Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases


Abstract
:1. Introduction
2. Methods
3. Oxidative Stress
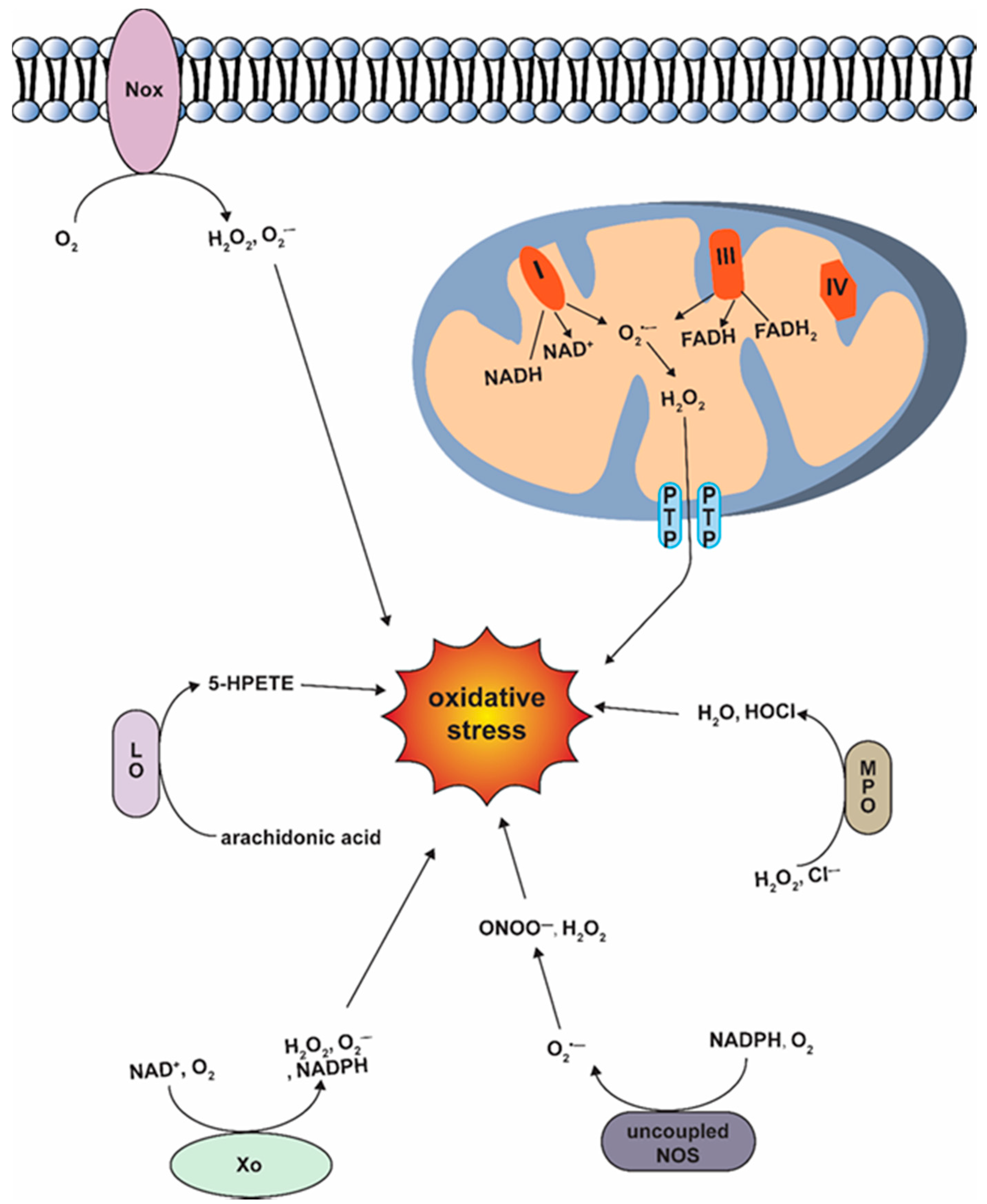
3.1. Generation of ROS
3.2. Antioxidant Defense Enzymes
3.3. Molecular Effects of Oxidative Stress
3.3.1. Oxidative Stress and Lipids, Protein, DNA Damage
3.3.2. Oxidative Stress and Inflammation
3.3.3. Oxidative Stress and Programmed Cell Death
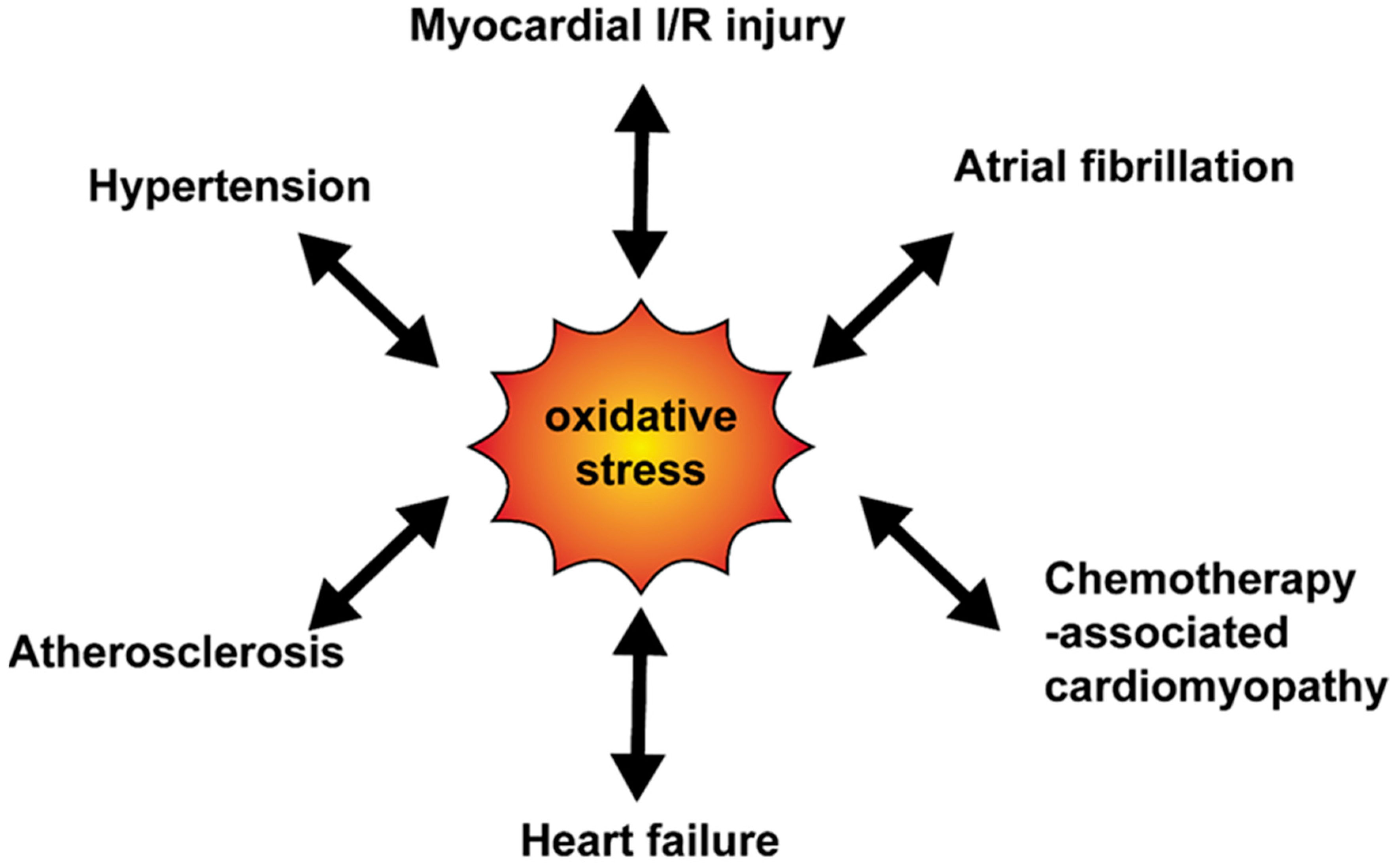
4. Oxidative Stress and Cardiovascular Disease
4.1. Oxidative Stress and Myocardial Ischemia-Reperfusion (I/R) Injury
4.2. Oxidative Stress and Heart Failure (HF)
4.3. Oxidative Stress and Atherosclerosis
4.4. Oxidative Stress and Atrial Fibrillation (AF)
4.5. Oxidative Stress and Hypertension
5. Therapies for Oxidative Stress-Associated Cardiovascular Diseases
5.1. Antioxidant Molecules
5.1.1. Nutritional Supplements
5.1.2. Novel Experimental Antioxidant-Based Therapies
5.1.3. Antioxidant Role of Clinical Drugs
5.2. miRNAs
5.3. Nanoparticles
5.4. Limitation
5.5. Novelty
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bisht, S.; Faiq, M.; Tolahunase, M.; Dada, R. Oxidative stress and male infertility. Nat. Rev. Urol. 2017, 14, 470–485. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Biesalski, H.K.; Grune, T.; Tinz, J.; Zöllner, I.; Blumberg, J.B. Reexamination of a meta-analysis of the effect of antioxidant supplementation on mortality and health in randomized trials. Nutrients 2010, 2, 929–949. [Google Scholar] [CrossRef]
- Kim, K.S.; Song, C.G.; Kang, P.M. Targeting Oxidative Stress Using Nanoparticles as a Theranostic Strategy for Cardiovascular Diseases. Antioxid. Redox Signal. 2019, 30, 733–746. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive Oxygen Species, Cellular Redox Systems and Apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Dighe, P.A.; Mezera, V.; Monternier, P.A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottenberg, H.; Hoek, J.B. The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 2017, 16, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol. Rev. 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Groemping, Y.; Lapouge, K.; Smerdon, S.J.; Rittinger, K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell 2003, 113, 343–355. [Google Scholar] [CrossRef]
- Pick, E. Role of the Rho GTPase Rac in the activation of the phagocyte NADPH oxidase: Outsourcing a key task. Small GTPases 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Paclet, M.-H.; Henderson, L.M.; Campion, Y.; Morel, F.; Dagher, M.-C. Localization of Nox2 N-terminus using polyclonal antipeptide antibodies. Biochem. J. 2004, 382, 981–986. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Prasad, S.; Gupta, S.C.; Tyagi, A.K. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett. 2017, 387. [Google Scholar] [CrossRef]
- Lei, X.G.; Zhu, J.-H.; Cheng, W.-H.; Bao, Y.; Ho, Y.-S.; Reddi, A.R.; Holmgren, A.; Arnér, E.S.J. Paradoxical Roles of Antioxidant Enzymes: Basic Mechanisms and Health Implications. Physiol. Rev. 2016, 96, 307–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Cramer-Morales, K.; Heer, C.D.; Mapuskar, K.A.; Domann, F.E. SOD2 targeted gene editing by CRISPR/Cas9 yields Human cells devoid of MnSOD. Free Radic. Biol. Med. 2015, 89, 379–386. [Google Scholar] [CrossRef] [Green Version]
- Miao, L.; Clair, D.K.S. Regulation of Superoxide Dismutase Genes: Implications in Diseases. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [Green Version]
- Sepasi Tehrani, H.; Moosavi-Movahedi, A.A. Catalase and its mysteries. Prog. Biophys. Mol. Biol. 2018, 140. [Google Scholar] [CrossRef]
- He, L.; He, T.; Farrar, S.; Ji, L.; Liu, T.; Ma, X. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell. Physiol. Biochem. 2017, 44, 532–553. [Google Scholar] [CrossRef]
- Margis, R.; Dunand, C.; Teixeira, F.K.; Margis-Pinheiro, M. Glutathione peroxidase family—An evolutionary overview. FEBS J. 2008, 275, 3959–3970. [Google Scholar] [CrossRef]
- Didion, S.P.; Ryan, M.J.; Didion, L.A.; Fegan, P.E.; Sigmund, C.D.; Faraci, F.M. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ. Res. 2002, 91, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Bolli, R.; Qiu, Y.; Tang, X.L.; Guo, Y.; French, B.A. Gene therapy with extracellular superoxide dismutase protects conscious rabbits against myocardial infarction. Circulation 2001, 103, 1893–1898. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Marcos, M.A.; Blázquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; García-Ortiz, L.; Martínez-Salgado, C. Serum Superoxide Dismutase Is Associated with Vascular Structure and Function in Hypertensive and Diabetic Patients. Oxidative Med. Cell. Longev. 2016, 2016. [Google Scholar] [CrossRef]
- Godin, N.; Liu, F.; Lau, G.J.; Brezniceanu, M.-L.; Chénier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.-L.; Chan, J.S.D. Catalase overexpression prevents hypertension and tubular apoptosis in angiotensinogen transgenic mice. Kidney Int. 2010, 77, 1086–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Maulik, N.; Engelman, R.M.; Ho, Y.S.; Magnenat, J.L.; Rousou, J.A.; Flack, J.E.; Deaton, D.; Das, D.K. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation 1997, 96, II-216-20. [Google Scholar] [PubMed]
- Torzewski, M.; Ochsenhirt, V.; Kleschyov, A.L.; Oelze, M.; Daiber, A.; Li, H.; Rossmann, H.; Tsimikas, S.; Reifenberg, K.; Cheng, F.; et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickremasinghe, D.; Peiris, H.; Chandrasena, L.G.; Senaratne, V.; Perera, R. Case control feasibility study assessing the association between severity of coronary artery disease with Glutathione Peroxidase-1 (GPX-1) and GPX-1 polymorphism (Pro198Leu). BMC Cardiovasc. Disord. 2016, 16, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Zuzak, E.; Horecka, A.; Kiełczykowska, M.; Dudek, A.; Musik, I.; Kurzepa, J.; Kurzepa, J. Glutathione level and glutathione reductase activity in serum of coronary heart disease patients. J. Pre-Clin. Clin. Res. 2017, 11, 103–105. [Google Scholar] [CrossRef]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1. [Google Scholar] [CrossRef] [Green Version]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006, 113, 1779–1786. [Google Scholar] [CrossRef] [Green Version]
