1. Introduction
Pulmonary arterial hypertension (PAH) is a rare disease that rapidly progresses by increased proliferation in pulmonary vascular cells causing irreversible vascular remodeling. This accounts for increased pulmonary arterial pressure and later cardiac dysfunction, the leading cause of patient mortality in PAH [
1]. According to Hoeper et al. [
2], the frequency of PAH is 1.1 to 17.6 per million adults per year and has an occurrence of 6.6 to 26 individuals per million. Even though the disease remains incurable, despite the ineffective existing therapy, the only alternative treatment for this disease is a heart and lung transplantation. Unfortunately, this option is only accessible in designated surgical centers, and only half of these patients survive the surgery with a lifespan of approximately five years [
3,
4].
Akt signaling plays an important role in the pathogenesis of hypoxia-induced PAH [
5]. In PAH, Akt activation results in the stimulation of cell survival by preventing apoptotic pathways and activating prosurvival mechanisms. Recently, we identified that the nitration of Akt1 at Tyr350 causes its activation, ensuing greater endothelial nitric oxide synthase (eNOS) phosphorylation, subsequent mitochondrial translocation, and affecting mitochondrial function [
6]. Increased nitric oxide (NO) production augmented high levels of peroxynitrite formation, nitrosative stress, and enhanced protein nitration, which was reported in PAH [
7]. These altered proteins may affect many important cytosolic and mitochondrial metabolic pathways; therefore, controlling nitrosative stress is an effective option in preventing PAH progression.
Mitochondria are an important source of reactive oxygen species (ROS), which accounts for vascular dysfunction in PAH [
8]. Under conditions of chronic hypoxia, hypoxia-inducible factor and eNOS regulate various cellular functions to meet increased cellular bioenergetic requirements in the pathogenesis of PAH [
9,
10]. Mitochondrial dysfunction limits ATP production, which results in a metabolic shift towards the Warburg effect often found in highly proliferative cancer cells and pulmonary vascular cells [
11]. Another factor inducing cellular proliferation is anaplerosis, which is the replenishment of tricarboxylic acid (TCA) cycle carbon intermediates via the glutaminase-mediated deamidation of glutamine or the carboxylation of pyruvate [
12]. Recent studies have linked anaplerosis and glutaminolysis (anabolic pathways that promote cellular biomass for highly proliferative tumor cells) to the hyperproliferative state of PAH, leading to hypertrophy and pulmonary vascular resistance [
13].
Antioxidant therapy has been studied for several decades, but its cellular effects are global, and it does not show any clinical significance. Recently, it was reported that in the sugen/hypoxic PAH model, the chemical antioxidant TEMPOL failed to attenuate right ventricular hypertrophy and pulmonary arterial remodeling [
14]. A targeted approach in antioxidant therapy is a promising technique and could maximize the potential of an antioxidant. Nitroxides are the family of free radical compounds, which are effective scavengers of ROS [
15]. Nitroxides function as a superoxide dismutase (SOD) mimetic, performing the interconversion of an oxammonium cation or hydroxylamine [
16]. These properties make nitroxide suitable for the development of defensive peptides for preventing Akt nitration in our study. The affinity peptide that is exclusively targeted to protect the Akt nitration site is conjugated with a nitroxide moiety, forming a nitroxide peptide (NP). Specifically, this would block the tyrosine (Tyr)-350 nitration of Akt, and the conjugated nitroxide antioxidant part would effectively alleviate free radical attacks. In this study, we infer that preventing Akt nitration with antioxidant NPs may be an effective remedy in controlling metabolic reprogramming, anaplerosis, and vascular remodeling events in PAH.
2. Materials and Methods
2.1. Development of Nitroxide-Conjugated Affinity Peptide
Nitroxide peptide (NP) was synthesized and purified by the 21st Century Biochemicals company (Marlboro, MA, USA) from two parts, one being the peptide part with the affinity to Akt near the Tyr 350 residue (Ser-Arg-Ile-Arg-Ser), and the other being a conjugated antioxidant, nitroxide (3-carboxy-2,2,5,5-tetramethyl-3-pyrroline-1-yloxy), covalently attached to the free N-terminal amine. The purified peptide was stored at −80 °C as a lyophilized powder. Samples were weighed and freshly prepared just before each experiment or injection.
