellular Apoptosis
3.2.2. GAPDH Translocation to the Mitochondria
3.3. Readers of Sulfhydrated GAPDH
4. Erasers of Redox-Associated Modifications
4.1. Key Features of Cellular Redox Erasers
4.2. Erasers of GAPDH Redox Modifications
4.2.1. Erasers of Intra- and Intermolecular Disulfide Bonds
4.2.2. Erasers of GAPDH S-Thiolation
4.2.3. Erasers of GAPDH Nitrosylation and Sulfhydration
5. Human Pathologies Associated with Dysregulated GAPDH Function
5.1. GAPDH in Neurodegeneration
5.2. GAPDH in Metabolic Disorders
5.3. Therapeutic Targeting of Redox-Modified GAPDH
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Michels, S.; Rogalska, E.; Branlant, G. Phosphate-binding sites in phosphorylating glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus. Eur. J. Biochem. 1996, 235, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.P.; Bernhard, S.A. Transfer of 1,3-diphosphoglycerate between glyceraldehyde-3-phosphate dehydrogenase and 3-phosphoglycerate kinase via an enzyme-substrate-enzyme complex. Biochemistry 1982, 21, 4189–4194. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Green, M.R. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 1993, 259, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, E.J. Glyceraldehyde-3-phosphate dehydrogenase is required for vesicular transport in the early secretory pathway. J. Biol. Chem. 2001, 276, 2480–2486. [Google Scholar] [CrossRef] [Green Version]
- Raje, C.I.; Kumar, S.; Harle, A.; Nanda, J.S.; Raje, M. The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J. Biol. Chem. 2007, 282, 3252–3261. [Google Scholar] [CrossRef] [Green Version]
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641. [Google Scholar] [CrossRef] [Green Version]
- Demarse, N.A.; Ponnusamy, S.; Spicer, E.K.; Apohan, E.; Baatz, J.E.; Ogretmen, B.; Davies, C. Direct binding of glyceraldehyde 3-phosphate dehydrogenase to telomeric DNA protects telomeres against chemotherapy-induced rapid degradation. J. Mol. Biol. 2009, 394, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674. [Google Scholar] [CrossRef]
- Sirover, M.A. On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta 2011, 1810, 741–751. [Google Scholar] [CrossRef]
- Ventura, M.; Mateo, F.; Serratosa, J.; Salaet, I.; Carujo, S.; Bachs, O.; Pujol, M.J. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int. J. Biochem. Cell Biol. 2010, 42, 1672–1680. [Google Scholar] [CrossRef]
- Baba, T.; Kobayashi, H.; Kawasaki, H.; Mineki, R.; Naito, H.; Ohmori, D. Glyceraldehyde-3-phosphate dehydrogenase interacts with phosphorylated Akt resulting from increased blood glucose in rat cardiac muscle. FEBS Lett 2010, 584, 2796–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laschet, J.J.; Minier, F.; Kurcewicz, I.; Bureau, M.H.; Trottier, S.; Jeanneteau, F.; Griffon, N.; Samyn, B.; Van Beeumen, J.; Louvel, J.; et al. Glyceraldehyde-3-phosphate dehydrogenase is a GABAA receptor kinase linking glycolysis to neuronal inhibition. J. Neurosci. 2004, 24, 7614–7622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Han, D.; Kim, K.; Kang, Y.; Kim, Y. O-GlcNAcylation disrupts glyceraldehyde-3-phosphate dehydrogenase homo-tetramer formation and mediates its nuclear translocation. Biochim. Biophys. Acta 2009, 1794, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Brune, B. Characterization of a nitric-oxide-catalysed ADP-ribosylation of glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 1992, 210, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Molina y Vedia, L.; McDonald, B.; Reep, B.; Brune, B.; Di Silvio, M.; Billiar, T.R.; Lapetina, E.G. Nitric oxide-induced S-nitrosylation of glyceraldehyde-3-phosphate dehydrogenase inhibits enzymatic activity and increases endogenous ADP-ribosylation. J. Biol. Chem. 1992, 267, 24929–24932. [Google Scholar]
- Cahuana, G.M.; Tejedo, J.R.; Jimenez, J.; Ramirez, R.; Sobrino, F.; Bedoya, F.J. Nitric oxide-induced carbonylation of Bcl-2, GAPDH and ANT precedes apoptotic events in insulin-secreting RINm5F cells. Exp. Cell Res. 2004, 293, 22–30. [Google Scholar] [CrossRef]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef] [Green Version]