- Kisucka, J.; Chauhan, A.K.; Patten, I.S.; Yesilaltay, A.; Neumann, C.; Van Etten, R.A.; Krieger, M.; Wagner, D.D. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circ. Res. 2008, 103, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Bárány, T.; Simon, A.; Szabó, G.; Benkő, R.; Mezei, Z.; Molnár, L.; Becker, D.; Merkely, B.; Zima, E.; Horváth, E.M. Oxidative Stress-Related Parthanatos of Circulating Mononuclear Leukocytes in Heart Failure. Oxidative Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Maupin-Furlow, J.A. Methionine Sulfoxide Reductases of Archaea. Antioxidants 2018, 7, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.-Y.; Du, F.; Meng, B.; Xie, G.-H.; Cao, J.; Fan, D.; Yu, H. Hepatic overexpression of methionine sulfoxide reductase A reduces atherosclerosis in apolipoprotein E-deficient mice. J. Lipid Res. 2015, 56, 1891–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picot, C.R.; Perichon, M.; Lundberg, K.C.; Friguet, B.; Szweda, L.I.; Petropoulos, I. Alterations in mitochondrial and cytosolic methionine sulfoxide reductase activity during cardiac ischemia and reperfusion. Exp. Gerontol. 2006, 41, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Urreizti, R.; Asteggiano, C.; Vilaseca, M.A.; Corbella, E.; Pintó, X.; Grinberg, D.; Balcells, S. A CBS haplotype and a polymorphism at the MSR gene are associated with cardiovascular disease in a Spanish case-control study. Clin. Biochem. 2007, 40, 864–868. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Hilgers, R.H.P.; Kundumani-Sridharan, V.; Subramani, J.; Chen, L.C.; Cuello, L.G.; Rusch, N.J.; Das, K.C. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Yang, G.; Hong, C.; Liu, J.; Holle, E.; Yu, X.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Inhibition of endogenous thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Investig. 2003, 112, 1395–1406. [Google Scholar] [CrossRef]
- Couchie, D.; Vaisman, B.; Abderrazak, A.; Mahmood, D.F.D.; Hamza, M.M.; Canesi, F.; Diderot, V.; El Hadri, K.; Nègre-Salvayre, A.; Le Page, A.; et al. Human Plasma Thioredoxin-80 Increases With Age and in ApoE Mice Induces Inflammation, Angiogenesis, and Atherosclerosis. Circulation 2017, 136, 464–475. [Google Scholar] [CrossRef]
- Ouyang, Y.; Peng, Y.; Li, J.; Holmgren, A.; Lu, J. Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems. Metallomics 2018, 10, 218–228. [Google Scholar] [CrossRef]
- Adluri, R.S.; Thirunavukkarasu, M.; Zhan, L.; Dunna, N.R.; Akita, Y.; Selvaraju, V.; Otani, H.; Sanchez, J.A.; Ho, Y.-S.; Maulik, N. Glutaredoxin-1 overexpression enhances neovascularization and diminishes ventricular remodeling in chronic myocardial infarction. PLoS ONE 2012, 7, e34790. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.A.; Kang, S.W.; Kim, H.K.; Yang, K.-S.; Chae, H.Z.; Rhee, S.G. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J. Biol. Chem. 2003, 278, 47361–47364. [Google Scholar] [CrossRef] [Green Version]
- Gianazza, E.; Brioschi, M.; Fernandez, A.M.; Banfi, C. Lipoxidation in cardiovascular diseases. Redox Biol. 2019, 23, 101119. [Google Scholar] [CrossRef]
- Shahidi, F.; Zhong, Y. Lipid oxidation and improving the oxidative stability. Chem. Soc. Rev. 2010, 39, 4067–4079. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef]
- Hauck, A.K.; Huang, Y.; Hertzel, A.V.; Bernlohr, D.A. Adipose oxidative stress and protein carbonylation. J. Biol. Chem. 2019, 294, 1083–1088. [Google Scholar] [CrossRef] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- England, K.; O’Driscoll, C.; Cotter, T.G. Carbonylation of glycolytic proteins is a key response to drug-induced oxidative stress and apoptosis. Cell Death Differ. 2004, 11, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Marnett, L.J. Oxyradicals and DNA damage. Carcinogenesis 2000, 21, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Menezo, Y.J.R.; Silvestris, E.; Dale, B.; Elder, K. Oxidative stress and alterations in DNA methylation: Two sides of the same coin in reproduction. Reprod. Biomed. Online 2016, 33, 668–683. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wu, X.; Hu, Q.; Wu, J.; Wang, G.; Hong, Z.; Ren, J. Mitochondrial DNA in liver inflammation and oxidative stress. Life Sci. 2019, 236, 116464. [Google Scholar] [CrossRef]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abimannan, T.; Peroumal, D.; Parida, J.R.; Barik, P.K.; Padhan, P.; Devadas, S. Oxidative stress modulates the cytokine response of differentiated Th17 and Th1 cells. Free Radic. Biol. Med. 2016, 99, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Federico, A.; Morgillo, F.; Tuccillo, C.; Ciardiello, F.; Loguercio, C. Chronic inflammation and oxidative stress in human carcinogenesis. Int. J. Cancer 2007, 121, 2381–2386. [Google Scholar] [CrossRef] [PubMed]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [Green Version]
- Green, D.R. The Coming Decade of Cell Death Research: Five Riddles. Cell 2019, 177, 1094–1107. [Google Scholar] [CrossRef]
- Del Re, D.P.; Amgalan, D.; Linkermann, A.; Liu, Q.; Kitsis, R.N. Fundamental Mechanisms of Regulated Cell Death and Implications for Heart Disease. Physiol. Rev. 2019, 99, 1765–1817. [Google Scholar] [CrossRef]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulda, S. Regulation of necroptosis signaling and cell death by reactive oxygen species. Biol. Chem. 2016, 397, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFα-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Non-Apoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—From mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef] [Green Version]
