Design of Light-Sensitive Triggers for Endothelial NO-Synthase Activation

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Methods and Procedures
2.1.1. Homology Modeling
2.1.2. Docking of the Nanotriggers into the Binding Site of eNOS
2.1.3. Molecular Dynamics
2.1.4. Calculation of the Interaction Energies
2.1.5. Statistics
2.2. Experimental Procedures
3. Results
3.1. Results
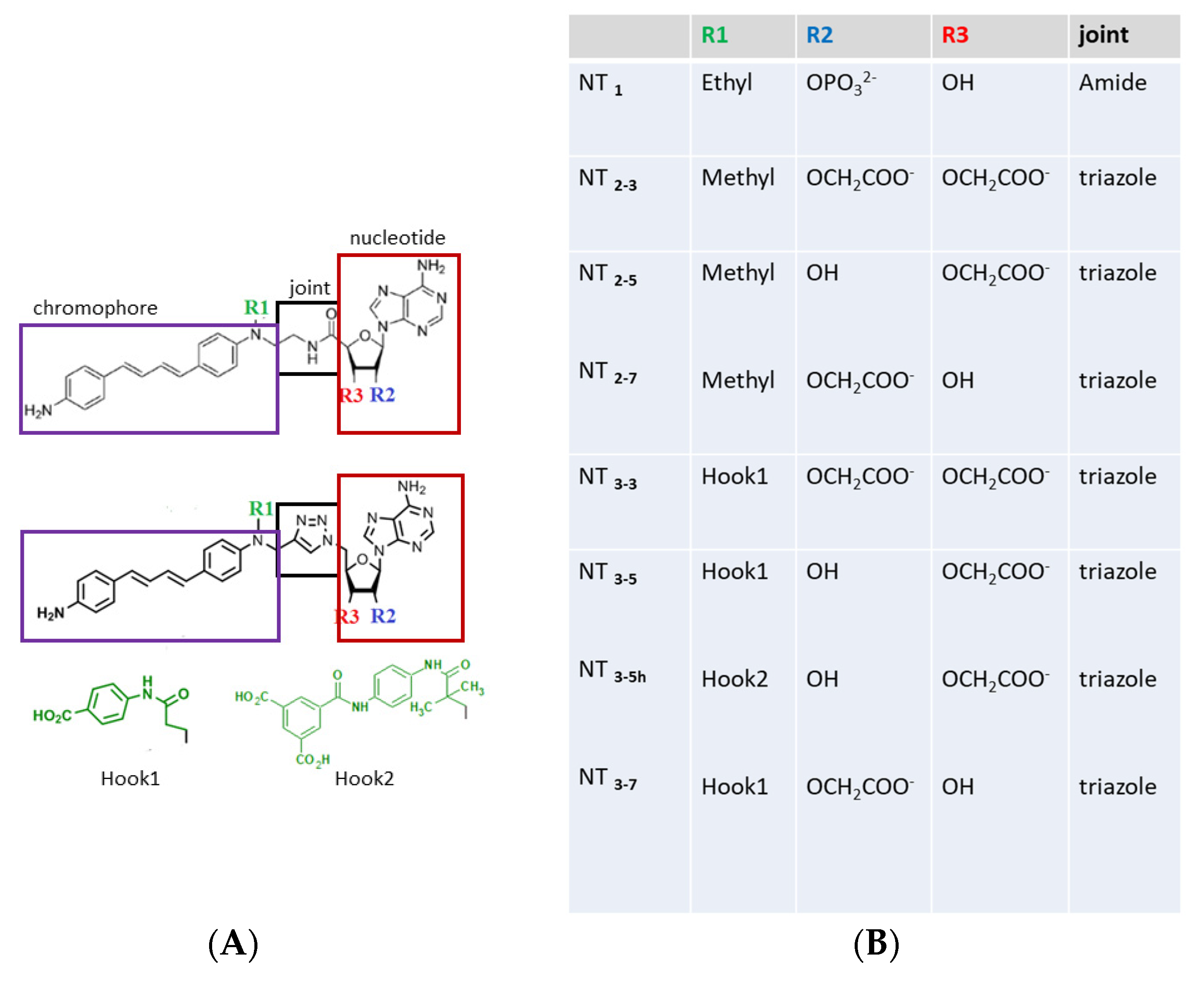
3.1.1. Structures of the NT1 Nanotrigger Prototype and Design of Derivatives
- Test the effect of charge(s) carried by R2 or/and R3 on the ribose in the recognition of the conserved arginines of eNOS. Previous experimental data clearly emphasized the importance of electrostatic effects on their binding to eNOS by comparing the binding affinity of NT2_3, NT2_5 and NT2_7 with their diol derivative bearing no charge because both 2’ and 3’ positions were substituted by OH groups [18]. It was therefore logical to further explore these electrostatic effects by replacing the 2’ phosphate group by carboxylate group(s), one or two at the 2’ and 3’ positions of the sugar (R2 and R3 in Figure 1). To do so, we generated a compound containing two carboxylates, with a total charge of -2, instead of a phosphate group in NT1; we also generated compounds carrying a single carboxylate group either at the 2’ or 3’ position of the ribose with a charge of –1 (Figure 1); in the case of the carboxylate-bearing NT, a new triazole joint replaced the amide bond as it is considered to be its biostere and it can be formed by click chemistry [18].
- Restore the loss of interaction energy inherent to the use of a single-charged carboxylate moiety (replacing the phosphate of NT1). To increase the affinity for e/nNOS, we re-engineered the previous NT [18] with an additional carboxylate substituted on a phenyl ring (R1 = hook 1) (Figure 1). Hook1 was expected to increase both the total charge of the compound and its hydrophobicity;
- Improve affinity and specificity for the endothelial NOS, eNOS, with hook 2 (Figure 1) targeting eNOS-specific variable residues.