- Grant, C.M.; Quinn, K.A.; Dawes, I.W. Differential protein S-thiolation of glyceraldehyde-3-phosphate dehydrogenase isoenzymes influences sensitivity to oxidative stress. Mol. Cell Biol. 1999, 19, 2650–2656. [Google Scholar] [CrossRef] [Green Version]
- Butera, G.; Mullappilly, N.; Masetto, F.; Palmieri, M.; Scupoli, M.T.; Pacchiana, R.; Donadelli, M. Regulation of autophagy by nuclear GAPDH and Its aggregates in cancer and neurodegenerative disorders. Int. J. Mol. Sci. 2019, 20, 2062. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Zhyvoloup, A.; Bakovic, J.; Thomas, N.; Yu, B.Y.K.; Das, S.; Orengo, C.; Newell, C.; Ward, J.; Saladino, G.; et al. Protein CoAlation and antioxidant function of coenzyme A in prokaryotic cells. Biochem. J. 2018, 475, 1909–1937. [Google Scholar] [CrossRef] [Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ralser, M.; Wamelink, M.M.; Kowald, A.; Gerisch, B.; Heeren, G.; Struys, E.A.; Klipp, E.; Jakobs, C.; Breitenbach, M.; Lehrach, H.; et al. Dynamic rerouting of the carbohydrate flux is key to counteracting oxidative stress. J. Biol. 2007, 6, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, D.; Bronowska, A.K.; Morgan, B.; Doka, E.; Van Laer, K.; Nagy, P.; Grater, F.; Dick, T.P. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015, 11, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yan, L.J. Protein oxidative modifications: Beneficial roles in disease and health. J. Biochem. Pharmacol. Res. 2013, 1, 15–26. [Google Scholar]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef]
- Sies, H.; Berndt, C.; Jones, D.P. Oxidative Stress. Annu. Rev. Biochem. 2017, 86, 715–748. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Bruce King, S. Potential biological chemistry of hydrogen sulfide (H2S) with the nitrogen oxides. Free Radic. Biol. Med. 2013, 55, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Gould, N.; Doulias, P.T.; Tenopoulou, M.; Raju, K.; Ischiropoulos, H. Regulation of protein function and signaling by reversible cysteine S-nitrosylation. J. Biol. Chem. 2013, 288, 26473–26479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmann, M.G.; Moras, D.; Olsen, K.W. Chemical and biological evolution of nucleotide-binding protein. Nature 1974, 250, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Olsen, K.W.; Moras, D.; Rossmann, M.G.; Harris, J.I. Sequence variability and structure of D-glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1975, 250, 9313–9321. [Google Scholar]
- Warizaya, M.; Kinoshita, T.; Kato, A.; Nakajima, H.; Fujii, T. Cloning, expression, purification, crystallization and preliminary X-ray analysis of human liver glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 567–568. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, J.L.; Tanner, J.J. High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 290–301. [Google Scholar] [CrossRef]
- Sirover, M.A. Structural analysis of glyceraldehyde-3-phosphate dehydrogenase functional diversity. Int. J. Biochem. Cell Biol. 2014, 7, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Polgar, L. Ion-pair formation as a source of enhanced reactivity of the essential thiol group of D-glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 1975, 51, 63–71. [Google Scholar] [CrossRef]
- Corbier, C.; Michels, S.; Wonacott, A.J.; Branlant, G. Characterization of the two anion-recognition sites of glyceraldehyde-3-phosphate dehydrogenase from Bacillus stearothermophilus by site-directed mutagenesis and chemical modification. Biochemistry 1994, 33, 3260–3265. [Google Scholar] [CrossRef]
- Hildebrandt, T.; Knuesting, J.; Berndt, C.; Morgan, B.; Scheibe, R. Cytosolic thiol switches regulating basic cellular functions: GAPDH as an information hub? Biol. Chem. 2015, 396, 523–537. [Google Scholar] [CrossRef]