- Binder, A.; Ali, A.; Chawla, R.; Aziz, H.A.; Abbate, A.; Jovin, I.S. Myocardial protection from ischemia-reperfusion injury post coronary revascularization. Expert Rev. Cardiovasc. Ther. 2015, 13, 1045–1057. [Google Scholar] [CrossRef]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Kahles, T.; Brandes, R.P. Which NADPH Oxidase Isoform Is Relevant for Ischemic Stroke? The Case for Nox 2. Antioxid. Redox Signal. 2013, 18, 1400–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Braunersreuther, V.; Montecucco, F.; Asrih, M.; Ashri, M.; Pelli, G.; Galan, K.; Frias, M.; Burger, F.; Quinderé, A.L.G.; Montessuit, C.; et al. Role of NADPH oxidase isoforms NOX1, NOX2 and NOX4 in myocardial ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2013, 64. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology 2009, 24. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Zhang, J.; Strom, J.; Lee, S.; Chen, Q.M. Myocardial ischemic reperfusion induces de novo Nrf2 protein translation. Biochim. Biophys. Acta 2014, 1842, 1638–1647. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-S.; Zhou, Y.-N.; Li, L.; Li, S.-F.; Long, D.; Chen, X.-L.; Zhang, J.-B.; Feng, L.; Li, Y.-P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef]
- Cadenas, S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic. Biol. Med. 2018, 117, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Dhanabalan, K.; Mzezewa, S.; Huisamen, B.; Lochner, A. Mitochondrial Oxidative Phosphorylation Function and Mitophagy in Ischaemic/Reperfused Hearts from Control and High-Fat Diet Rats: Effects of Long-Term Melatonin Treatment. Cardiovasc. Drugs Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Amanakis, G.; Sun, J.; Fergusson, M.M.; McGinty, S.; Liu, C.; Molkentin, J.D.; Murphy, E. Cysteine 202 of Cyclophilin D is a site of multiple post-translational modifications and plays a role in cardioprotection. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.-K.; Blackwood, E.A.; Azizi, K.; Thuerauf, D.J.; Fahem, A.G.; Hofmann, C.; Kaufman, R.J.; Doroudgar, S.; Glembotski, C.C. ATF6 Decreases Myocardial Ischemia/Reperfusion Damage and Links ER Stress and Oxidative Stress Signaling Pathways in the Heart. Circ. Res. 2017, 120, 862–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kazzi, M.; Rayner, B.S.; Chami, B.; Dennis, J.M.; Thomas, S.R.; Witting, P.K. Neutrophil-Mediated Cardiac Damage after Acute Myocardial Infarction: Significance of Defining a New Target Cell Type for Developing Cardioprotective Drugs. Antioxid. Redox Signal. 2020, 33, 689–712. [Google Scholar] [CrossRef] [PubMed]
- Neri, M.; Fineschi, V.; Di Paolo, M.; Pomara, C.; Riezzo, I.; Turillazzi, E.; Cerretani, D. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr. Vasc. Pharmacol. 2015, 13, 26–36. [Google Scholar] [CrossRef]
- Berg, K.; Jynge, P.; Bjerve, K.; Skarra, S.; Basu, S.; Wiseth, R. Oxidative stress and inflammatory response during and following coronary interventions for acute myocardial infarction. Free Radic. Res. 2005, 39, 629–636. [Google Scholar] [CrossRef]
- Akila; D’Souza, B.; Vishwanath, P.; D’Souza, V. Oxidative injury and antioxidants in coronary artery bypass graft surgery: Off-pump CABG significantly reduces oxidative stress. Clin. Chim. Acta 2007, 375, 147–152. [Google Scholar] [CrossRef]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Compr. Physiol. 2015, 6, 187–214. [Google Scholar] [CrossRef]
- LeLeiko, R.M.; Vaccari, C.S.; Sola, S.; Merchant, N.; Nagamia, S.H.; Thoenes, M.; Khan, B.V. Usefulness of elevations in serum choline and free F2)-isoprostane to predict 30-day cardiovascular outcomes in patients with acute coronary syndrome. Am. J. Cardiol. 2009, 104, 638–643. [Google Scholar] [CrossRef]
- Paulus, W.J.; Tschöpe, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzel, T.; Gori, T.; Keaney, J.F.; Maack, C.; Daiber, A. Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur. Heart J. 2015, 36, 2555–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, D.B.; Colucci, W.S. Mitochondrial oxidative stress in heart failure: “oxygen wastage” revisited. Circ. Res. 2000, 86, 119–120. [Google Scholar] [CrossRef] [Green Version]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.-L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol. 2003, 41, 2164–2171. [Google Scholar] [CrossRef] [Green Version]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.-H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Sabri, A.; Hughie, H.H.; Lucchesi, P.A. Regulation of hypertrophic and apoptotic signaling pathways by reactive oxygen species in cardiac myocytes. Antioxid. Redox Signal. 2003, 5, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Kameda, K.; Matsunaga, T.; Abe, N.; Hanada, H.; Ishizaka, H.; Ono, H.; Saitoh, M.; Fukui, K.; Fukuda, I.; Osanai, T.; et al. Correlation of oxidative stress with activity of matrix metalloproteinase in patients with coronary artery disease. Possible role for left ventricular remodelling. Eur. Heart J. 2003, 24, 2180–2185. [Google Scholar] [CrossRef]
- Cai, A.W.; Taylor, M.H.; Ramu, B. Treatment of chemotherapy-associated cardiomyopathy. Curr. Opin. Cardiol. 2019, 34, 296–302. [Google Scholar] [CrossRef]
- Yu, A.F.; Steingart, R.M.; Fuster, V. Cardiomyopathy Associated with Cancer Therapy. J. Card. Fail. 2014, 20, 841–852. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef] [PubMed]