2.2. SIN-1 Induced Akt Nitration Assay
Human pulmonary artery endothelial cells (HPAECs) purchased from Lonza, Greenwood, SC, were cultured using an endothelium media specific for HPAEC with 10% FBS. Cells were used from passages 3–6. All experiments were performed on 80%–90% confluent cells. Akt nitration was induced by treatment with 3-morpholinosyndnomine (SIN-1) (1 mM) for 1 h. NP pretreatment groups were treated with 1 µM NP for 30 min, and then cells were washed in PBS and treated with SIN-1 for 1 h. All treatments were accompanied by including controls with the corresponding vehicle. After treatment, the cells were washed in PBS and lysed in RIPA buffer containing protease and phosphatase inhibitor cocktail [
17].
2.3. Human Subject
Deidentified lung samples consisted of patients with a diagnosis of group I PAH (idiopathic pulmonary arterial hypertension (IPAH) group, N = 10) and healthy controls (control group, N = 10) and were obtained through Pulmonary Hypertension Breakthrough Initiative (PHBI). The PHBI study protocol was approved by the Institutional Review Boards of the participating sites in the network, and all sites were adherent to the requirements of the U.S. Federal Policy for the Protection of Human Subjects (45 CFR, Part 46), and supported the general ethical principles of the Declaration of Helsinki.
2.4. Rat Model of PH
Female Sprague Dawley rats (200–250 g) obtained from Charles River (Wilmington, MA, USA) were used in this study. Animals were housed at 22 °C, 12 h light/dark cycle, and had access to standard rodent food and water ad libitum. All experimental procedures were approved by the University of Arizona Institutional Animal Care and Use Committee. Generally, PAH was induced by a single injection of SU5416 (50 mg/kg) subcutaneously, followed by 3 weeks of hypoxia (10% O2) and 2 weeks of normoxia. This study included five animal groups: control group; SU1, rats were analyzed after 1 week of SU5416 and hypoxia treatment; SU2, rats were analyzed after 2 weeks of SU5416 and hypoxia treatment; SU5, rats were analyzed after 5 weeks of SU5416 treatment (3 weeks of hypoxia with a following 2 weeks of normoxia); and SU2+NP, peptide treatment 0.1 mg/kg/d intravenous (i.v.) injection started with SU5416 treatment for 2 weeks.
2.5. Hemodynamic Measurement
Animals were anesthetized with Inactin 100 mg/kg intraperitoneally (i.p.) (T133, Sigma-Aldrich, St. Louis, MO, USA). A customized pressure transducer catheter (SPR-513, Millar Instruments, Houston, TX, USA) was inserted into the RV via the right jugular vein and advanced into the right ventricle to monitor right ventricular systolic pressure (RVSP), as described previously [
7,
18]. Briefly, the pressure transducer catheter was connected to a Millar Transducer Control Unit TC-510 and PL3504 PowerLab 4/35 data acquisition system (AD Instruments, Colorado Springs, CO, USA) to monitor RV pressure for 30 min. After this, a tracheal catheter was connected to a ventilator system (Harvard Rodent Ventilator-683; Harvard Apparatus, South Natick, MA, USA), the thorax was opened, and the lungs flushed with 0.9% sodium chloride through the right ventricle (RV). Heart and lungs were collected from animals; the RV free wall was separated from the left ventricle (LV) and the septum (S). Fulton index (RV/ LV + S ratio) as a parameter of RV hypertrophy was calculated. The total wet lung weight was measured and normalized by the body weight of the animal. The left lung was fixed in formalin and embedded in paraffin for histological examination. The other portion of the lung was stored at −80 °C for further biochemical studies.