- Little, C.; O’Brien, P.J. (Mechanism of peroxide-inactivation of the sulphydryl enzyme glyceraldehyde-3-phosphate dehydrogenase. Eur. J. Biochem. 1969, 10, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Hancock, J.T.; Henson, D.; Nyirenda, M.; Desikan, R.; Harrison, J.; Lewis, M.; Hughes, J.; Neill, S.J. Proteomic identification of glyceraldehyde 3-phosphate dehydrogenase as an inhibitory target of hydrogen peroxide in Arabidopsis. Plant. Physiol. Biochem. 2005, 43, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Baty, J.W.; Hampton, M.B.; Winterbourn, C.C. Proteomic detection of hydrogen peroxide-sensitive thiol proteins in Jurkat cells. Biochem. J. 2005, 389, 785–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, Y.; Peak-Chew, S.Y.; Newell, C.; Miller-Aidoo, S.; Mangal, S.; Zhyvoloup, A.; Bakovic, J.; Malanchuk, O.; Pereira, G.C.; Kotiadis, V.; et al. Protein CoAlation: A redox-regulated protein modification by coenzyme A in mammalian cells. Biochem. J. 2017, 474, 2489–2508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, D.P.; Skipsey, M.; Grundy, N.M.; Edwards, R. Stress-induced protein S-glutathionylation in Arabidopsis. Plant. Physiol. 2005, 138, 2233–2244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imber, M.; Huyen, N.T.T.; Pietrzyk-Brzezinska, A.J.; Loi, V.V.; Hillion, M.; Bernhardt, J.; Tharichen, L.; Kolsek, K.; Saleh, M.; Hamilton, C.J.; et al. Protein S-bacillithiolation functions in thiol protection and redox regulation of the glyceraldehyde-3-phosphate dehydrogenase Gap in Staphylococcus aureus under hypochlorite stress. Antioxid. Redox Signal. 2018, 28, 410–430. [Google Scholar] [CrossRef] [Green Version]
- Hillion, M.; Imber, M.; Pedre, B.; Bernhardt, J.; Saleh, M.; Loi, V.V.; Maass, S.; Becher, D.; Astolfi Rosado, L.; Adrian, L.; et al. The glyceraldehyde-3-phosphate dehydrogenase GapDH of Corynebacterium diphtheriae is redox-controlled by protein S-mycothiolation under oxidative stress. Sci. Rep. 2017, 7, 5020. [Google Scholar] [CrossRef] [Green Version]
- Shenton, D.; Grant, C.M. Protein S-thiolation targets glycolysis and protein synthesis in response to oxidative stress in the yeast Saccharomyces cerevisiae. Biochem. J. 2003, 374, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Akter, S.; Huang, J.; Bodra, N.; De Smet, B.; Wahni, K.; Rombaut, D.; Pauwels, J.; Gevaert, K.; Carroll, K.; Van Breusegem, F.; et al. DYn-2 Based Identification of Arabidopsis Sulfenomes. Mol. Cell Proteom. 2015, 14, 1183–1200. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Gupta, V.; Carroll, K.S.; Liebler, D.C. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 2014, 5, 4776. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Willems, P.; Van Breusegem, F.; Messens, J. Pathways crossing mammalian and plant sulfenomic landscapes. Free Radic. Biol. Med. 2018, 122, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Yang, J.; Liebler, D.C.; Carroll, K.S. Diverse redoxome reactivity profiles of carbon nucleophiles. J. Am. Chem. Soc. 2017, 39, 5588–5595. [Google Scholar] [CrossRef] [PubMed]
- Waszczak, C.; Akter, S.; Eeckhout, D.; Persiau, G.; Wahni, K.; Bodra, N.; Van Molle, I.; De Smet, B.; Vertommen, D.; Gevaert, K.; et al. Sulfenome mining in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2014, 111, 11545–11550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez, L.V.; Allison, W.S. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J. Biol. Chem. 1974, 249, 6234–6243. [Google Scholar]
- Zaffagnini, M.; Fermani, S.; Calvaresi, M.; Orru, R.; Iommarini, L.; Sparla, F.; Falini, G.; Bottoni, A.; Trost, P. Tuning cysteine reactivity and sulfenic acid stability by protein microenvironment in glyceraldehyde-3-phosphate dehydrogenases of Arabidopsis thaliana. Antioxid. Redox Signal. 2016, 24, 502–517. [Google Scholar] [CrossRef]
- Cseke, E.; Boross, L. Factors affecting the reactivity of the activated SH-group of D-glyceraldehyde 3-phosphate dehydrogenase. Acta Biochim. Biophys. Acad. Sci. Hung. 1970, 5, 385–397. [Google Scholar]
- Nakajima, H.; Amano, W.; Fujita, A.; Fukuhara, A.; Azuma, Y.T.; Hata, F.; Inui, T.; Takeuchi, T. The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J. Biol. Chem. 2007, 282, 26562–26574. [Google Scholar] [CrossRef]
- Barinova, K.V.; Serebryakova, M.V.; Muronetz, V.I.; Schmalhausen, E.V. S-glutathionylation of glyceraldehyde-3-phosphate dehydrogenase induces formation of C150–C154 intrasubunit disulfide bond in the active site of the enzyme. Biochim. Biophys. Acta Gen. Subj. 2007, 1861, 3167–3177. [Google Scholar] [CrossRef]