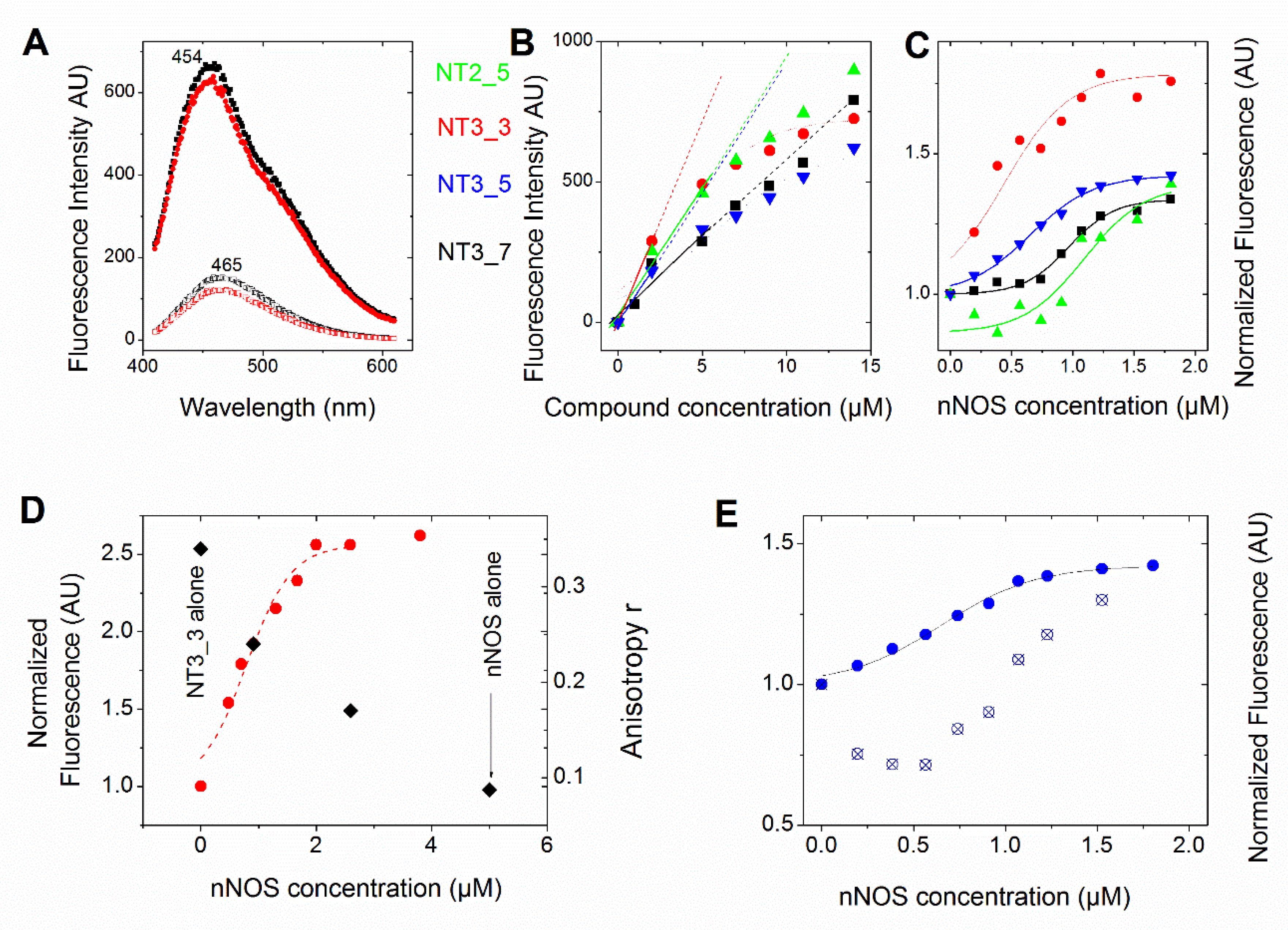
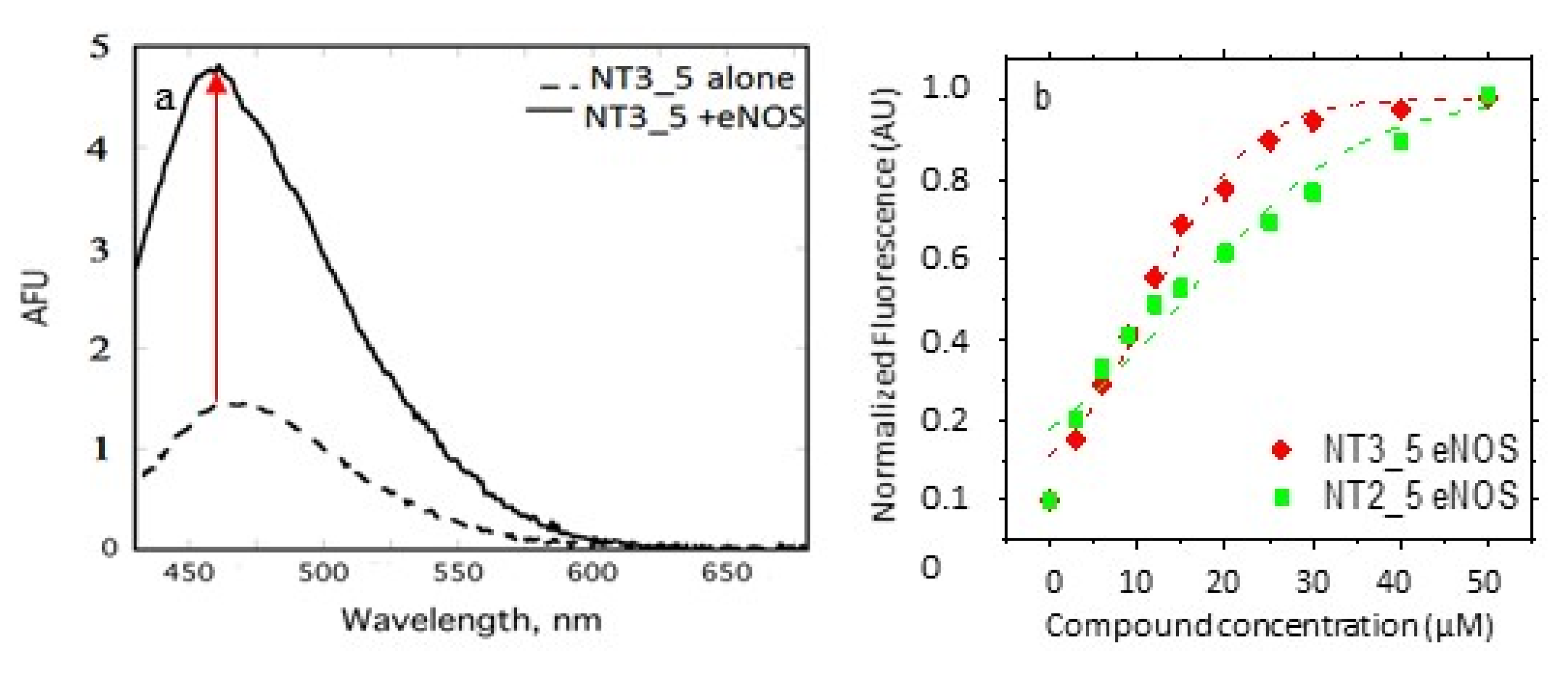
3.1.2. Evaluation of the Substitution of the 2’ Phosphate Group by (a) Carboxylate Group(s) on the Nanotrigger Affinity for e/nNOS Determined by Fluorescence Measurements
3.1.3. Interactions of NTs with eNOS as Determined by MD Simulations; Impact of the Carboxylate Group(s) Versus the 2’ Phosphate Group
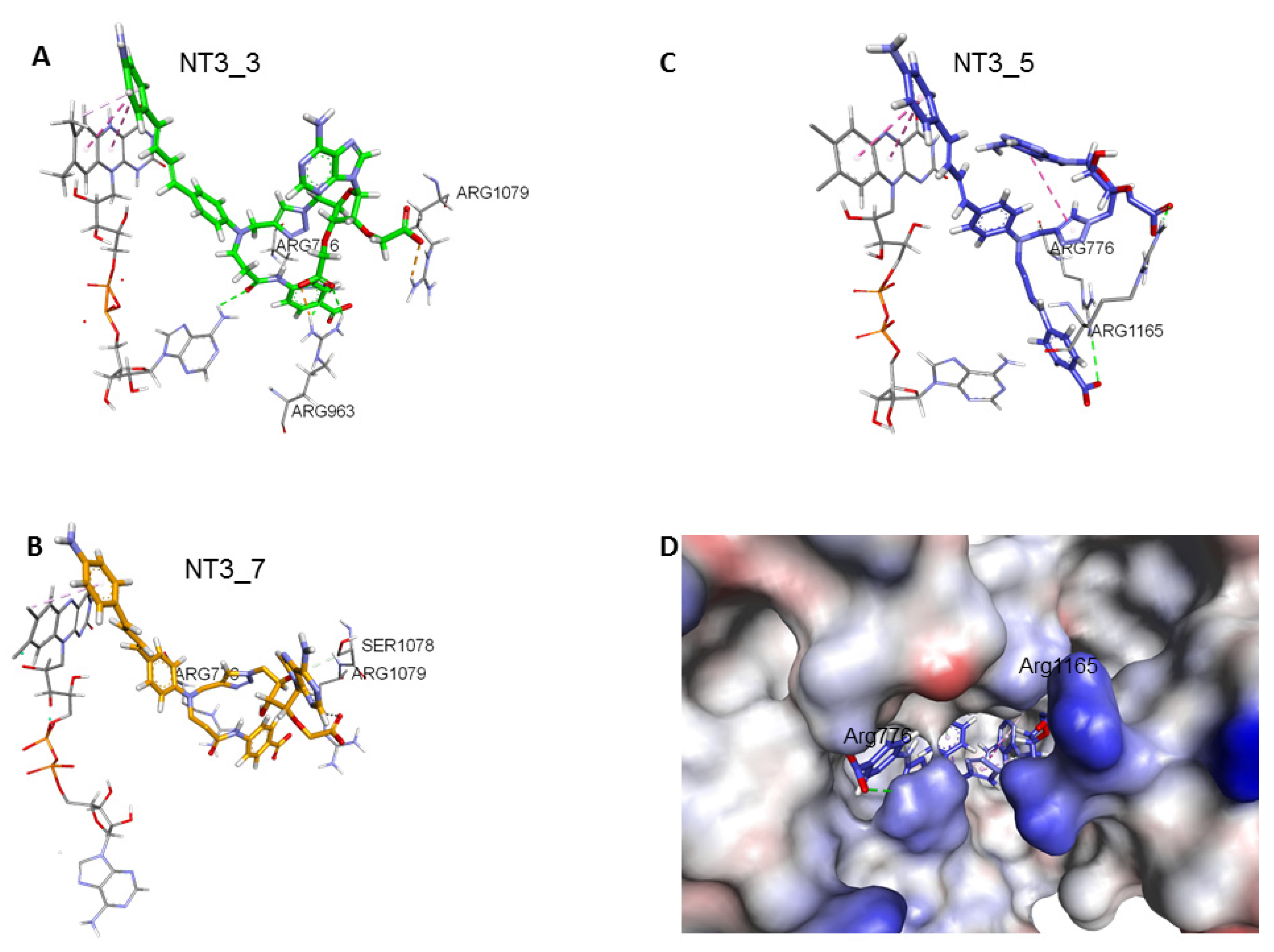
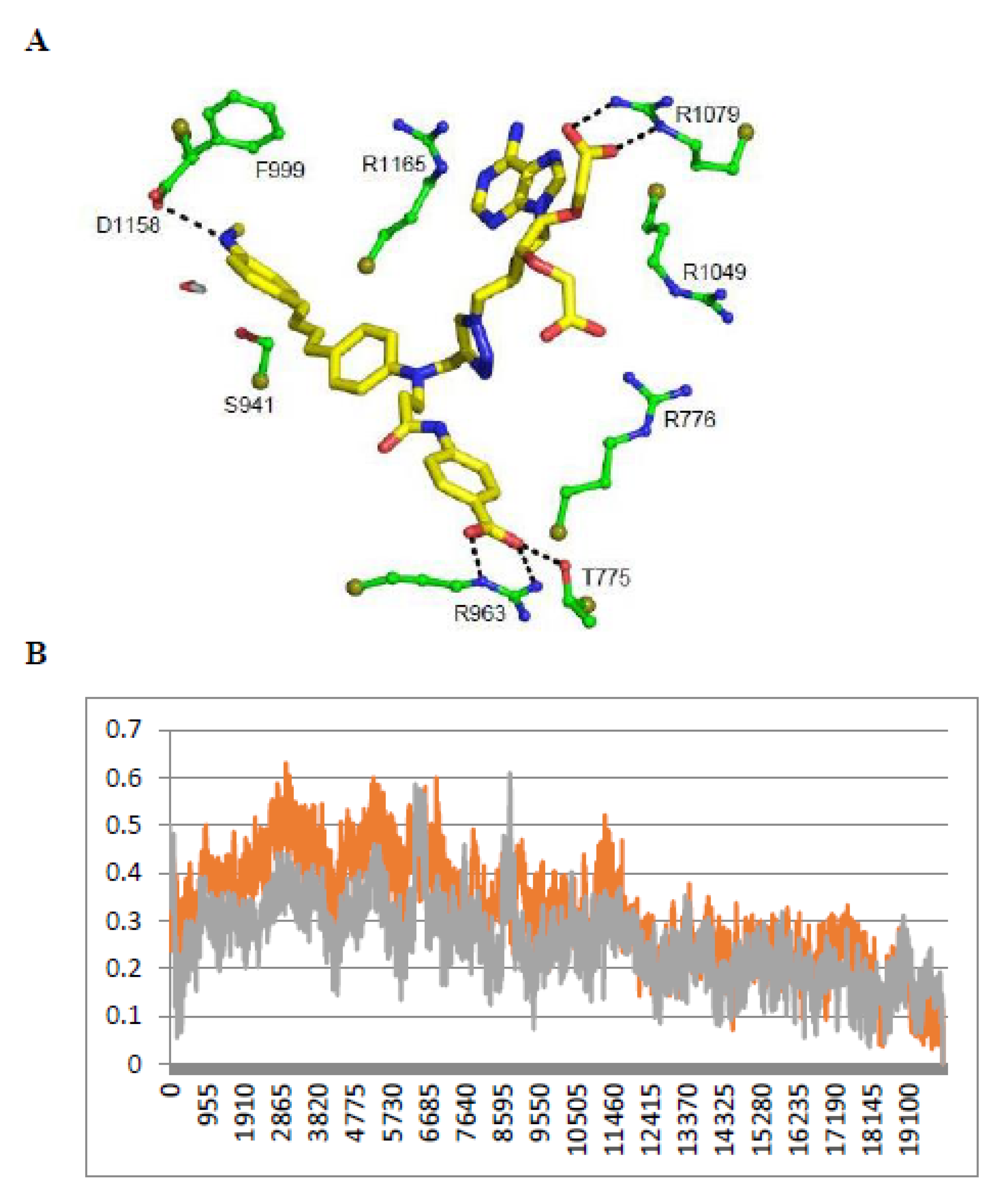
- Stacking with FAD: As shown in Figure 3 and Table 2, the chromophores of the various NTs were close to the FAD isoalloxazine ring, with various degrees of stacking on FAD isoalloxazine rings. The terminal phenyl ring carrying the NH2 donor group of NT3_3 stacked on FAD (see the dotted pink lines emphasizing the hydrophobic contacts, Figure 3A). This was also the case for NT3_5 (see below). In contrast, NT3_7 did not stack well with FAD as its chromophore was relatively tilted to the side (Figure 3B).