- Songbo, M.; Lang, H.; Xinyong, C.; Bin, X.; Ping, Z.; Liang, S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicol. Lett. 2019, 307, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Philip, I.; Lebret, M.; Chatel, D.; Maclouf, J.; Tedgui, A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure: A potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation 1998, 97, 1536–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belch, J.J.; Bridges, A.B.; Scott, N.; Chopra, M. Oxygen free radicals and congestive heart failure. Br. Heart J. 1991, 65, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scicchitano, P.; Cortese, F.; Gesualdo, M.; De Palo, M.; Massari, F.; Giordano, P.; Ciccone, M.M. The role of endothelial dysfunction and oxidative stress in cerebrovascular diseases. Free Radic. Res. 2019, 53, 579–595. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Lassègue, B.; Sorescu, D.; Szöcs, K.; Yin, Q.; Akers, M.; Zhang, Y.; Grant, S.L.; Lambeth, J.D.; Griendling, K.K. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ. Res. 2001, 88, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Ellmark, S.H.M.; Dusting, G.J.; Ng Tang Fui, M.; Guzzo-Pernell, N.; Drummond, G.R. The contribution of Nox4 to NADPH oxidase activity in mouse vascular smooth muscle. Cardiovasc. Res. 2005, 65, 495–504. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Brandes, R.P.; Nguyen, K.; Amidi, M.; Dehghani, F.; Busse, R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ. Res. 2000, 87, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Ago, T.; Kitazono, T.; Ooboshi, H.; Iyama, T.; Han, Y.H.; Takada, J.; Wakisaka, M.; Ibayashi, S.; Utsumi, H.; Iida, M. Nox4 as the major catalytic component of an endothelial NAD(P)H oxidase. Circulation 2004, 109, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohara, Y.; Peterson, T.E.; Harrison, D.G. Hypercholesterolemia increases endothelial superoxide anion production. J. Clin. Investig. 1993, 91, 2546–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, K.A.; Yuan Yuan, D.; Nawaz, W.; Ze, H.; Zhuo, C.X.; Talal, B.; Taleb, A.; Mais, E.; Qilong, D. Antioxidant therapy for management of oxidative stress induced hypertension. Free Radic. Res. 2017, 51, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [Green Version]
- Guzik, T.J.; West, N.E.; Black, E.; McDonald, D.; Ratnatunga, C.; Pillai, R.; Channon, K.M. Vascular superoxide production by NAD(P)H oxidase: Association with endothelial dysfunction and clinical risk factors. Circ. Res. 2000, 86, E85–E90. [Google Scholar] [CrossRef] [Green Version]
- Azarsiz, E.; Kayikcioglu, M.; Payzin, S.; Yildirim Sözmen, E. PON1 activities and oxidative markers of LDL in patients with angiographically proven coronary artery disease. Int. J. Cardiol. 2003, 91, 43–51. [Google Scholar] [CrossRef]
- Lau, D.H.; Nattel, S.; Kalman, J.M.; Sanders, P. Modifiable Risk Factors and Atrial Fibrillation. Circulation 2017, 136, 583–596. [Google Scholar] [CrossRef]
- Kalani, R.; Judge, S.; Carter, C.; Pahor, M.; Leeuwenburgh, C. Effects of caloric restriction and exercise on age-related, chronic inflammation assessed by C-reactive protein and interleukin-6. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Samman Tahhan, A.; Sandesara, P.B.; Hayek, S.S.; Alkhoder, A.; Chivukula, K.; Hammadah, M.; Mohamed-Kelli, H.; O’Neal, W.T.; Topel, M.; Ghasemzadeh, N.; et al. Association between oxidative stress and atrial fibrillation. Heart Rhythm 2017, 14, 1849–1855. [Google Scholar] [CrossRef]
- Jalife, J.; Kaur, K. Atrial remodeling, fibrosis, and atrial fibrillation. Trends Cardiovasc. Med. 2015, 25, 475–484. [Google Scholar] [CrossRef] [Green Version]
- Morita, N.; Lee, J.H.; Xie, Y.; Sovari, A.; Qu, Z.; Weiss, J.N.; Karagueuzian, H.S. Suppression of Re-Entrant and Multifocal Ventricular Fibrillation by the Late Sodium Current Blocker Ranolazine. J. Am. Coll. Cardiol. 2011, 57, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.P.; Sims, S.M.; Cook, M.A.; Karmazyn, M. Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. J. Pharmacol. Exp. Ther. 1998, 286, 1208–1214. [Google Scholar] [PubMed]
- Anzai, K.; Ogawa, K.; Kuniyasu, A.; Ozawa, T.; Yamamoto, H.; Nakayama, H. Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem. Biophys. Res. Commun. 1998, 249, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A. Cellular and Molecular Mechanisms of Arrhythmia by Oxidative Stress. Cardiol. Res. Pract. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Ishii, Y.; Schuessler, R.B.; Gaynor, S.L.; Yamada, K.; Fu, A.S.; Boineau, J.P.; Damiano, R.J. Inflammation of atrium after cardiac surgery is associated with inhomogeneity of atrial conduction and atrial fibrillation. Circulation 2005, 111, 2881–2888. [Google Scholar] [CrossRef] [Green Version]
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.F.; Chen, P.S.; Xie, L.H.; Chen, F.; et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1594–H1605. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.; Wang, X.; Yuan, M.; Tian, C.; Li, H.; Yang, X.; Li, X.; Li, Y.; Yang, Y.; Liu, N.; et al. Mechanisms and Treatments of Oxidative Stress in Atrial Fibrillation. Curr. Pharm. Des. 2018, 24, 3062–3071. [Google Scholar] [CrossRef]
- Rodrigo, R.; Prat, H.; Passalacqua, W.; Araya, J.; Guichard, C.; Bächler, J.P. Relationship between Oxidative Stress and Essential Hypertension. Hypertens. Res. 2007, 30, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Schiffrin, E.L. Increased generation of superoxide by angiotensin II in smooth muscle cells from resistance arteries of hypertensive patients: Role of phospholipase D-dependent NAD(P)H oxidase-sensitive pathways. J. Hypertens. 2001, 19, 1245–1254. [Google Scholar] [CrossRef]
- Konukoglu, D.; Uzun, H. Endothelial Dysfunction and Hypertension. Adv. Exp. Med. Biol. 2017, 956, 511–540. [Google Scholar] [CrossRef] [PubMed]
- Dinh, Q.N.; Drummond, G.R.; Sobey, C.G.; Chrissobolis, S. Roles of Inflammation, Oxidative Stress, and Vascular Dysfunction in Hypertension. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashi, Y.; Sasaki, S.; Nakagawa, K.; Matsuura, H.; Oshima, T.; Chayama, K. Endothelial function and oxidative stress in renovascular hypertension. N. Engl. J. Med. 2002, 346, 1954–1962. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.H.; Edmunds, E.; Nuttall, S.L.; Landray, M.J.; Blann, A.D.; Beevers, D.G. Oxidative stress in malignant and non-malignant phase hypertension. J. Hum. Hypertens. 2002, 16, 333–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, S.-K.; Ju, W.; Cho, B.; Oh, S.-W.; Park, S.M.; Koo, B.-K.; Park, B.-J. Efficacy of vitamin and antioxidant supplements in prevention of cardiovascular disease: Systematic review and meta-analysis of randomised controlled trials. BMJ 2013, 346. [Google Scholar] [CrossRef] [Green Version]