- Bedhomme, M.; Adamo, M.; Marchand, C.H.; Couturier, J.; Rouhier, N.; Lemaire, S.D.; Zaffagnini, M.; Trost, P. Glutathionylation of cytosolic glyceraldehyde-3-phosphate dehydrogenase from the model plant Arabidopsis thaliana is reversed by both glutaredoxins and thioredoxins in vitro. Biochem. J. 2012, 445, 337–347. [Google Scholar] [CrossRef] [Green Version]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1642–1653. [Google Scholar] [CrossRef]
- Loi, V.V.; Rossius, M.; Antelmann, H. Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 2015, 6, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuji, K.; Yoon, K.S.; Ogo, S. Glyceraldehyde-3-phosphate dehydrogenase from Citrobacter sp. S-77 is post-translationally modified by CoA (protein CoAlation) under oxidative stress. FEBS Open Bio 2019, 9, 53–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillig, C.H.; Berndt, C. Cellular functions of glutathione. Biochim. Biophys. Acta 2013, 1830, 3137–3138. [Google Scholar] [CrossRef] [PubMed]
- Brandes, N.; Schmitt, S.; Jakob, U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signal. 2009, 11, 997–1014. [Google Scholar] [CrossRef]
- Pan, S.; Berk, B.C. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: Key role for glutaredoxin in the death pathway. Circ. Res. 2007, 100, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Cross, J.V.; Templeton, D.J. Oxidative stress inhibits MEKK1 by site-specific glutathionylation in the ATP-binding domain. Biochem. J. 2004, 381, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Haendeler, J. Thioredoxin-1 and posttranslational modifications. Antioxid. Redox Signal. 2006, 8, 1723–1728. [Google Scholar] [CrossRef]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [Green Version]
- Greetham, D.; Vickerstaff, J.; Shenton, D.; Perrone, G.G.; Dawes, I.W.; Grant, C.M. Thioredoxins function as deglutathionylase enzymes in the yeast Saccharomyces cerevisiae. BMC Biochem. 2010, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Kehr, S.; Jortzik, E.; Delahunty, C.; Yates, J.R., 3rd; Rahlfs, S.; Becker, K. Protein S-glutathionylation in malaria parasites. Antioxid. Redox Signal. 2011, 15, 2855–2865. [Google Scholar] [CrossRef] [Green Version]
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brune, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffagnini, M.; Morisse, S.; Bedhomme, M.; Marchand, C.H.; Festa, M.; Rouhier, N.; Lemaire, S.D.; Trost, P. Mechanisms of nitrosylation and denitrosylation of cytoplasmic glyceraldehyde-3-phosphate dehydrogenase from Arabidopsis thaliana. J. Biol. Chem. 2013, 288, 22777–22789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustarini, D.; Milzani, A.; Aldini, G.; Carini, M.; Rossi, R.; Dalle-Donne, I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid. Redox Signal. 2005, 7, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hausladen, A.; Zeng, M.; Que, L.; Heitman, J.; Stamler, J.S. A metabolic enzyme for S-nitrosothiol conserved from bacteria to humans. Nature 2010, 410, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Jothivasan, V.K.; Hamilton, C.J. Mycothiol: Synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat. Prod. Rep. 2008, 25, 1091–1117. [Google Scholar] [CrossRef] [PubMed]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, H.S.; Steenkamp, D.J. Thiols of intracellular pathogens. Identification of ovothiol A in Leishmania donovani and structural analysis of a novel thiol from Mycobacterium bovis. Eur. J. Biochem. 1994, 224, 203–213. [Google Scholar] [CrossRef]
- Sakuda, S.; Zhou, Z.Y.; Yamada, Y. Structure of a novel disulfide of 2-(N-acetylcysteinyl)amido-2-deoxy-alpha-D-glucopyranosyl-myo-inositol produced by Streptomyces sp. Biosci. Biotechnol. Biochem. 1994, 58, 1347–1348. [Google Scholar] [CrossRef]
- Newton, G.L.; Rawat, M.; La Clair, J.J.; Jothivasan, V.K.; Budiarto, T.; Hamilton, C.J.; Claiborne, A.; Helmann, J.D.; Fahey, R.C. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009, 5, 625–627. [Google Scholar] [CrossRef]
- Gout, I. Coenzyme A: A protective thiol in bacterial antioxidant defence. Biochem. Soc. Trans. 2019, 47, 469–476. [Google Scholar] [CrossRef]