- 2’ and/or 3’ carboxylate(s) interactions with eNOS: The 3’ carboxylate of NT3_5 bound to the conserved R1165. A view of NT3_5 fitted into its binding site is shown in Figure 3C,D, emphasizing the interactions with both R776 and R1165, which are residues located close to the protein- solvent interface, thus forming a charged “aperture of the site”. In NT3_7, the 2’ carboxylate moiety linked to the ribose ring bound to a conserved arginine of eNOS, R1079. The comparison of NT3_7 and NT3_5 interaction energies with eNOS showed a lower stability for the 2’ carboxylate in NT3_7 as compared to the 3’ carboxylate in NT3_5 (Table 3), in agreement with the experimental results. The total energy of interaction followed the electrostatic energies of the complexes (Table 3), namely NT3_3 > NT3_5 > NT3_7. Within error ranges, the interaction energies of NT1 and NT3_5 with eNOS were found to be comparable.As for NT3_3, the 2’ and 3’ carboxylate groups usually bound the conserved R1079 and the variable R963, respectively. To get more insight about a possible steric clash between the adjacent carboxylates on the ribose, hence a relative decreased stability, we computed the stabilities of both carboxylates along a number of MD runs (Figure A3). The 2’ carboxylate was found to be less stable than the 3’ carboxylate of NT3_3. Consequently, structures with only one carboxylate bound to eNOS co-existed (Figure A3) along with structures with both carboxylates bound (Figure 3A) during the three dynamics runs.