2.6. Histological Analysis
For the morphometric assessment of pulmonary vessels, 5 μm tissue sections were dewaxed and stained with Verhoeff–Van Gieson (EVG) elastic fibers stain, by HistoWiz Inc. (Brooklyn, NY, USA), using standard operating procedures and a fully automated workflow system. Ten transversely sectioned pulmonary arteries (diameter < 300 μm) within each category per each animal (
N = 6 per group) were randomly selected from the whole-slide 40× digitized image Aperio AT2 scanner (Leica Biosystems, IL, USA). Immunohistochemistry was performed using standard protocols. The sections were deparaffinized and incubated with primary antibodies against Akt Y350 NO2 (1:100) and Ki67 (1:800) (ab15580 Abcam) and Dab Rabbit H1 (pH 6) for 20 min. The morphometric analysis was done by an investigator in a blinded grouping fashion. The wall thickness of the pulmonary artery (PA) was measured using the publicly available software Fiji ImageJ (
http://fiji.sc/Fiji; in the public domain) [
19].
2.7. Western Blot Analysis
For an analysis of the total lung protein, lung tissues were lysed as previously described [
20]. Briefly, 20–40 mg of lung tissue (human/rat) was lysed in permeabilization buffer mixed with protease and phosphatase inhibitor cocktails (78429, Thermo Scientific, Rockford, IL, USA) using a Fisher Homogenizer 850. The homogenate was centrifuged at 10,000×
g for 10 min, and the supernatant was carefully collected. Cell membrane and cytosolic fractions were isolated using the FractionPREPTM cell fraction kit (Biovision, Milpitas, CA, USA) according to the manufacturer’s instruction. The protein concentration was measured using the BCA protein assay kit (Thermo Scientific, Rockford, IL, USA). Samples were incubated with 6x Laemmli sample buffer (Boston Bioproducts Inc., Ashland, MA, USA) for 5 min at 95 °C, loaded on the 4%–20% SDS-PAGE Mini-PROTEAN TGX Stain-FreeTM gels (Bio-Rad Laboratories Inc., Hercules, CA, USA), and separated by electrophoresis. Protein bands were transferred using the Trans-Blot Turbo transferring system (Bio-Rad Laboratories Inc.) and then blocked with 5% bovine serum albumin (37525, Thermo Scientific, Rockford, IL, USA) in Tris-buffered saline. Membranes were probed using antibodies against Glut4 (07-1404, 1:1000) from Millipore; pyruvate carboxylase (PC) (Ab229267, 1:1000) and peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) (Ab191838, 1:1000) from Abcam; and pyruvate dehydrogenase (PDH) (45-6600, 1:1000) from Invitrogen; phospho Akt (Ser 473) (4060S, 1:2000), Akt (9272S, 1:1000), hexokinase 1 (HK1) (C35C4, 1:1000), glyceraldehyde-3 phosphate dehydrogenase (GAPDH) (2118S, 1:1000), lactate dehydrogenase-A (LDHA) (2012S, 1:1000), phospho glycogen synthase kinase (GSK) 3 β (Ser9) (9336S, 1:1000), GSK3 β (9315S, 1:1000), and glucose 6-phosphate dehydrogenase (G6PD) (8866S, 1:1000) from Cell Signaling Technology. Mouse monoclonal anti-nitrotyrosine antibodies were obtained from Calbiochem, La Jolla, CA, USA (487923, 1:500). Akt Y350 NO2 antibody was obtained as a custom-made antibody against Akt1-nitro (341-353), and the sequence was CGRLPF-nitroY-NQDHEK. Two peptides were developed with and without Y350 nitration. The antibody developed against nitrated peptide was cleaned using non-nitrated peptide (Akt1 (341-353): CGRLPFYNQDHEK) to remove unspecific antibodies. Affinity purification was done using large, reusable affinity purification columns against the unmodified and nitrated peptides. ELISA validation showed specificity towards the nitrated sequence and, therefore, selectively purified antibodies raised to nitro-Akt (Pacific Immunology Corp. Ramona, CA, USA). Since the quantification of Akt nitration was not fully established by mass spectrometry, we used a common method of a newly developed antibody for the quantification of the specific Y350 nitration of Akt. The reactive bands were visualized by the chemiluminescent ChemiDoc
TM MP Imaging System (Bio-Rad Laboratories Inc., Hercules, CA, USA) and analyzed using Image Lab
TM software. The protein loading was normalized per total sample of protein as a fold control using stain-free gels as previously described [
21]. This normalization is equal to housekeeping genes normalization and has been rigorously evaluated by Bio-Rad company (
http://www.bio-rad.com/en-us/applications-technologies/stain-free-imaging-technology?ID=NZ0G1815) and by our lab in comparison with beta-actin normalization.