- Delcardayre, S.B.; Davies, J.E. Staphylococcus aureus coenzyme A disulfide reductase, a new subfamily of pyridine nucleotide-disulfide oxidoreductase. Sequence, expression, and analysis of cdr. J. Biol. Chem. 1998, 273, 5752–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, R.; Zhang, Y.M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Malanchuk, O.M.; Panasyuk, G.G.; Serbin, N.M.; Gout, I.T.; Filonenko, V.V. Generation and characterization of monoclonal antibodies specific to coenzyme A. Biopolym. Cell 2015, 31, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Anand, P.; Hausladen, A.; Wang, Y.J.; Zhang, G.F.; Stomberski, C.; Brunengraber, H.; Hess, D.T.; Stamler, J.S. Identification of S-nitroso-CoA reductases that regulate protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2014, 111, 18572–18577. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Hogg, N. The biochemistry and physiology of S-nitrosothiols. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 585–600. [Google Scholar] [CrossRef]
- Zhang, D.; Macinkovic, I.; Devarie-Baez, N.O.; Pan, J.; Park, C.M.; Carroll, K.S.; Filipovic, M.R.; Xian, M. Detection of protein S-sulfhydration by a tag-switch technique. Angew. Chem. Int. Ed. Engl. 2014, 53, 575–581. [Google Scholar] [CrossRef] [Green Version]
- Jarosz, A.P.; Wei, W.; Gauld, J.W.; Auld, J.; Ozcan, F.; Aslan, M.; Mutus, B. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) is inactivated by S-sulfuration in vitro. Free Radic. Biol. Med. 2015, 89, 512–521. [Google Scholar] [CrossRef]
- Gao, X.H.; Krokowski, D.; Guan, B.J.; Bederman, I.; Majumder, M.; Parisien, M.; Diatchenko, L.; Kabil, O.; Willard, B.; Banerjee, R.; et al. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. Elife 2015, 4, e10067. [Google Scholar] [CrossRef]
- Cumming, R.C.; Schubert, D. Amyloid-beta induces disulfide bonding and aggregation of GAPDH in Alzheimer’s disease. FASEB J. 2005, 19, 2060–2062. [Google Scholar] [CrossRef]
- Samson, A.L.; Knaupp, A.S.; Sashindranath, M.; Borg, R.J.; Au, A.E.; Cops, E.J.; Saunders, H.M.; Cody, S.H.; McLean, C.A.; Nowell, C.J.; et al. Nucleocytoplasmic coagulation: An injury-induced aggregation event that disulfide crosslinks proteins and facilitates their removal by plasmin. Cell Rep. 2012, 2, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, H.; Amano, W.; Fukuhara, A.; Kubo, T.; Misaki, S.; Azuma, Y.T.; Inui, T.; Takeuchi, T. An aggregate-prone mutant of human glyceraldehyde-3-phosphate dehydrogenase augments oxidative stress-induced cell death in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2009, 390, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.L.; Knaupp, A.S.; Kass, I.; Kleifeld, O.; Marijanovic, E.M.; Hughes, V.A.; Lupton, C.J.; Buckle, A.M.; Bottomley, S.P.; Medcalf, R.L. Oxidation of an exposed methionine instigates the aggregation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 2014, 289, 26922–26936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaffagnini, M.; Michelet, L.; Massot, V.; Trost, P.; Lemaire, S.D. Biochemical characterization of glutaredoxins from Chlamydomonas reinhardtii reveals the unique properties of a chloroplastic CGFS-type glutaredoxin. J. Biol. Chem. 2008, 283, 8868–8876. [Google Scholar] [CrossRef] [Green Version]
- Zaffagnini, M.; Michelet, L.; Marchand, C.; Sparla, F.; Decottignies, P.; Le Marechal, P.; Miginiac-Maslow, M.; Noctor, G.; Trost, P.; Lemaire, S.D. The thioredoxin-independent isoform of chloroplastic glyceraldehyde-3-phosphate dehydrogenase is selectively regulated by glutathionylation. FEBS J. 2007, 274, 212–226. [Google Scholar] [CrossRef]
- Wu, C.; Parrott, A.M.; Liu, T.; Jain, M.R.; Yang, Y.; Sadoshima, J.; Li, H. Distinction of thioredoxin transnitrosylation and denitrosylation target proteins by the ICAT quantitative approach. J. Proteom. 2011, 74, 2498–2509. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Liu, T.; Chen, W.; Oka, S.; Fu, C.; Jain, M.R.; Parrott, A.M.; Baykal, A.T.; Sadoshima, J.; Li, H. Redox regulatory mechanism of transnitrosylation by thioredoxin. Mol. Cell Proteom. 2010, 9, 2262–2275. [Google Scholar] [CrossRef] [Green Version]