- Contribution of the hook R1: The 2’ phosphate of NT1 interacting with three conserved arginines R1049, R1079, and R1165, similar to the binding of the 2’ phosphate of NADPH [16,23]. As electrostatic interactions largely contributed to overall stability of the complexes, replacing the methyl groups of NT2_3, NT2_5, and NT2_7 with a charged hook R1 was expected to result in increased affinity of the compounds for NOS binding. The hook R1 formed an electrostatic interaction mainly with R776 in all three compounds; in NT3_3 and NT3_5; additional interactions of polar residues further stabilized hook1 (Table 2). Thus, hook1 was predicted to contribute to an increased stability of the protein-ligand complex as shown in Table 3, in agreement with experimental data (Table 1).
- Interactions induced by the triazole linker: We chose a triazole linker between the chromophore and the nucleotide (Figure 1) for its ease of generation by click chemistry [16,18]. The triazole was relatively rigid and participated in NT3_5 binding to NOS, as shown in Figure 3C. Extended stacking interactions were observed in NT3_5 between the triazole ring, the adenine ring, and P1014 that formed an “hydrophobic hub”. To adapt and contributing to this hydrophobic hub, one chromophore phenyl ring was somewhat twisted with respect to the first phenyl-NH2 moiety. This hub was further stabilized by I1018 and L1164, F1160 from the other side. In addition, R776 interacted with both the triazole and the benzoate ring and carboxylate group of hook1. In contrast, in NT3_7, the conformation of the triazole ribose was rather extended, the adenine being weakly stabilized by M1119. In NT3_3, the conformation of the triazole ribose was also rather extended. The adenine showed hydrophobic interactions with P1014 and L1164, as well as cation-π interaction with R1165.