- Theodosiou, M.; Laudet, V.; Schubert, M. From carrot to clinic: An overview of the retinoic acid signaling pathway. Cell. Mol. Life Sci. 2010, 67, 1423–1445. [Google Scholar] [CrossRef]
- Generoso, G.; Bittencourt, M.S. Vitamin A: An enhanced vision of the relationship between apolipoproteins and cardiovascular risk? Atherosclerosis 2017, 265, 256–257. [Google Scholar] [CrossRef]
- Pashkow, F.J.; Watumull, D.G.; Campbell, C.L. Astaxanthin: A novel potential treatment for oxidative stress and inflammation in cardiovascular disease. Am. J. Cardiol. 2008, 101, 58D–68D. [Google Scholar] [CrossRef]
- Fassett, R.G.; Coombes, J.S. Astaxanthin, oxidative stress, inflammation and cardiovascular disease. Future Cardiol. 2009, 5, 333–342. [Google Scholar] [CrossRef]
- Visioli, F.; Artaria, C. Astaxanthin in cardiovascular health and disease: Mechanisms of action, therapeutic merits, and knowledge gaps. Food Funct. 2017, 8, 39–63. [Google Scholar] [CrossRef]
- May, J.M.; Harrison, F.E. Role of vitamin C in the function of the vascular endothelium. Antioxid. Redox Signal. 2013, 19, 2068–2083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farías, J.G.; Molina, V.M.; Carrasco, R.A.; Zepeda, A.B.; Figueroa, E.; Letelier, P.; Castillo, R.L. Antioxidant Therapeutic Strategies for Cardiovascular Conditions Associated with Oxidative Stress. Nutrients 2017, 9, 966. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.V.; Mistry, B.M.; Shinde, S.K.; Syed, R.; Singh, V.; Shin, H.-S. Therapeutic potential of quercetin as a cardiovascular agent. Eur. J. Med. Chem. 2018, 155, 889–904. [Google Scholar] [CrossRef]
- Cuadrado, A.; Manda, G.; Hassan, A.; Alcaraz, M.J.; Barbas, C.; Daiber, A.; Ghezzi, P.; León, R.; López, M.G.; Oliva, B.; et al. Transcription Factor NRF2 as a Therapeutic Target for Chronic Diseases: A Systems Medicine Approach. Pharmacol. Rev. 2018, 70, 348–383. [Google Scholar] [CrossRef] [Green Version]
- Meili-Butz, S.; Niermann, T.; Fasler-Kan, E.; Barbosa, V.; Butz, N.; John, D.; Brink, M.; Buser, P.T.; Zaugg, C.E. Dimethyl fumarate, a small molecule drug for psoriasis, inhibits Nuclear Factor-kappaB and reduces myocardial infarct size in rats. Eur. J. Pharmacol. 2008, 586, 251–258. [Google Scholar] [CrossRef]
- Kuang, Y.; Zhang, Y.; Xiao, Z.; Xu, L.; Wang, P.; Ma, Q. Protective effect of dimethyl fumarate on oxidative damage and signaling in cardiomyocytes. Mol. Med. Rep. 2020, 22, 2783–2790. [Google Scholar] [CrossRef]
- Luo, M.; Sun, Q.; Zhao, H.; Tao, J.; Yan, D. The Effects of Dimethyl Fumarate on Atherosclerosis in the Apolipoprotein E-Deficient Mouse Model with Streptozotocin-Induced Hyperglycemia Mediated by the Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element (Nrf2/ARE) Signaling Pathway. Med. Sci. Monit. 2019, 25, 7966–7975. [Google Scholar] [CrossRef]
- Okafor, O.N.; Farrington, K.; Gorog, D.A. Allopurinol as a therapeutic option in cardiovascular disease. Pharmacol. Ther. 2017, 172, 139–150. [Google Scholar] [CrossRef]
- Agarwal, V.; Hans, N.; Messerli, F.H. Effect of allopurinol on blood pressure: A systematic review and meta-analysis. J. Clin. Hypertens. 2013, 15, 435–442. [Google Scholar] [CrossRef]
- Altenhöfer, S.; Radermacher, K.A.; Kleikers, P.W.M.; Wingler, K.; Schmidt, H.H.H.W. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yang, G.; Zhang, X.; Wang, P.; Weng, X.; Yang, Y.; Li, Z.; Fang, M.; Xu, Y.; Sun, A.; et al. Megakaryocytic Leukemia 1 Bridges Epigenetic Activation of NADPH Oxidase in Macrophages to Cardiac Ischemia-Reperfusion Injury. Circulation 2018, 138, 2820–2836. [Google Scholar] [CrossRef] [PubMed]
- Roth Flach, R.J.; Su, C.; Bollinger, E.; Cortes, C.; Robertson, A.W.; Opsahl, A.C.; Coskran, T.M.; Maresca, K.P.; Keliher, E.J.; Yates, P.D.; et al. Myeloperoxidase inhibition in mice alters atherosclerotic lesion composition. PLoS ONE 2019, 14, e0214150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tita, C.; Gilbert, E.M.; Van Bakel, A.B.; Grzybowski, J.; Haas, G.J.; Jarrah, M.; Dunlap, S.H.; Gottlieb, S.S.; Klapholz, M.; Patel, P.C.; et al. A Phase 2a dose-escalation study of the safety, tolerability, pharmacokinetics and haemodynamic effects of BMS-986231 in hospitalized patients with heart failure with reduced ejection fraction. Eur. J. Heart Fail. 2017, 19, 1321–1332. [Google Scholar] [CrossRef] [Green Version]
- Dao, V.T.-V.; Casas, A.I.; Maghzal, G.J.; Seredenina, T.; Kaludercic, N.; Robledinos-Anton, N.; Di Lisa, F.; Stocker, R.; Ghezzi, P.; Jaquet, V.; et al. Pharmacology and Clinical Drug Candidates in Redox Medicine. Antioxid. Redox Signal. 2015, 23, 1113–1129. [Google Scholar] [CrossRef] [Green Version]
- Khalaf, D.; Krüger, M.; Wehland, M.; Infanger, M.; Grimm, D. The Effects of Oral l-Arginine and l-Citrulline Supplementation on Blood Pressure. Nutrients 2019, 11, 1679. [Google Scholar] [CrossRef] [Green Version]
- Claustrat, B.; Leston, J. Melatonin: Physiological effects in humans. Neurochirurgie 2015, 61, 77–84. [Google Scholar] [CrossRef]
- Sun, H.; Gusdon, A.M.; Qu, S. Effects of melatonin on cardiovascular diseases: Progress in the past year. Curr. Opin. Lipidol. 2016, 27, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Ding, Z.; Liu, S.; Wang, X.; Deng, X.; Fan, Y.; Sun, C.; Wang, Y.; Mehta, J.L. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid. Redox Signal. 2015, 22, 760–771. [Google Scholar] [CrossRef] [Green Version]
- Carreira, R.S.; Monteiro, P.; Gon Alves, L.M.; Providência, L.A. Carvedilol: Just another Beta-blocker or a powerful cardioprotector? Cardiovasc. Hematol. Disord. Drug Targets 2006, 6, 257–266. [Google Scholar] [CrossRef]