2.8. Seahorse Assays
For the glycostress Seahorse assay, human pulmonary artery smooth muscle cells (HPASMCs) (purchased from Lonza, Greenwood, SC, USA) were seeded in a 24-well Seahorse cell culture microplate at 50,000/well and kept overnight to form a monolayer. On the day of experiment, media were aspirated and cells were incubated at 37 °C in a non-CO2 incubator for 1 h with 0.5 mL XF base medium (cat# 102353-100, Agilent, Santa Clara, CA, USA) supplemented with 2 mM glutamine. Glucose (56 µL, 100 mM), oligomycin (62 µL, 100 µM) and 2-deoxyglucose (69 µL, 500 mM) were added to the flux pack wells (cat# 102342-100, Agilent, Santa Clara, CA, USA). ECAR (extracellular acidification rate) was then measured using the Seahorse Bioscience XFe24 extracellular flux analyzer (Agilent, Santa Clara, CA, USA) according to the manufacturer’s protocol. For the mitostress assay, cells were prepared similarly to the glycostress assay, with the exception that the XF base incubation media was supplemented with pyruvate (1 mM), glutamine (2 mM), and glucose (10 mM). Oligomycin (56 µL, 10 µM), FCCP (62 µL, 10 µM) and rotenone + antimycin-A (69 µL, 5 µM) were added to the flux pack wells. OCR (oxygen consumption rate) was then identified according to the manufacturer’s protocol. Data were normalized to cell number as determined by flow cytometry. All the chemicals used for these experiments, unless specified, were procured from Sigma-Aldrich, Saint Louis, MO.
2.9. Lactate Dehydrogenase Assay
LDH activity in lung tissue was assessed using the Lactate Dehydrogenase Activity Colorimetric Assay Kit (K726, BioVision Inc. CA, USA) according to the manufacturer’s instruction. All values were normalized with respect to protein concentrations as determined by the PierceTM BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA).
2.10. Pyruvate Dehydrogenase Assay
Lung tissue was lysed using RIPA buffer, and the lysate was used for the PDH activity. PDH activity was measured with the PDH Activity Assay Kit (MAK183, Sigma-Aldrich, Saint Louis, MO, USA) per manufacturer protocol. All values were normalized with respect to protein concentrations as determined by the PierceTM BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA).
2.11. Metabolic Intermediates Analysis
Data were acquired using the West Coast Metabolomics Center at UC Davis. Briefly, a Restek corporation Rtx-5Sil MS column was used with a helium mobile phase at the temperature interval 50–330 °C and flow-rate 1 mL min−1. Injection volume was 0.5 μL at 50 °C ramped to 250 °C by 12 °C s−1. Oven temperature started at 50 °C for 1 min, then ramped at 20 °C min−1 to 330 °C, and held constant for 5 min. Mass spectrometry analysis was done on a Leco Pegasus IV mass spectrometer with unit mass resolution at 17 spectra s−1 from 80 to 500 Da at −70 eV ionization energy and 1800 V detector voltage with a 230 °C transfer line and a 250 °C ion source. Raw data files were preprocessed directly after data acquisition and stored as ChromaTOF-specific *.peg files, as generic *.txt result files, and additionally as generic ANDI MS *.cdf files. ChromaTOF ver. 2.32 was used for data preprocessing without smoothing, 3 s peak width, baseline subtraction just above the noise level, and automatic mass spectral deconvolution and peak detection at signal/noise levels of 5:1 throughout the chromatogram. Apex masses were reported for use in the BinBase algorithm. Resulting *.txt files were exported to a data server with absolute spectra intensities and further processed by a filtering algorithm implemented in the metabolomics BinBase database. Significantly changed metabolites were processed with principal component analysis (PCA).