- Morigasaki, S.; Shimada, K.; Ikner, A.; Yanagida, M.; Shiozaki, K. Glycolytic enzyme GAPDH promotes peroxide stress signaling through multistep phosphorelay to a MAPK cascade. Mol. Cell 2008, 30, 108–113. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, A.N.; Lee, A.; Place, W.; Shiozaki, K. Multistep phosphorelay proteins transmit oxidative stress signals to the fission yeast stress-activated protein kinase. Mol. Biol. Cell 2000, 11, 1169–1181. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Mitsubayashi, Y.; Aiba, H.; Mizuno, T. Spy1, a histidine-containing phosphotransfer signaling protein, regulates the fission yeast cell cycle through the Mcs4 response regulator. J. Bacteriol. 2000, 182, 4868–4874. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, S.; Park, J.B.; Lee, S.D.; Kim, J.H.; Ha, S.H.; Hasumi, K.; Endo, A.; Suh, P.G.; Ryu, S.H. Hydrogen peroxide induces association between glyceraldehyde 3-phosphate dehydrogenase and phospholipase D2 to facilitate phospholipase D2 activation in PC12 cells. J. Neurochem. 2003, 85, 1228–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.D.; Lee, B.D.; Han, J.M.; Kim, J.H.; Kim, Y.; Suh, P.G.; Ryu, S.H. Phospholipase D2 activity suppresses hydrogen peroxide-induced apoptosis in PC12 cells. J. Neurochem. 2000, 75, 1053–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exton, J.H. Phospholipase D. Ann. N. Y. Acad. Sci. 2000, 905, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Hara, M.R.; Ahmad, A.S.; Cascio, M.B.; Kamiya, A.; Ehmsen, J.T.; Agrawal, N.; Hester, L.; Dore, S.; Snyder, S.H.; et al. GOSPEL: A neuroprotective protein that binds to GAPDH upon S-nitrosylation. Neuron 2009, 63, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, N.; Hara, M.R.; Kornberg, M.D.; Cascio, M.B.; Bae, B.I.; Shahani, N.; Thomas, B.; Dawson, T.M.; Dawson, V.L.; Snyder, S.H.; et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 2008, 10, 866–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Tristan, C.A.; Ramos, A.; Shahani, N.; Emiliani, F.E.; Nakajima, H.; Noeh, C.C.; Kato, Y.; Takeuchi, T.; Noguchi, T.; Kadowaki, H.; et al. Role of apoptosis signal-regulating kinase 1 (ASK1) as an activator of the GAPDH-Siah1 stress-signaling cascade. J. Biol. Chem. 2015, 290, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Richardson, D.R. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J. Biol. Chem. 2011, 286, 15413–15427. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Lee, B.I.; Cheon, S.Y.; Kim, H.W.; Kim, H.J.; Kim, G.W. Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease. Neuroscience 2009, 163, 1128–1134. [Google Scholar] [CrossRef]
- Tarze, A.; Deniaud, A.; Le Bras, M.; Maillier, E.; Molle, D.; Larochette, N.; Zamzami, N.; Jan, G.; Kroemer, G.; Brenner, C. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene 2007, 26, 2606–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohr, M.J.; Murphy, E.; Steenbergen, C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS ONE 2014, 9, e111448. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Morgan, M.; Shen, R.F.; Steenbergen, C.; Murphy, E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res. 2007, 101, 1155–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mir, S.; Sen, T.; Sen, N. Cytokine-induced GAPDH sulfhydration affects PSD95 degradation and memory. Mol. Cell 2014, 56, 786–795. [Google Scholar] [CrossRef] [Green Version]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–1368. [Google Scholar]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Nordberg, J.; Arner, E.S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Collet, J.F.; Messens, J. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef]
- Williams, C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995, 9, 1267–1276. [Google Scholar] [CrossRef] [Green Version]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [Green Version]
- Deponte, M.; Lillig, C.H. Enzymatic control of cysteinyl thiol switches in proteins. Biol. Chem. 2015, 396, 401–413. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.K.; Gronau, K.; Mader, U.; Hessling, B.; Becher, D.; Antelmann, H. S-bacillithiolation protects against hypochlorite stress in Bacillus subtilis as revealed by transcriptomics and redox proteomics. Mol. Cell Proteom. 2011, 10, M111009506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landino, L.M.; Hagedorn, T.D.; Kennett, K.L. Evidence for thiol/disulfide exchange reactions between tubulin and glyceraldehyde-3-phosphate dehydrogenase. Cytoskeleton (Hoboken) 2014, 71, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Hwang, N.R.; Yim, S.H.; Kim, Y.M.; Jeong, J.; Song, E.J.; Lee, Y.; Lee, J.H.; Choi, S.; Lee, K.J. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 2009, 423, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.J. The structure and amount of tubulin in cells and tissues. J. Biol. Chem. 1979, 254, 2168–2171. [Google Scholar]