- Dandona, P.; Ghanim, H.; Brooks, D.P. Antioxidant activity of carvedilol in cardiovascular disease. J. Hypertens. 2007, 25, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-M.; Kuo, H.-C.; Hsu, C.-N.; Huang, L.-T.; Tain, Y.-L. Metformin reduces asymmetric dimethylarginine and prevents hypertension in spontaneously hypertensive rats. Transl. Res. 2014, 164, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Nesti, L.; Natali, A. Metformin effects on the heart and the cardiovascular system: A review of experimental and clinical data. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Song, X.; Sun, W.; Wang, Y.; Liu, B. Effect of Hypoxia-Induced MicroRNA-210 Expression on Cardiovascular Disease and the Underlying Mechanism. Oxidative Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jia, Q.; Xinnong, C.; Xie, Y.; Yang, Y.; Zhang, A.; Liu, R.; Zhuo, Y.; Zhang, J. Role of cardiac progenitor cell-derived exosome-mediated microRNA-210 in cardiovascular disease. J. Cell. Mol. Med. 2019, 23, 7124–7131. [Google Scholar] [CrossRef] [Green Version]
- Mutharasan, R.K.; Nagpal, V.; Ichikawa, Y.; Ardehali, H. microRNA-210 is upregulated in hypoxic cardiomyocytes through Akt- and p53-dependent pathways and exerts cytoprotective effects. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1519–H1530. [Google Scholar] [CrossRef] [Green Version]
- Merlo, A.; Bernardo-Castiñeira, C.; Sáenz-de-Santa-María, I.; Pitiot, A.S.; Balbín, M.; Astudillo, A.; Valdés, N.; Scola, B.; Del Toro, R.; Méndez-Ferrer, S.; et al. Role of VHL, HIF1A and SDH on the expression of miR-210: Implications for tumoral pseudo-hypoxic fate. Oncotarget 2017, 8, 6700–6717. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Pan, X.; Fan, Y.; Hu, X.; Liu, X.; Xiang, M.; Wang, J.A. Dysregulated expression of microRNAs and mRNAs in myocardial infarction. Am. J. Transl. Res. 2015, 7, 2291–2304. [Google Scholar]
- Hu, S.; Huang, M.; Li, Z.; Jia, F.; Ghosh, Z.; Lijkwan, M.A.; Fasanaro, P.; Sun, N.; Wang, X.; Martelli, F.; et al. MicroRNA-210 as a novel therapy for treatment of ischemic heart disease. Circulation 2010, 122, S124–S131. [Google Scholar] [CrossRef] [Green Version]
- Karakas, M.; Schulte, C.; Appelbaum, S.; Ojeda, F.; Lackner, K.J.; Münzel, T.; Schnabel, R.B.; Blankenberg, S.; Zeller, T. Circulating microRNAs strongly predict cardiovascular death in patients with coronary artery disease-results from the large AtheroGene study. Eur. Heart J. 2017, 38, 516–523. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tan, N.; Yang, J.; Liu, X.; Cao, X.; He, P.; Dong, X.; Qin, S.; Zhang, C. A translational study of circulating cell-free microRNA-1 in acute myocardial infarction. Clin. Sci. 2010, 119, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.; Xiong, X.; Liu, Y.; Wang, J. miRNA-1: Functional roles and dysregulation in heart disease. Mol. Biosyst. 2014, 10, 2775–2782. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Zhang, M.-F.; Wen, H.-Y.; Hu, C.-L.; Liu, R.; Wei, H.-Y.; Ai, C.-M.; Wang, G.; Liao, X.-X.; Li, X. Comparing the diagnostic values of circulating microRNAs and cardiac troponin T in patients with acute myocardial infarction. Clinics 2013, 68, 75–80. [Google Scholar] [CrossRef]
- Katare, R.; Riu, F.; Mitchell, K.; Gubernator, M.; Campagnolo, P.; Cui, Y.; Fortunato, O.; Avolio, E.; Cesselli, D.; Beltrami, A.P.; et al. Transplantation of Human Pericyte Progenitor Cells Improves the Repair of Infarcted Heart through Activation of an Angiogenic Program Involving Micro-RNA-132. Circ. Res. 2011, 109, 894–906. [Google Scholar] [CrossRef] [Green Version]
- Gray, W.D.; French, K.M.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of Therapeutic Covariant microRNA Clusters in Hypoxia Treated Cardiac Progenitor Cell Exosomes using Systems Biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Pan, Y.; Li, X.H.; Yang, X.Y.; Feng, Y.L.; Tan, H.H.; Jiang, L.; Feng, J.; Yu, X.Y. Cardiac progenitor cell-derived exosomes prevent cardiomyocytes apoptosis through exosomal miR-21 by targeting PDCD4. Cell Death Dis. 2016, 7, e2277. [Google Scholar] [CrossRef] [Green Version]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100. [Google Scholar] [CrossRef]
- Polyakova, E.A.; Zaraiskii, M.I.; Mikhaylov, E.N.; Baranova, E.I.; Galagudza, M.M.; Shlyakhto, E.V. Association of myocardial and serum miRNA expression patterns with the presence and extent of coronary artery disease: A cross-sectional study. Int. J. Cardiol. 2020. [Google Scholar] [CrossRef]
- Zampetaki, A.; Dudek, K.; Mayr, M. Oxidative stress in atherosclerosis: The role of microRNAs in arterial remodeling. Free Radic. Biol. Med. 2013, 64, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Yin, Y.L.; Guo, T.; Sun, X.Y.; Ma, H.; Zhu, M.L.; Zhao, F.R.; Xu, P.; Chen, Y.; Wan, G.R.; et al. Inhibition of Aberrant MicroRNA-133a Expression in Endothelial Cells by Statin Prevents Endothelial Dysfunction by Targeting GTP Cyclohydrolase 1 in vivo. Circulation 2016, 134, 1752–1765. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-Y.; Chiu, J.-J. Atherosclerosis and flow: Roles of epigenetic modulation in vascular endothelium. J. Biomed. Sci. 2019, 26, 56. [Google Scholar] [CrossRef] [PubMed]
- Loyer, X.; Potteaux, S.; Vion, A.-C.; Guérin, C.L.; Boulkroun, S.; Rautou, P.-E.; Ramkhelawon, B.; Esposito, B.; Dalloz, M.; Paul, J.-L.; et al. Inhibition of microRNA-92a prevents endothelial dysfunction and atherosclerosis in mice. Circ. Res. 2014, 114, 434–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Chen, Y.; Shi, J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem. Rev. 2019, 119, 4881–4985. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Bae, S.; Hong, D.; Lim, H.; Yoon, J.H.; Hwang, O.; Park, S.; Ke, Q.; Khang, G.; Kang, P.M. H2O2-responsive molecularly engineered polymer nanoparticles as ischemia/reperfusion-targeted nanotherapeutic agents. Sci. Rep. 2013, 3, 2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Park, M.; Kang, C.; Dilmen, S.; Kang, T.H.; Kang, D.G.; Ke, Q.; Lee, S.U.; Lee, D.; Kang, P.M. Hydrogen Peroxide-Responsive Nanoparticle Reduces Myocardial Ischemia/Reperfusion Injury. J. Am. Heart Assoc. 2016, 5. [Google Scholar] [CrossRef]