2.12. Statistical Analysis
Statistical analysis was performed using GraphPad Prism version 5.01 (GraphPad Software, San Diego, CA, USA). Outliers were identified with Grubbs’ test using GraphPad outlier calculator (alpha = 0.5) at
https://www.graphpad.com/quickcalcs/Grubbs1.cfm. The mean value (± SE) was calculated for all samples, and significance was determined by the unpaired t-test, Mann–Whitney U t-test, or analysis of variance (ANOVA). For one-way ANOVA, Bonferroni multiple comparison tests to compare the selected pairs of columns was used. The significance was calculated using a 95% confidence interval.
4. Discussion
Pulmonary arterial hypertension (PAH) is a progressive and fatal condition characterized by narrowing of the pulmonary arteries because of muscular thickening and intimal proliferation [
23]. Abnormal Akt activation causes normal cells to undergo proliferative conversion [
24], but in PAH, the cause and activation of Akt are not fully elucidated. The nitration of Akt plays a crucial role in its activation, and we found increased Akt nitration in patients with PAH as well as in the sugen/hypoxia model. Akt nitration can activate eNOS phosphorylation, and this, in turn, increases peroxynitrite formation, nitrosative stress, and mitochondrial dysfunction. Prolonged mitochondrial dysfunction is responsible for PAH development with vascular damage and metabolic disorder [
6,
7]. The study of metabolic alterations can be performed either by metabolomics or proteomics analyses. In our present study, we followed protein expression and compared metabolite profiles between groups. This approach clearly demonstrated PAH-specific alterations in the metabolic enzymes and metabolites, unveiling the protective mechanism of the antioxidant peptide in the context of Akt nitration. Interestingly, during the two-week sugen/hypoxic model, we identified that activation of Akt upregulated the glucose transporter Glut4 to the membrane, and this augmented cellular glucose influx. This resulted in increased aerobic glycolysis and anaplerosis, and a reduction in glycogenesis and PPP, similar to highly proliferative cancer cells. Histological studies identified cellular proliferation and vascular remodeling in lung tissue of early PAH. These pathological modifications recapitulate increased RV systolic pressure and Fulton index in early PAH. NP inhibited Akt nitration at Tyr-350 and controlled metabolic derangement in early PAH. NP treatment balanced cellular glucose influx, glycogen synthesis, and PPP. These events controlled glycolysis, oxidative phosphorylation, and inhibited anaplerotic reprogramming. Thus, with NP treatment, proliferative pathological changes in PAH were found to be prevented, and RV functioning improved with normal RVSP. The summary of observed metabolic alterations and effects of NP treatment is combined in
Figure 7.
As the incidence of PAH is high in females, we used female rats to study the anaplerotic reprogramming and Akt nitration events in PAH. The high incidence of PAH in females could perhaps be due to increased nitric oxide production and nitrosative stress development [
25]. NO, and the superoxide anion produced in hypoxic stress, potentiates a variety of biological processes including peroxynitrite formation, nitration of many tyrosine residues of proteins, and a decrease in antioxidant status [
26]. Recently, we unveiled that Akt1 nitration at the Tyr-350 residue enhanced the phosphorylation of eNOS at Ser617/1179 and its translocation to mitochondria in pulmonary artery endothelial cells [
6]. Excitingly, in our present investigation, using the newly developed nitro Akt (Y350-NO2) antibody, we discovered that nitration of Akt at Tyr-350 residue was an early molecular modification possibly responsible for the pathogenesis and progression of PAH. Immunohistochemistry and protein expression studies authenticated that Akt nitration started early, from one week of the disease, and trailed up in two and five weeks of the sugen/hypoxic model of PAH. Increased oxidative and nitrosative stress in early sugen/hypoxic PAH could have potentiated these nitration events. Pathological instigation of the Akt pathway has been described in many cancers, specifically in lung carcinogenesis. Importantly, nitrosative stress-induced tyrosine nitration potentiated cellular proliferation, and metastasis [
27,
28].