- Landino, L.M.; Robinson, S.H.; Skreslet, T.E.; Cabral, D.M. Redox modulation of tau and microtubule-associated protein-2 by the glutathione/glutaredoxin reductase system. Biochem. Biophys. Res. Commun. 2004, 323, 112–117. [Google Scholar] [CrossRef]
- Landino, L.M.; Skreslet, T.E.; Alston, J.A. Cysteine oxidation of tau and microtubule-associated protein-2 by peroxynitrite: Modulation of microtubule assembly kinetics by the thioredoxin reductase system. J. Biol. Chem. 2004, 279, 35101–35105. [Google Scholar] [CrossRef] [Green Version]
- Landino, L.M.; Iwig, J.S.; Kennett, K.L.; Moynihan, K.L. Repair of peroxynitrite damage to tubulin by the thioredoxin reductase system. Free Radic. Biol. Med. 2004, 36, 497–506. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef]
- Shenton, D.; Perrone, G.; Quinn, K.A.; Dawes, I.W.; Grant, C.M. Regulation of protein S-thiolation by glutaredoxin 5 in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2002, 277, 16853–16859. [Google Scholar] [CrossRef] [Green Version]
- Linzner, N.; Loi, V.V.; Fritsch, V.N.; Tung, Q.N.; Stenzel, S.; Wirtz, M.; Hell, R.; Hamilton, C.J.; Tedin, K.; Fulde, M.; et al. Staphylococcus aureus uses the bacilliredoxin (BrxAB)/bacillithiol disulfide reductase (YpdA) redox pathway to defend against oxidative stress under infections. Front. Microbiol. 2019, 10, 1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wan, A.; Xu, G.; Ye, D. Small changes huge impact: The role of thioredoxin 1 in the regulation of apoptosis by S-nitrosylation. Acta Biochim. Biophys. Sin. 2013, 45, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, Y.; Wu, L.; Yang, G. Thioredoxin 1 regulation of protein S-desulfhydration. Biochem. Biophys. Rep. 2016, 5, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Woltjer, R.L.; Cimino, P.J.; Pan, C.; Montine, K.S.; Zhang, J.; Montine, T.J. Proteomic analysis of neurofibrillary tangles in Alzheimer disease identifies GAPDH as a detergent-insoluble paired helical filament tau binding protein. FASEB J. 2005, 19, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Itakura, M.; Nakajima, H.; Kubo, T.; Semi, Y.; Kume, S.; Higashida, S.; Kaneshige, A.; Kuwamura, M.; Harada, N.; Kita, A.; et al. Glyceraldehyde-3-phosphate Dehydrogenase Aggregates Accelerate Amyloid-beta Amyloidogenesis in Alzheimer Disease. J. Biol. Chem. 2015, 290, 26072–26087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchiya, K.; Tajima, H.; Kuwae, T.; Takeshima, T.; Nakano, T.; Tanaka, M.; Sunaga, K.; Fukuhara, Y.; Nakashima, K.; Ohama, E.; et al. Pro-apoptotic protein glyceraldehyde-3-phosphate dehydrogenase promotes the formation of Lewy body-like inclusions. Eur. J. Neurosci. 2005, 21, 317–326. [Google Scholar] [CrossRef]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef]
- Sultana, R.; Poon, H.F.; Cai, J.; Pierce, W.M.; Merchant, M.; Klein, J.B.; Markesbery, W.R.; Butterfield, D.A. Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol. Dis. 2006, 22, 76–87. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Tajima, H.; Yamada, M.; Takahashi, H.; Kuwae, T.; Sunaga, K.; Katsube, N.; Ishitani, R. Disclosure of a pro-apoptotic glyceraldehyde-3-phosphate dehydrogenase promoter: Anti-dementia drugs depress its activation in apoptosis. Life Sci. 2004, 74, 3245–3258. [Google Scholar] [CrossRef]
- Tatton, N.A. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson’s disease. Exp. Neurol. 2000, 166, 29–43. [Google Scholar] [CrossRef]
- Senatorov, V.V.; Charles, V.; Reddy, P.H.; Tagle, D.A.; Chuang, D.M. Overexpression and nuclear accumulation of glyceraldehyde-3-phosphate dehydrogenase in a transgenic mouse model of Huntington’s disease. Mol. Cell Neurosci. 2003, 22, 285–297. [Google Scholar] [CrossRef]