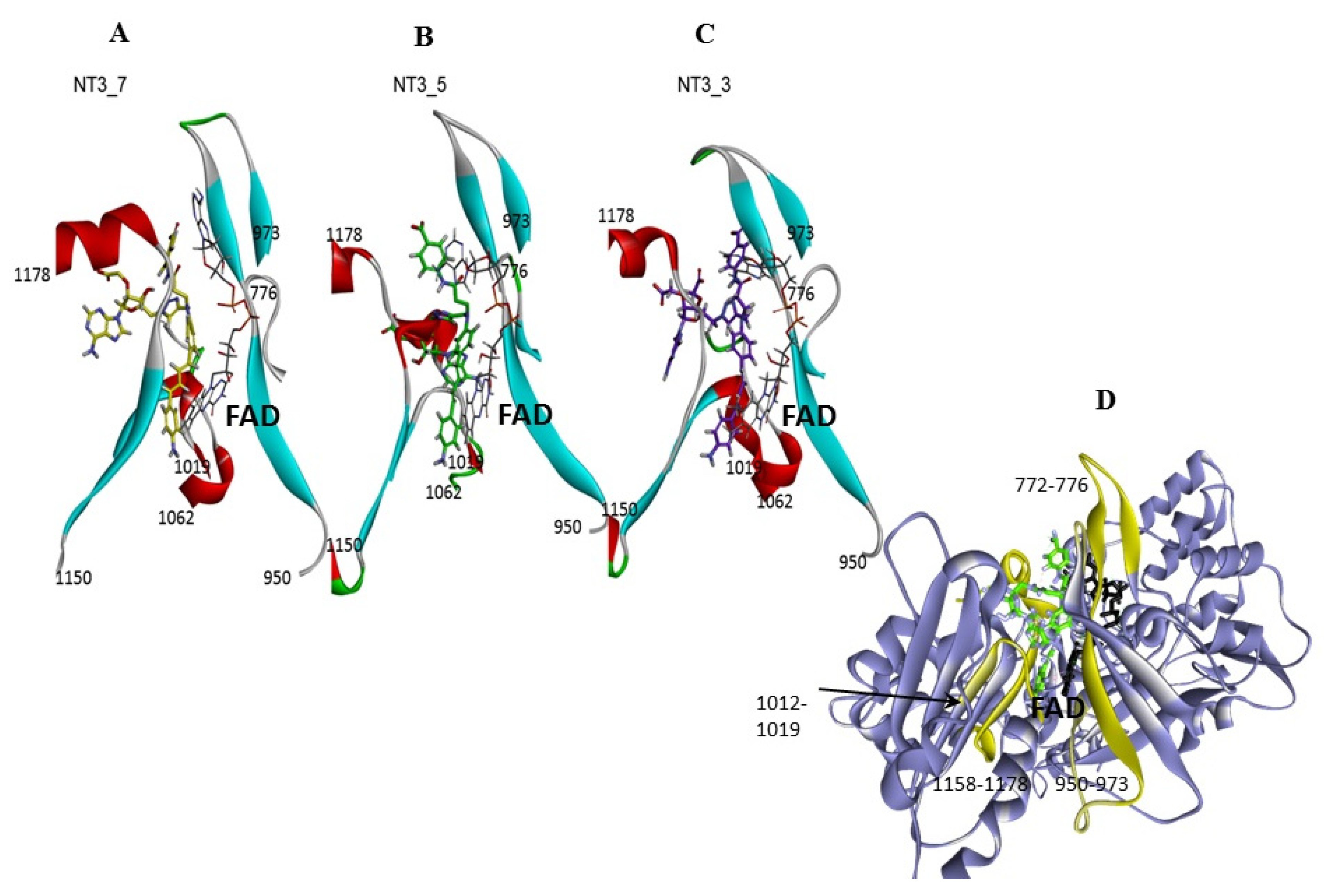
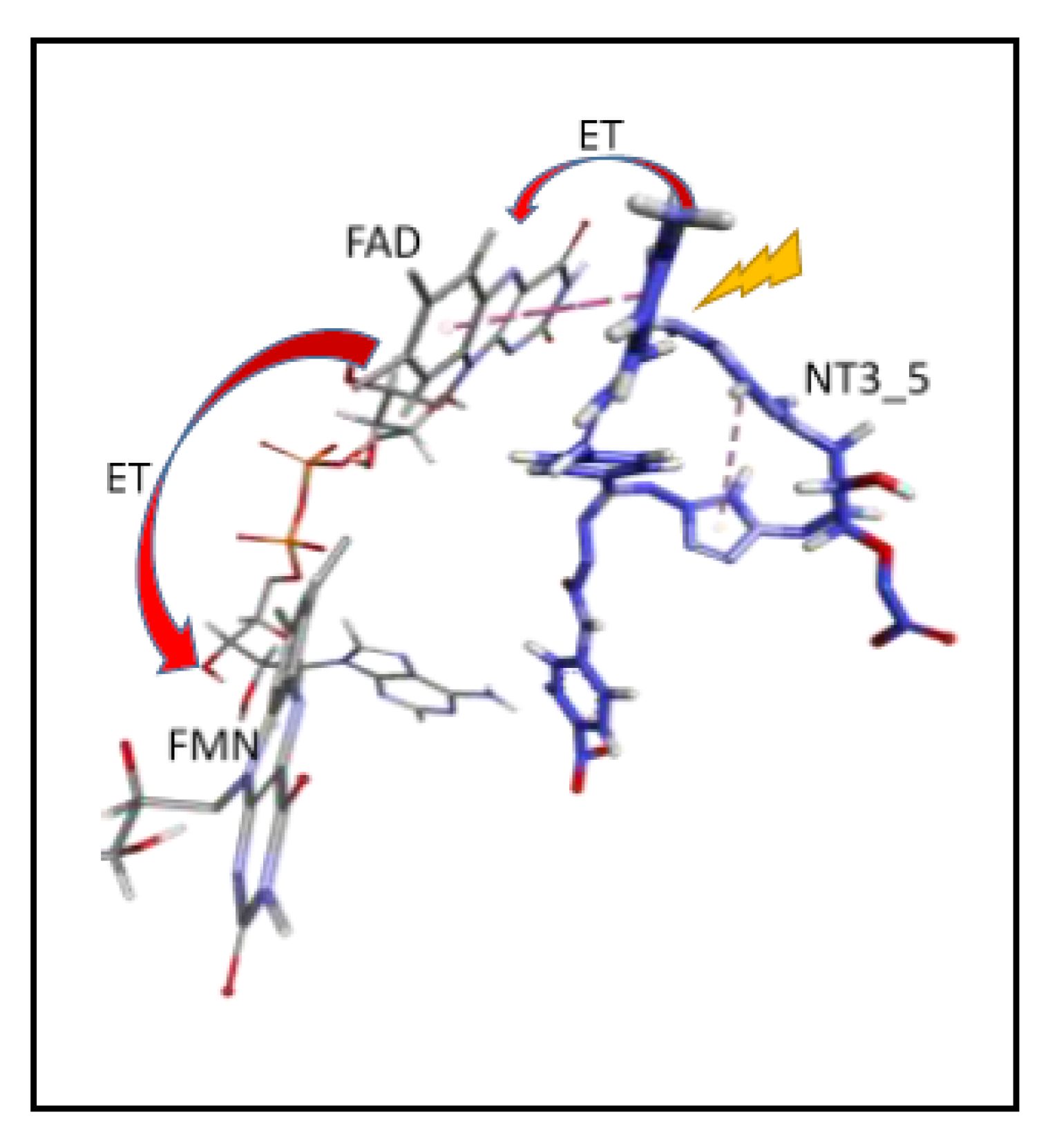
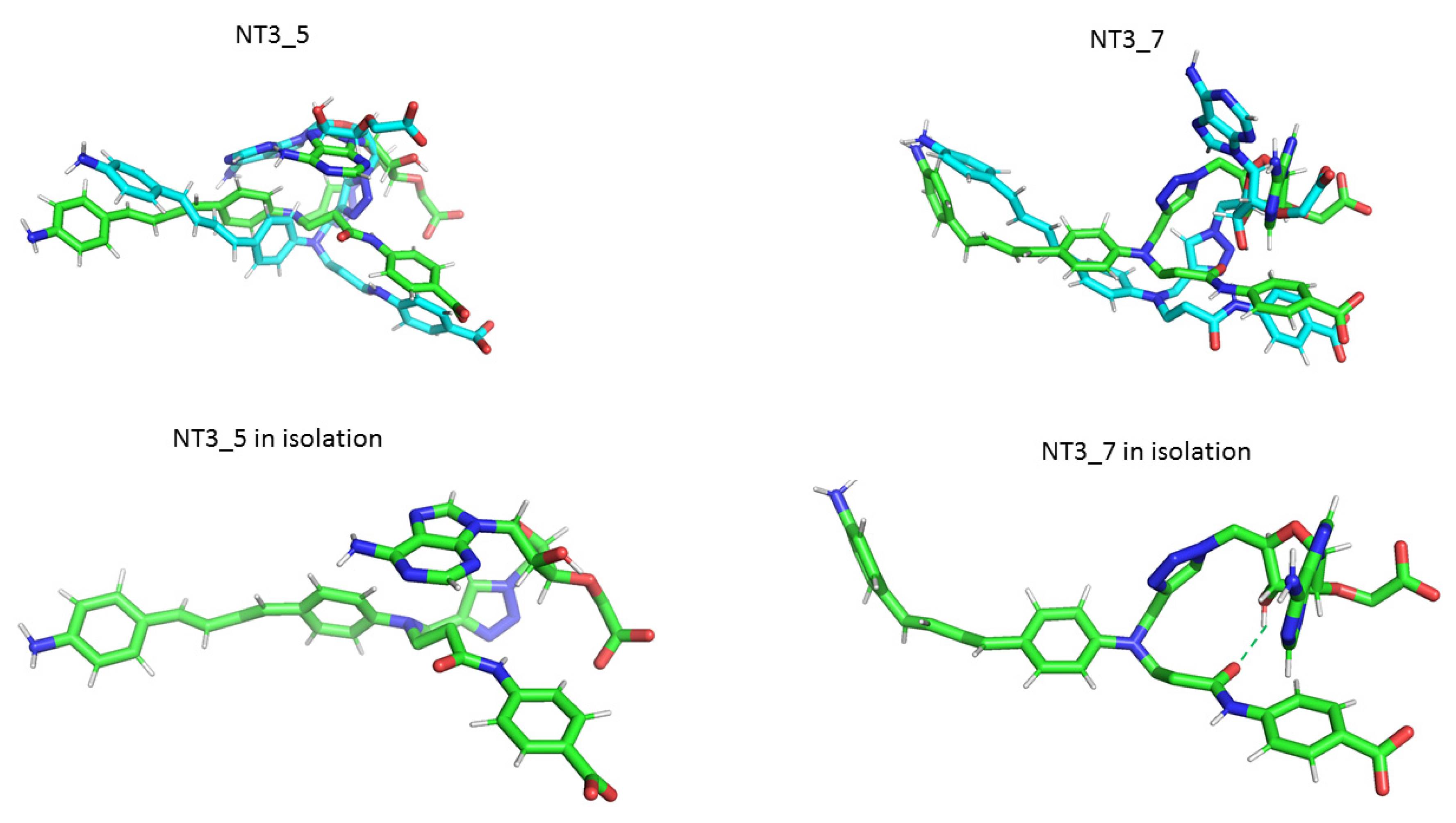
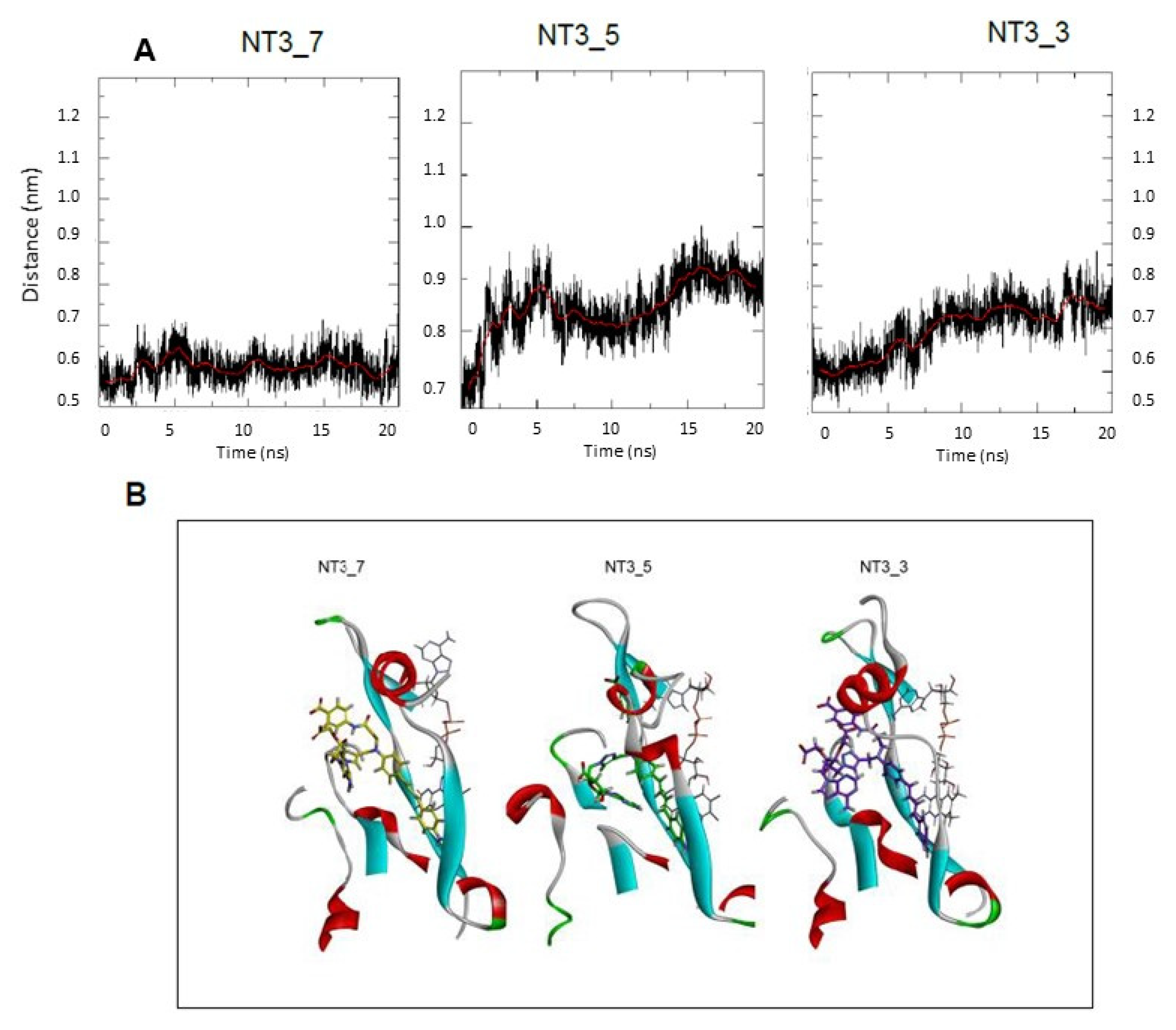
- Protein-induced fit to NT3_5 binding conformation: Figure 3C shows that strong interactions between the triazole, the last phenyl ring of the chromophore, and the adenine ring contributed to stabilize NT3_5 in its binding pocket. This hydrophobic “hub”, only found in NT3_5, required the protein to adapt, with a β sheet (950–973) being pushed apart from a linker (1012–1019). The distance between A958 and T1016 thus increased to 9.5 Ǻ as compared to the same distance in NT3_3 and NT3_7 (6 and 8 Ǻ, respectively) (Figure 4 and Figure A4). The differences in interactions at the level of the ribose carboxylate and of the triazole–chromophore–adenine dictated the orientation of the chromophore and its ability to stack with the isoalloxazine ring of FAD. Table 2 and Figure 3 show that NT3_5 stacked better with the protein than NT3_7 did. It is likely that these structural features enhanced the ability of NT3_5 to transfer electrons upon light activation, as observed experimentally.
- Interestingly, the conformation of the NT 3_5 in isolation was similar to the eNOS-bound state, in contrast with NT 3_7 for which both conformations largely differed (Figure A4; see also Figure A5 for a view with a 90° rotation). A conformation close to the bound state of the nanotrigger in isolation reduces the energy necessary to unfold the nanotrigger and, also, the entropic barrier between the isolated and bound states, making NT3_5 a better ligand for binding to the protein than NT3_7.