- Park, S.; Yoon, J.; Bae, S.; Park, M.; Kang, C.; Ke, Q.; Lee, D.; Kang, P.M. Therapeutic use of H2O2-responsive anti-oxidant polymer nanoparticles for doxorubicin-induced cardiomyopathy. Biomaterials 2014, 35, 5944–5953. [Google Scholar] [CrossRef]
- Somasuntharam, I.; Boopathy, A.V.; Khan, R.S.; Martinez, M.D.; Brown, M.E.; Murthy, N.; Davis, M.E. Delivery of Nox2-NADPH oxidase siRNA with polyketal nanoparticles for improving cardiac function following myocardial infarction. Biomaterials 2013, 34, 7790–7798. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, G.; Sy, J.C.; Brown, M.; Dikalov, S.; Yang, S.C.; Murthy, N.; Davis, M.E. The delivery of superoxide dismutase encapsulated in polyketal microparticles to rat myocardium and protection from myocardial ischemia-reperfusion injury. Biomaterials 2010, 31, 1372–1379. [Google Scholar] [CrossRef] [Green Version]
- Gray, W.D.; Che, P.; Brown, M.; Ning, X.; Murthy, N.; Davis, M.E. N-acetylglucosamine conjugated to nanoparticles enhances myocyte uptake and improves delivery of a small molecule p38 inhibitor for post-infarct healing. J. Cardiovasc. Transl. Res. 2011, 4, 631–643. [Google Scholar] [CrossRef] [Green Version]
- Soumya, R.S.; Vineetha, V.P.; Raj, P.S.; Raghu, K.G. Beneficial properties of selenium incorporated guar gum nanoparticles against ischemia/reperfusion in cardiomyoblasts (H9c2). Metallomics 2014. [Google Scholar] [CrossRef]
- Gross, G.J.; Lockwood, S.F. Acute and chronic administration of disodium disuccinate astaxanthin (Cardax) produces marked cardioprotection in dog hearts. Mol. Cell. Biochem. 2005, 272, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Rössig, L.; Hoffmann, J.; Hugel, B.; Mallat, Z.; Haase, A.; Freyssinet, J.M.; Tedgui, A.; Aicher, A.; Zeiher, A.M.; Dimmeler, S. Vitamin C inhibits endothelial cell apoptosis in congestive heart failure. Circulation 2001, 104, 2182–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sozen, E.; Demirel, T.; Ozer, N.K. Vitamin E: Regulatory role in the cardiovascular system. IUBMB Life 2019, 71, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.L.; Xia, J.; Ye, D.; Liu, J.; Chen, J.; Wang, G. Effects of quercetin on gene and protein expression of NOX and NOS after myocardial ischemia and reperfusion in rabbit. Cardiovasc. Ther. 2009, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Felicioni, L.; Sarzani, R.; Ucchino, S.; Spigonardo, F.; Mandolini, C.; Malatesta, S.; Bucci, M.; Mammarella, C.; Santovito, D.; et al. A unique microRNA signature associated with plaque instability in humans. Stroke 2011, 42, 2556–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, N.R.; Albert, C.M.; Gaziano, J.M.; Zaharris, E.; MacFadyen, J.; Danielson, E.; Buring, J.E.; Manson, J.E. A randomized factorial trial of vitamins C and E and beta carotene in the secondary prevention of cardiovascular events in women: Results from the Women’s Antioxidant Cardiovascular Study. Arch. Intern. Med. 2007, 167, 1610–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivekananthan, D.P.; Penn, M.S.; Sapp, S.K.; Hsu, A.; Topol, E.J. Use of antioxidant vitamins for the prevention of cardiovascular disease: Meta-analysis of randomised trials. Lancet 2003, 361, 2017–2023. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Compound: Isoforms | Effects | Research Type: Subjects | Main Findings | Ref |
|---|---|---|---|---|
| SOD: Cu/ZnSOD, MnSOD, ECSOD | Accelerates the reaction of superoxide anion to form H2O2 and oxide. | Preclinical: mice | Cu/ZnSOD-deficiency resulted in altered responsiveness in both large arteries and microvessels. | [22,28] |
| Preclinical: Rabbits | Gene transfer of ECSOD reduced infarct size. | [29] | ||
| Clinical: HTN patients | Serum levels of SOD were associated with alterations in vascular structure and function. | [30] | ||
| Catalase | Lower H2O2 concentration: accelerate the reaction of H2O2 with hydrogen donors to produce water | Preclinical: mice | Overexpression of catalase prevented HTN. | [25,31] |
| GPx: GPx 1–8 | Catalyze H2O2 or organic hydroperoxides to water or corresponding alcohols. | Preclinical: mice | GPX knockout mice were more susceptible to I/R injury. | [27,32] |
| Preclinical: mice | Deficiency of GPX accelerated atherosclerotic lesion progression. | [33] | ||
| Clinical: CAD patients | GPX-1 Pro198Leu polymorphism was higher in patients with CAD. | [34] | ||
| GR | Clear the oxidized dimer form of glutathione to reduced glutathione. | Clinical: CAD patients | Highest GR activity was associated with myocardial infarction. | [35,36] |
| Prx: 2-Cys, atypical 2-Cys, and 1-Cys Prx | Catalyze H2O2 or organic hydroperoxides to water or corresponding alcohols. | Preclinical: mice | Overexpression of Prx-3 inhibited left ventricular remodeling and HF after myocardial infarction. | [37,38] |
| Preclinical: mice | Prx1 protected against excessive endothelial activation and atherosclerosis. | [39] | ||
| Clinical: HF patients | Plasma PRX was higher in HF patients. | [40] | ||
| MSR: (1) MSRA and MSRB (2) fRMSR and MSRP (3) MPT/WPT OR enzymes | Reduce methionine sulfoxide residues in oxidatively damaged proteins to methionine residues. | Preclinical: mice | Hepatic overexpression of MSRA reduced dyslipidemia and atherosclerosis. | [41,42] |
| Preclinical: mice | Cytosolic MsrA protected the heart from I/R injury. | [43] | ||
| Clinical: CAD patients | MSR was associated with etiology of CAD. | [44] | ||
| Trx | Transfer electrons to Prxs, MSRs, other redox-sensitive transcription factors. | Preclinical: mice | Overexpression of Trx reversed aged-related HTN. | [45,46] |
| Preclinical: mice | Inhibition of endogenous cardiac Trx1 stimulated hypertrophy. | [47] | ||
| Clinical: General population | Trx80 increased in aged people. | [48] | ||