- Stehouwer, C.D.A. Microvascular dysfunction and hyperglycemia: A vicious cycle with widespread consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolm-Litty, V.; Sauer, U.; Nerlich, A.; Lehmann, R.; Schleicher, E.D. High glucose-induced transforming growth factor beta1 production is mediated by the hexosamine pathway in porcine glomerular mesangial cells. J. Clin. Investig. 1998, 101, 160–169. [Google Scholar] [CrossRef]
- Koya, D.; King, G.L. Protein kinase C activation and the development of diabetic complications. Diabetes 1998, 47, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengeller, Z.; Szabo, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 7, 12222–12226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yego, E.C.; Mohr, S. siah-1 protein is necessary for high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation and cell death in Muller cells. J. Biol. Chem. 2010, 285, 3181–3190. [Google Scholar] [CrossRef] [Green Version]
- Suarez, S.; McCollum, G.W.; Jayagopal, A.; Penn, J.S. High glucose-induced retinal pericyte apoptosis depends on association of GAPDH and Siah1. J. Biol. Chem. 2015, 290, 28311–28320. [Google Scholar] [CrossRef] [Green Version]
- Kusner, L.L.; Sarthy, V.P.; Mohr, S. Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase: A role in high glucose-induced apoptosis in retinal Muller cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 1553–1561. [Google Scholar]
- Feenstra, D.J.; Yego, E.C.; Mohr, S. Modes of retinal cell death in diabetic retinopathy. J. Clin. Exp. Ophthalmol. 2013, 4, 298. [Google Scholar]
- Yego, E.C.; Vincent, J.A.; Sarthy, V.; Busik, J.V.; Mohr, S. Differential regulation of high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation in Muller cells by IL-1beta and IL-6. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, M.; Kowluru, R.A. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes 2009, 58, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, M.R.; Thomas, B.; Cascio, M.B.; Bae, B.I.; Hester, L.D.; Dawson, V.L.; Dawson, T.M.; Sawa, A.; Snyder, S.H. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc. Natl. Acad. Sci. USA 2006, 103, 3887–3889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, S.; Pinto, A.; Paredi, G.; Tamborini, L.; De Micheli, C.; La Pietra, V.; Marinelli, L.; Novellino, E.; Conti, P.; Mozzarelli, A. Discovery of covalent inhibitors of glyceraldehyde-3-phosphate dehydrogenase, a target for the treatment of malaria. J. Med. Chem. 2014, 57, 7465–7471. [Google Scholar] [CrossRef]
- Dando, I.; Pacchiana, R.; Pozza, E.D.; Cataldo, I.; Bruno, S.; Conti, P.; Cordani, M.; Grimaldi, A.; Butera, G.; Caraglia, M.; et al. UCP2 inhibition induces ROS/Akt/mTOR axis: Role of GAPDH nuclear translocation in genipin/everolimus anticancer synergism. Free Radic. Biol. Med. 2017, 113, 176–189. [Google Scholar] [CrossRef]
- Gerszon, J.; Serafin, E.; Buczkowski, A.; Michlewska, S.; Bielnicki, J.A.; Rodacka, A. Functional consequences of piceatannol binding to glyceraldehyde-3-phosphate dehydrogenase. PLoS ONE 2018, 13, e0190656. [Google Scholar] [CrossRef] [Green Version]
- Lazarev, V.F.; Nikotina, A.D.; Semenyuk, P.I.; Evstafyeva, D.B.; Mikhaylova, E.R.; Muronetz, V.I.; Shevtsov, M.A.; Tolkacheva, A.V.; Dobrodumov, A.V.; Shavarda, A.L.; et al. Small molecules preventing GAPDH aggregation are therapeutically applicable in cell and rat models of oxidative stress. Free Radic. Biol. Med. 2016, 92, 29–38. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tossounian, M.-A.; Zhang, B.; Gout, I. The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants 2020, 9, 1288. https://doi.org/10.3390/antiox9121288
Tossounian M-A, Zhang B, Gout I. The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants. 2020; 9(12):1288. https://doi.org/10.3390/antiox9121288
Chicago/Turabian StyleTossounian, Maria-Armineh, Bruce Zhang, and Ivan Gout. 2020. "The Writers, Readers, and Erasers in Redox Regulation of GAPDH" Antioxidants 9, no. 12: 1288. https://doi.org/10.3390/antiox9121288
APA StyleTossounian, M. -A., Zhang, B., & Gout, I. (2020). The Writers, Readers, and Erasers in Redox Regulation of GAPDH. Antioxidants, 9(12), 1288. https://doi.org/10.3390/antiox9121288