3.1.4. Toward an eNOS-Specific NT
3.2. Discussion
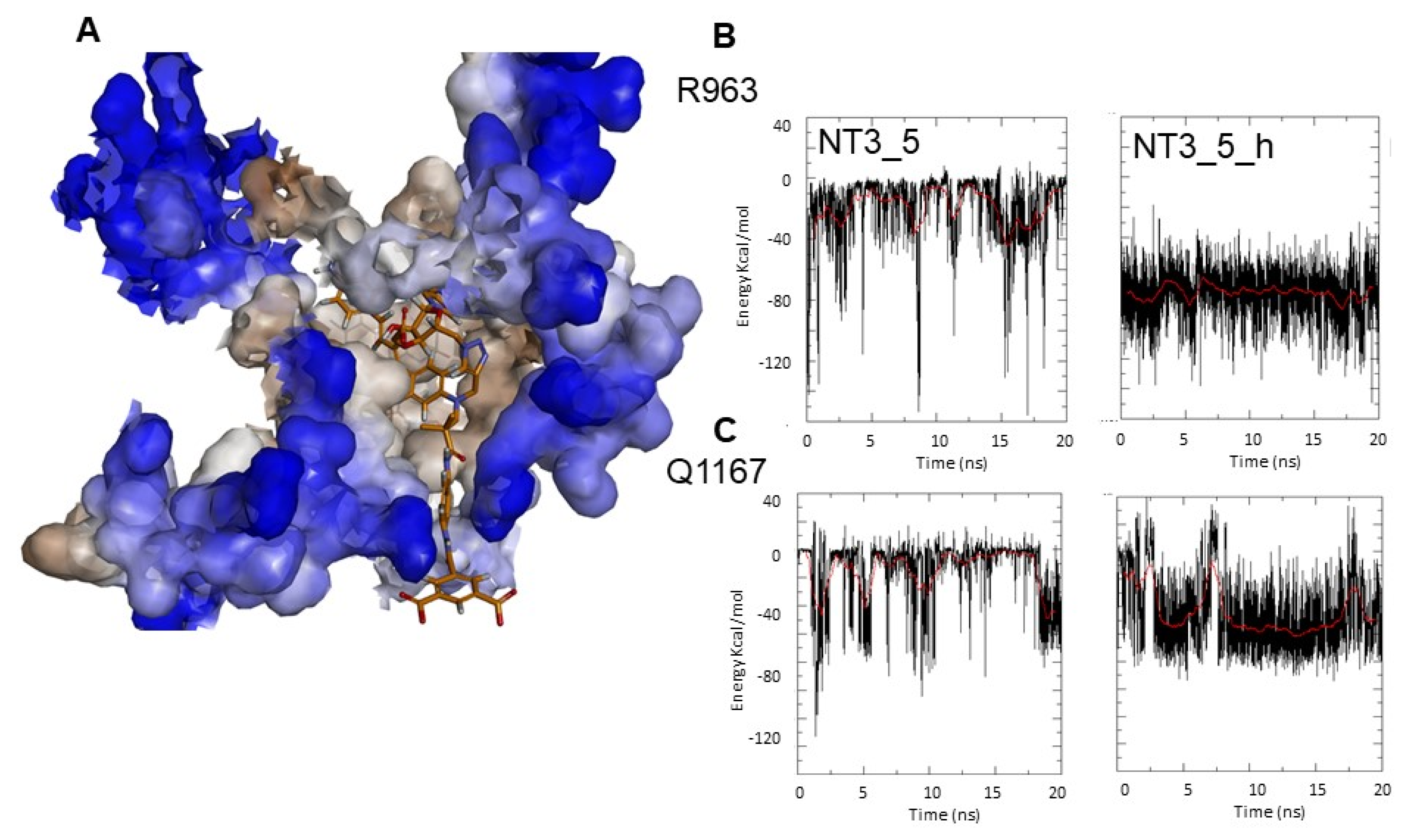
- Gate to the channel of the NADPH site: By inspecting Figure 3D, it is obvious that electrostatic interactions play an important role in the binding of any ligand to that site. The analysis of the dynamics (Figure 4 and Figure A4) shows that two of the three loops that were in the proximity of the NTs in eNOS (772–776 and 1012–1019) together with a β-sheet (950–978) and the C-terminal linker 1158-1178 formed a flexible aperture of the channel. These loops/linker movements guided the initial steps of the binding of the NTs and constituted a “gate keeper” to the channel. Along the entire path of NT binding, hydrophobic interactions guided the NTs to the conserved arginine region where the electrostatic interactions were the strongest. In addition to the electrostatic interactions with conserved arginines of eNOS, the chromophore and the adenine of NT3_5 contribute to binding by hydrophobic or polar contacts (A958, P1014, T1016, G1017, F1160, T1163, Table 2) and cation-π interactions (R776 and R963). The hook 1 in NT3_5 and hook2 in NT3_5h made additional electrostatic and hydrophobic interactions with one of the gate keeper loop (771–776), the β-sheet (950–978) and with C-terminal residues (Q1167) (Figure 5).
- Comparison between experimental and calculated interactions between NOS and the NTs: The interaction energies of NT1 and its monocarboxylate analogue at the 3’ position, NT3_5, were similar within the given error despite the fact that NT3_5 had a single charge at the level of the ribose and a charged hook1 contributing to binding while NT1 had two charges at the 2’ position of the ribose (Table 2). This is in agreement with the expected stabilization implemented by hook1, as observed experimentally (Table 1). Nevertheless, the interaction energies did not predict the experimental differences in Kd of about 20-fold given by the comparison of Kd = 360 ± 140 nM and 7 ± 3 µM for NT3_5 and NT1 binding to nNOS and eNOS, respectively [16]. Based on the anisotropy measurements (Figure 2D), we assume the following mechanism, whereby self-associated (NT)n needs to dissociate