Identification of early Akt nitration encouraged the design of a targeted NP for its prevention and to study the implication of Akt nitration in PAH pathogenesis. We developed an antioxidant NP having two moieties: one a peptide part, designed complimentary to the Tyr-350 residue of Akt to obtain affinity, and the second, an antioxidant nitroxide that helps to scavenge free radicals/electrons. Nitroxide is a stable free radical, containing a nitroxyl group and an unpaired electron. This can easily diffuse through the cell membranes. In biological settings, they demonstrate SOD and catalase-like activities, preventing the Fenton and Haber–Weiss reactions and mitigating protein and lipid damage [
29]. The efficacy of the antioxidant peptide (NP) was screened in HPAECs by inducing nitrosative stress with the peroxynitrite donor SIN-1. Extracellular treatment with NP alleviated (55%) nitration of Akt in endothelial cells. The dose of NP used in this study was 100 times lower than the concentration of the antioxidant, TEMPOL, normally used in treating PAH, and it did not affect the redox homeostasis of the cellular environment. It was also recently reported that TEMPOL treatment was not effective in preventing pulmonary arterial remodeling and, in fact, increased the severity of PAH [
14].
In accordance with our initial observation, we found that Akt nitration was an early event, and NP was tested in the sugen/hypoxic PAH model at week two of disease progression. Interestingly, we found that NP was effective in blocking the nitration of Akt in early PAH. In late PAH, activation of Akt at Ser-473 plays a crucial role in the reprogramming of metabolic pathways and PAH pathogenesis [
5]. Intriguingly, in early PAH, there was neither any phosphorylation at Ser-473 of Akt nor any change in the expression of total Akt. This observation strengthens the idea that the activation of Akt occurring in early PAH might be through nitration of Akt. Akt activation is strongly associated with cellular proliferation and vascular remodeling [
30]. Our results suggest that the inhibition of Akt nitration with NP could mitigate early Akt activation and vascular modification.
Pulmonary vascular remodeling is among the major factors in initiating higher pulmonary vascular resistance and pulmonary arterial pressure. Akt plays a crucial role in hypoxia-induced pulmonary vascular remodeling [
5]. In our early sugen/hypoxic PAH models, we found increased RV systolic pressure and right ventricular hypertrophy (Fulton index). RV hypertrophy instigated by increased vascular resistance is primarily a compensatory mechanism, but this often leads to RV dysfunction [
31]. In accordance with the increased pressure, the lung tissue of high-pressure rats showed perivascular fibrosis and vasoconstriction in pulmonary arteries. Ki-67, a sensitive protein marker strongly associated with cell proliferation [
32], was found to be highly expressed in the pulmonary vasculature. This proliferation in the vascular wall and vasoconstriction explains the reason for elevated RV pressure in early PAH. Fascinatingly, NP treatment improved RV function and pulmonary vascular remodeling in the two-week sugen/hypoxic model. Similarly, in a previous report, inhibition of Akt inhibited muscularization of pulmonary arterioles and reduced right ventricular hypertrophy [
33]. These findings suggest the idea that inhibition of Akt nitration with NP could regulate its activation and could prevent downstream cellular proliferative and structural changes in RV and lung vasculature. Moreover, unlike the small molecule inhibitors of Akt, NP can diminish pathological events of nitration-mediated Akt activation without affecting normal Akt function. Thus, we consider this to be an important benefit of the targeted antioxidant approach.
Previous reports identified that glycolytic shift (Warburg effect) is a major pathogenic cause, in extreme cases, of PAH [
10].
Figure 4F,G showed increased glycolytic and decreased oxidative phosphorylation rates in HPASMCs with SIN-1 treatment. This explains Akt nitration induced metabolic reprogramming from oxidative phosphorylation to glycolysis. NP treatment inhibited Akt nitration and attenuated the metabolic reprogramming. Akt activation by peroxynitrite can enhance cellular glucose uptake through the glucose transporter, Glut4, and can increase glycolysis in PAH [
34]. In the present study, we profiled Glut4 expression in different cellular fractions and found it was increased in the membrane/cytosolic fraction. In the early two weeks of PAH, nitrated Akt could perhaps aggravate glucose uptake through increased membrane Glut4 translocation. High levels of intracellular glucose influx could upregulate glycolytic metabolism to meet the demands of increased cellular proliferation. This signifies the Seahorse analysis that PAH shifts glycolysis and mitochondrial respiration in the sugen model. Proliferating cells tend to have an increased expression of glucose transporters and glycolytic enzymes to compensate for the increased metabolic needs [
35]. Inhibition of Akt nitration with NP maintained glucose influx by downregulating the translocation of Glut4 to the membrane fraction.