| Grx: Grx 1–5 | Catalyze the reduction of protein disulfides or mixed disulfides, and maintain the intracellular redox status. | Preclinical: mice | Grx-1 diminished ventricular remodeling in chronic myocardial infarction | [49,50] |
| Compound | Research Type: Subjects | Main Finding | Ref |
|---|---|---|---|
| Nutritional Supplements | |||
| Vitamin A | Clinical: Stable angina patients | Modified the effect of apolipoproteins on the risk of MI | [147] |
| Astaxanthin | Preclinical: Dogs | Astaxanthin protected from MI | [202] |
| Preclinical: Rats | Astaxanthin reduced HTN in spontaneously hypertensive rats | [149] | |
| Clinical: Obese adults | The supplemental of astaxanthin decreased oxidative stress | [150] | |
| Vitamin C | Clinical: CHF patients | Vitamin C inhibited endothelial cells apoptosis in CHF patients | [203] |
| Vitamin E | Clinical: General population | The intake of vitamin E reduced risk of coronary heart disease | [204] |
| Vitamin C+ vitamin E | Meta-analysis: general population | Vitamin E and vitamin c combination inhibited the rate of coronary heart disease. | [152] |
| Omega-3 | Preclinical: Rats | The supplement of omega-3 was associated with lower infarct size | [153] |
| Flavanols | Preclinical: Rats | Flavanols reduced the MI size and fat peroxidation | [205] |
| Clinical: HTN patients | Flavanols reduced the mean blood pressure in HTN patients | [154] | |
| Clinical: CVD patients | Flavonoid reduced coronary heart disease mortality. | [152] | |
| Multiple supplements | Meta-analysis: Cancer or CVD patients | Nutritional supplements showed protective in malnutrition patients. | [7] |
| Novel Experimental Antioxidant-Based Therapies | |||
| NRF2 activators | Preclinical: Knockout mice | In Nrf2 knockout mice, cardiac structure and function were impaired. | [155] |
| DMF | Preclinical: Rats | DMF reduced MI size. | [156] |
| Preclinical: Mice | DMF reduced development of atherosclerosis in diabetes mice model | [158] | |
| Allopurinol | Meta-analysis: HTN patients | Allopurinol showed a modest reduction of blood pressure | [160] |
| Clinical: CABG patients | Allopurinol showed reduced in-hospital mortality and cardiac complications | [159] | |
| GKT137831 | Preclinical: Mice | GKT137831 resulted in anti-atherosclerotic effect | [161] |
| Preclinical: Mice | GKT137831 rescued cardiac function after I/R injury | [162] | |
| MPO inhibitors | Preclinical: Mice | MPO inhibitors showed utility to stabilize atherosclerotic lesion | [163] |
| CXL-1427 | Clinical: HF patients | CXL-1427 showed a favorable safety and hemodynamic effect | [164] |
| L-citrulline | Meta-analysis: HTN patients | Administration of L-citrulline lowered blood pressure | [166] |
| L-arginine | Meta-analysis: HTN patients | Administration of L-arginine lowered blood pressure | [166] |
| Clinical Drugs | |||
| Melatonin | Clinical: CAD patients | Melatonin decreased CK-MB in patients undergoing primary percutaneous procedure | [168] |
| PCSK9 inhibitor | Preclinical: Mice | PCSK9 inhibition decreased ROS | [169] |
| Carvedilol | Clinical: General population | Carvedilol significantly inhibited ROS generation | [171] |
| Metformin | Preclinical: Rats | Metformin showed antihypertensive effect in spontaneously hypertensive rats by restoring ADMA-NO balance | [172] |
| miRNAs | |||
| miRNA-210 | Preclinical: Knockout mice | miRNA-210 was decreased by HIF-1α knockout | [177] |
| Preclinical: Mice | The intramyocardial injection of miRNA-210 improved cardiac function after MI | [179] | |
| Clinical: Acute MI patients | miRNA-210 level was increased patients with MI | [178] | |
| Clinical: ACS patients | miRNA-210 level was associated with cardiovascular-related mortality | [180] | |
| miRNA-1 | Preclinical: Transgenic mice | miRNA increased ROS and decreased production of SOD | [181] |
| Preclinical: Rat | miRNA-1 was associated with MI size | [182] | |
| Preclinical: Mice | The post-infarction transplantation of miRNA-1 improved cardiac function | [183] | |
| Clinical: MI patients | Serum levels of miRNA-1 in patients with acute coronary syndrome correlated with the circulating troponin T | [184] | |
| miRNA-133 | Preclinical: Mice | Inhibition of miRNA-133 prevented endothelial dysfunction | [191] |
| Clinical: CAD patients | miRNA-133 was higher in CAD patients | [189] | |
| Clinical: Patients undergoing carotid endarterectomy | miRNA-133 level was increased in symptomatic plaques | [206] | |
| Nanoparticles | |||
| H2O2-responsive nanoparticles | Preclinical: Mice | H2O2-responsive nanoparticles showed anti-apoptotic role in hind-limb I/R and liver I/R models | [195] |
| Preclinical: Mice | H2O2-responsive nanoparticles showed protective role in myocardial I/R injury | [196] | |
| Preclinical: Mice | H2O2-responsive nanoparticles showed protective role in doxorubicin-induced cardiomyopathy | [197] | |
| Nanoparticles/NOX2 siRNA | Preclinical: Mice | Nanoparticles coated with NOX2 siRNA improved cardiac function 3 days after surgery | [198] |
| Nanoparticles/SOD1 | Preclinical: Rat | Nanoparticles carrying SOD1 decreased myocyte apoptosis and improved cardiac function | [199] |
| Nanoparticles/N-acetylcysteine | Preclinical: Rat | Nanoparticles carrying N-acetylcysteine attenuated cardiac fibrosis after I/R injury | [200] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Kang, P.M. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants 2020, 9, 1292. https://doi.org/10.3390/antiox9121292
Wang W, Kang PM. Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants. 2020; 9(12):1292. https://doi.org/10.3390/antiox9121292
Chicago/Turabian StyleWang, Wenjun, and Peter M. Kang. 2020. "Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases" Antioxidants 9, no. 12: 1292. https://doi.org/10.3390/antiox9121292
APA StyleWang, W., & Kang, P. M. (2020). Oxidative Stress and Antioxidant Treatments in Cardiovascular Diseases. Antioxidants, 9(12), 1292. https://doi.org/10.3390/antiox9121292