In the present study, glycolytic enzymes HK-1 and GAPDH showed increased expression in lung tissue of the two-week sugen/hypoxia model. HK1 is the first rate-limiting enzyme of glycolysis, and its overexpression can initiate downstream pathways, amplifying reactions that offer increased ATP to proliferating cells [
36]. Also, Akt activation can exacerbate glycolysis by GAPDH phosphorylation and activation in hypoxic stress conditions [
37]. Here, we speculate that Akt activation by nitration played a cardinal role in cellular glucose uptake and increased glycolysis in proliferating cells of early PAH. LDHA converts pyruvate to more lactate in rapid flux metabolism through glycolysis. Increased LDH expression and activity is characteristic of fast-growing cells, and its suppression stimulates mitochondrial respiration and decreases proliferation [
38]. Similarly, in our study, nitration of Akt intersects with the aerobic glycolysis pathway in the two-week hypoxic/sugen model with increased LDHA expression. Increased lactic acid production may result in lactic acidosis, a serious physiological event in PAH [
39]. Pyruvate generated in the latter phases of glycolysis is converted to either lactate, via aerobic glycolytic metabolism, or acetyl-CoA, through the oxidation of glucose by PDH. PDH is often considered as the “mitochondrial gate-keeping enzyme”, promoting pyruvate entry into the OXPHOS [
40,
41]. Interestingly, inhibition of Akt activation with NP increases mitochondrial enzyme PDH, signifying upregulation of oxidative phosphorylation, and a reduction of lactate accumulation. Thus, in our early PAH model, NP treatment efficiently prevented the metabolic reprogramming from glycolysis to oxidative phosphorylation and prevented cellular proliferative changes.
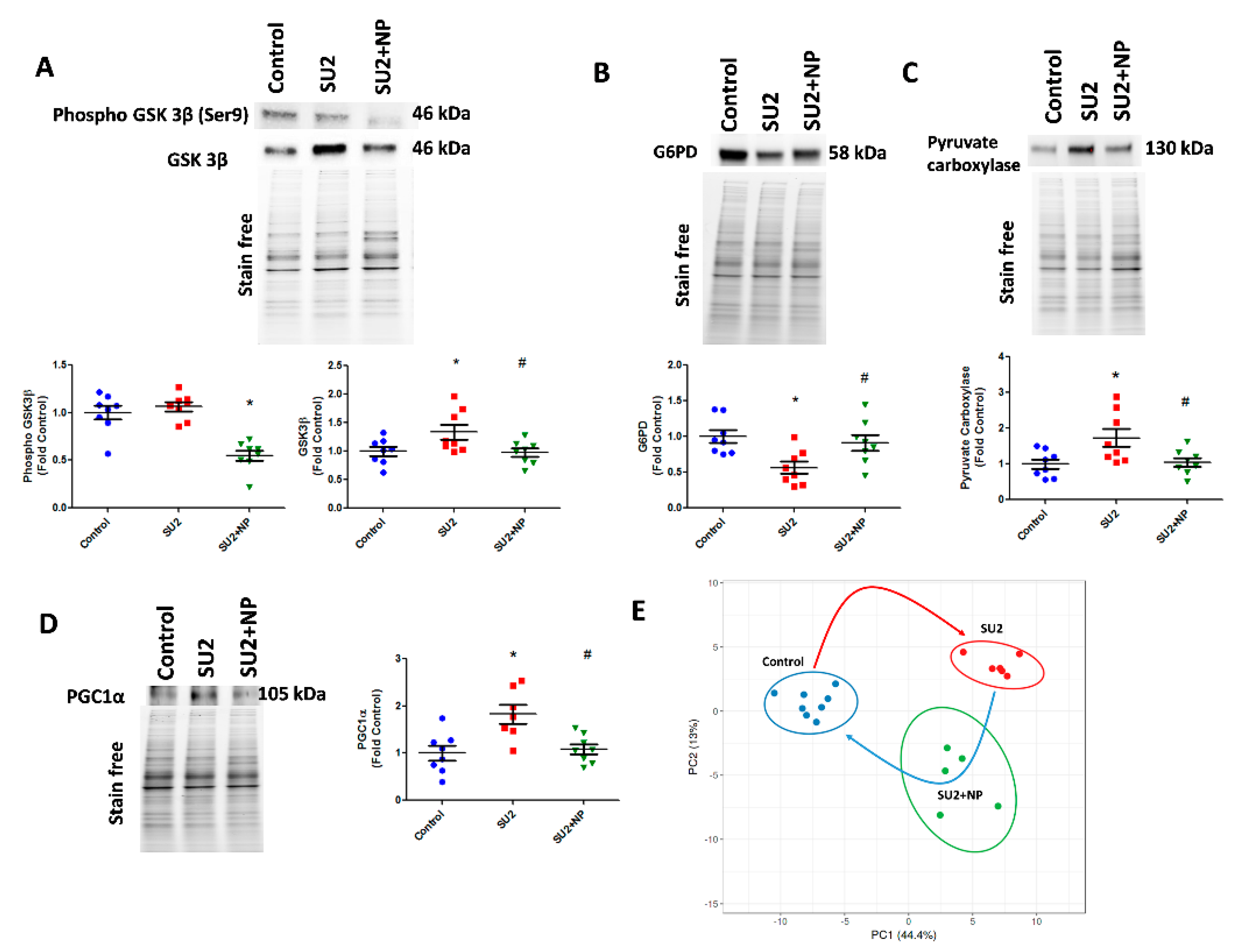
GSK3β is a serine-threonine kinase, regulating a variety of cellular functions like cell division, proliferation, and differentiation. Akt is one of the critical regulators of GSK-3, and phosphorylation and inactivation of GSK-3 represent the anti-apoptotic effects of Akt [
42]. In the PAH model, Akt nitration did not upregulate phosphorylation of Ser9 in GSK3β, but the total expression of GSK3β was increased. GSK3β phosphorylation is controlled by phosphorylated Akt, but in early PAH, Akt phosphorylation was found not activated, and this could perhaps be the reason for no change in phosphor-GSK3β expression. Elevation of GSK3β inversely correlates with glycogen synthase [
43]. Inhibition of glycogen synthase downregulates glycogen synthesis and could produce more LDH as well as oxaloacetate for aerobic respiration and anaplerosis. However, NP by preventing Akt activation decreased GSK3β expression and maintained glycogen synthesis. On the other hand, we found that G6PD, the rate-limiting enzyme of PPP [
44], was decreased. This could downregulate PPP and redirect the metabolism towards increased glycolysis, similar to what we found in a recent complex III-deficient model [
7]. In accordance with this, our recent report in PAH patients suggests that G6PD deficiency potentiates hemolysis and disease progression [
45]. In the two-week sugen/hypoxic model, nitro Akt activation maintained G6PD expression and corrected the PPP, thus balancing glycolytic metabolism to normal.
Downregulation of glycogen synthesis and PPP in early PAH lungs shifted the metabolism towards glycolysis and perhaps produced increased pyruvate. As observed in our study, pyruvate could have activated PC and produced increased oxaloacetate for the TCA cycle. This could result in increased anaplerosis. Anaplerosis by PC and glutaminolysis are the reasons for pulmonary vascular constriction and increased proliferation in PAH [
13]. Increased glycolysis and anaplerosis provides supplementary biomass for cellular proliferation in early PAH. Increased anaplerosis could lead to a reduction in oxidative phosphorylation and cause mitochondrial dysfunction [
11]. Two weeks of sugen/hypoxia increased PGC-1α expression in lung tissue, representing impairment in mitochondrial function and mitochondria biogenesis demand. PGC-1α might be increased for compensating the metabolic reprogramming and mitochondrial dysfunction in early hypoxic stress [
46]. Activation of Akt is associated with the regulation of PGC-1α [
47]. In our early PAH model, activation of Akt by Tyr-350 nitration may have upregulated PGC-1α expression. Excitingly, the prevention of Akt nitration with NP downregulated PGC-1α expression, representing improved mitochondrial functioning and a reduction in cellular anaplerosis.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