before binding to nNOS as a monomer. Such a self-association was not observed for NT1 [11,12] but was also observed for NT2_5 (Figure 2B). We hypothesize that these self-associated structures are at the origin of the cytotoxicities of the second generation of NT (as NT2_5) above 5 µM [14,15] in HUVEC cells, while NT1 was not toxic to HUVEC up to 20 µM.
- NT + nNT ⇋ (NT)n and NT + nNOS → NT-nNOS
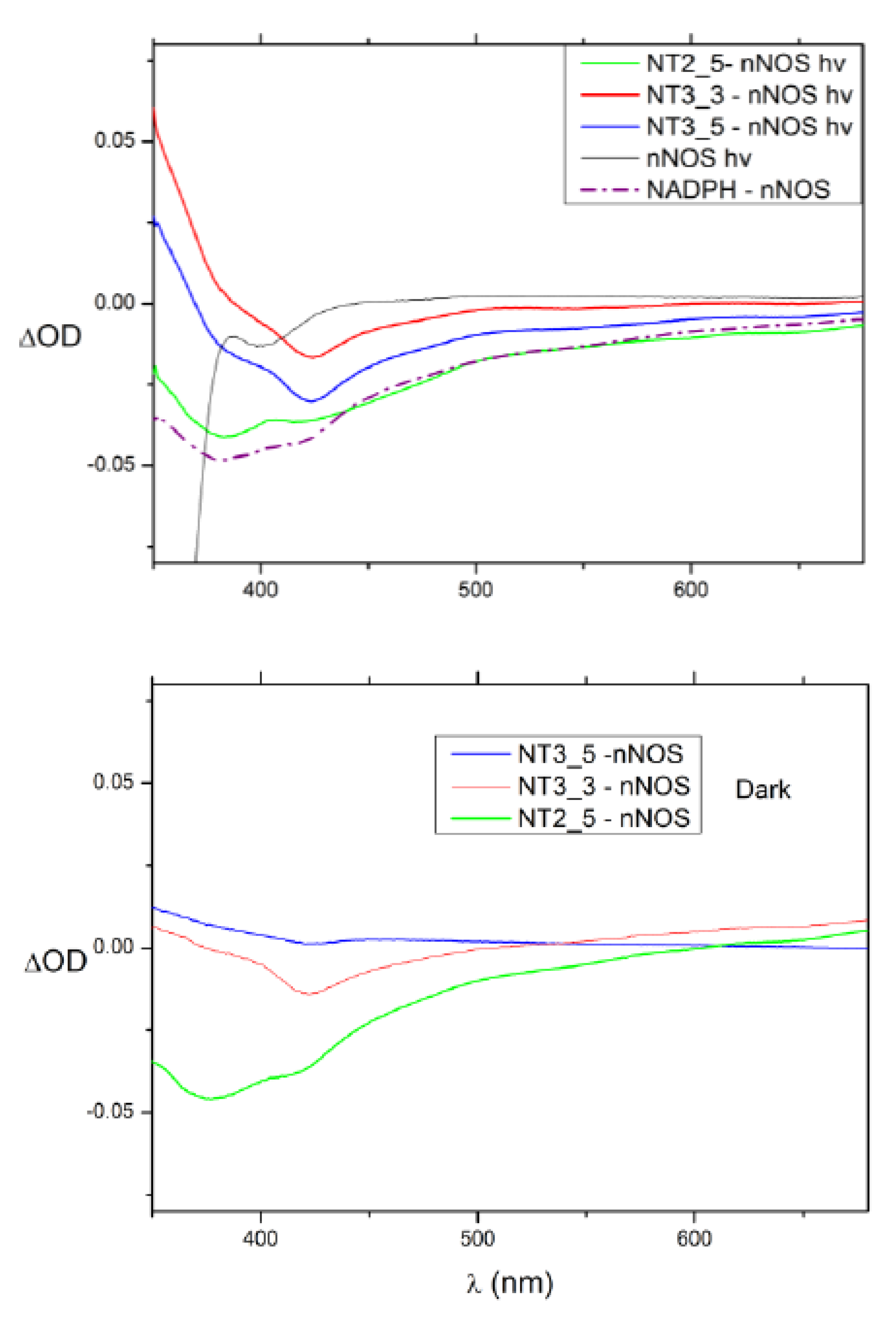
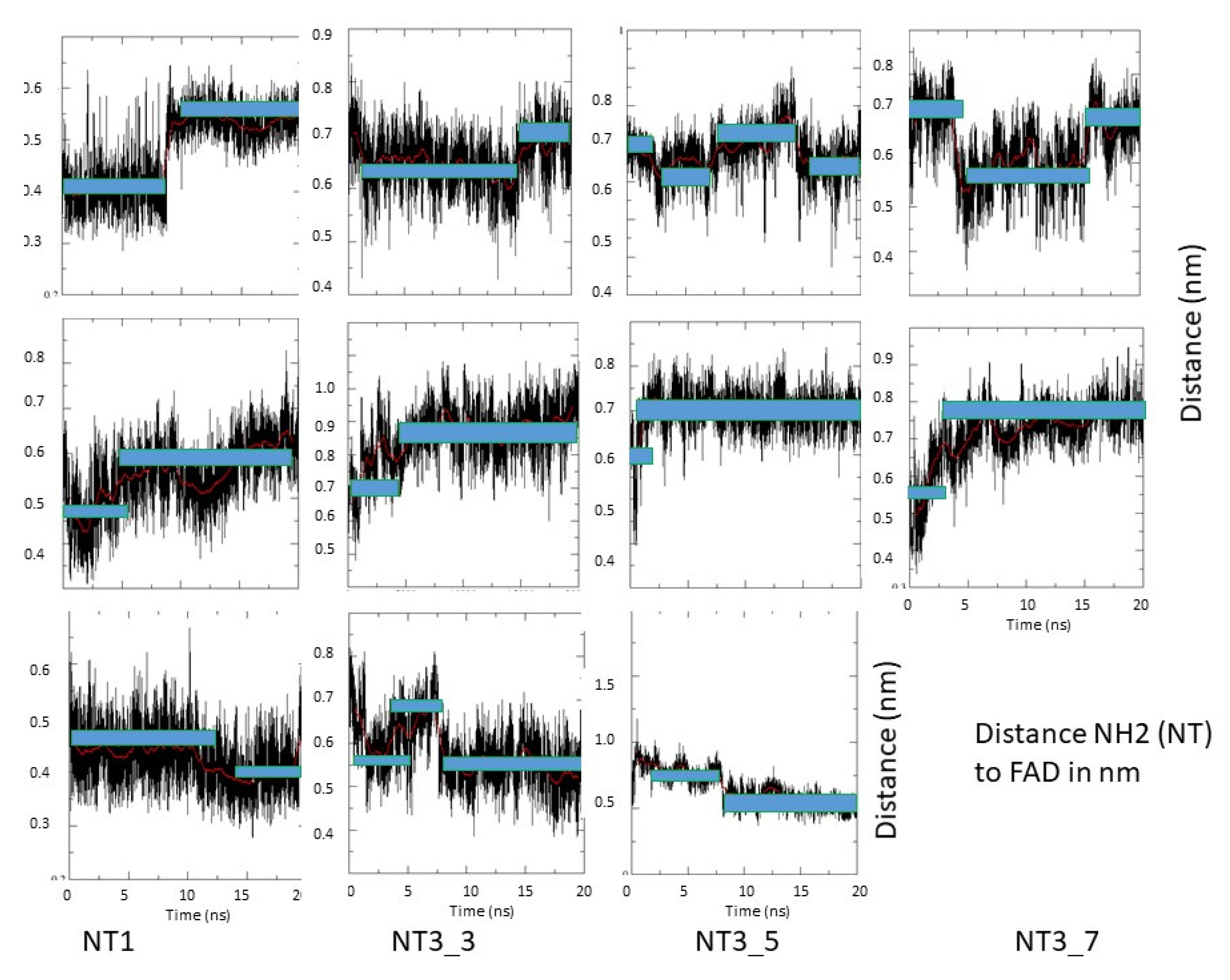
- Electron transfer to e/nNOS (Scheme 1): NT1 and NT3_5 transferred electrons to the protein after light activation, which led to initiation of the electron flow in e/nNOS and experimental detection of NO [12,21]. In contrast, NT3_7, which bears a single carboxylate at the 2’ position, had a smaller interaction energy with eNOS than had NT3_5. NT3_7 chromophore did not stack well on the FAD isoalloxazine ring, suggesting a reduced electron transfer ability in the excited state, in agreement with its parent NT2_7 being unable to generate NO [18,19]. This is in agreement with previous studies on the effect of stacking interactions between flavin derivatives and aromatic hydride-donor model compounds [32]. We also computed the distance between the electron-donating NH2 terminal of the NTs and the electron acceptor FAD at the N5 position. It fluctuated between two levels in the various replica (Supplementary Figure A7): While in NT1, this distance fluctuated between 4.5 and 5.6 Ǻ, the levels ranged between 5.6 and 7.2 Ǻ for NT3_5, 6.2 and 7.5 and 5.7 and 7.7 Ǻ for NT3_3 and NT3_7. Although the distances were shorter for NT1 than NT3_5, favorable entropic (ring–ring) interactions in NT3_5 provided stabilization of the adenine by the first phenyl ring of the chromophore and further interactions with the triazole. Consequently, the two phenyl rings became twisted one with respect to the other. We cannot rule out that the excited state(s) may thus possess some character of a twisted intramolecular charge transfer state (TICT) with a high dipole moment, that would support the ability of NT3_5 or NT2_5 to transfer electrons to FAD. TICT was described for push–pull polyenes and merocyanines [33,34].
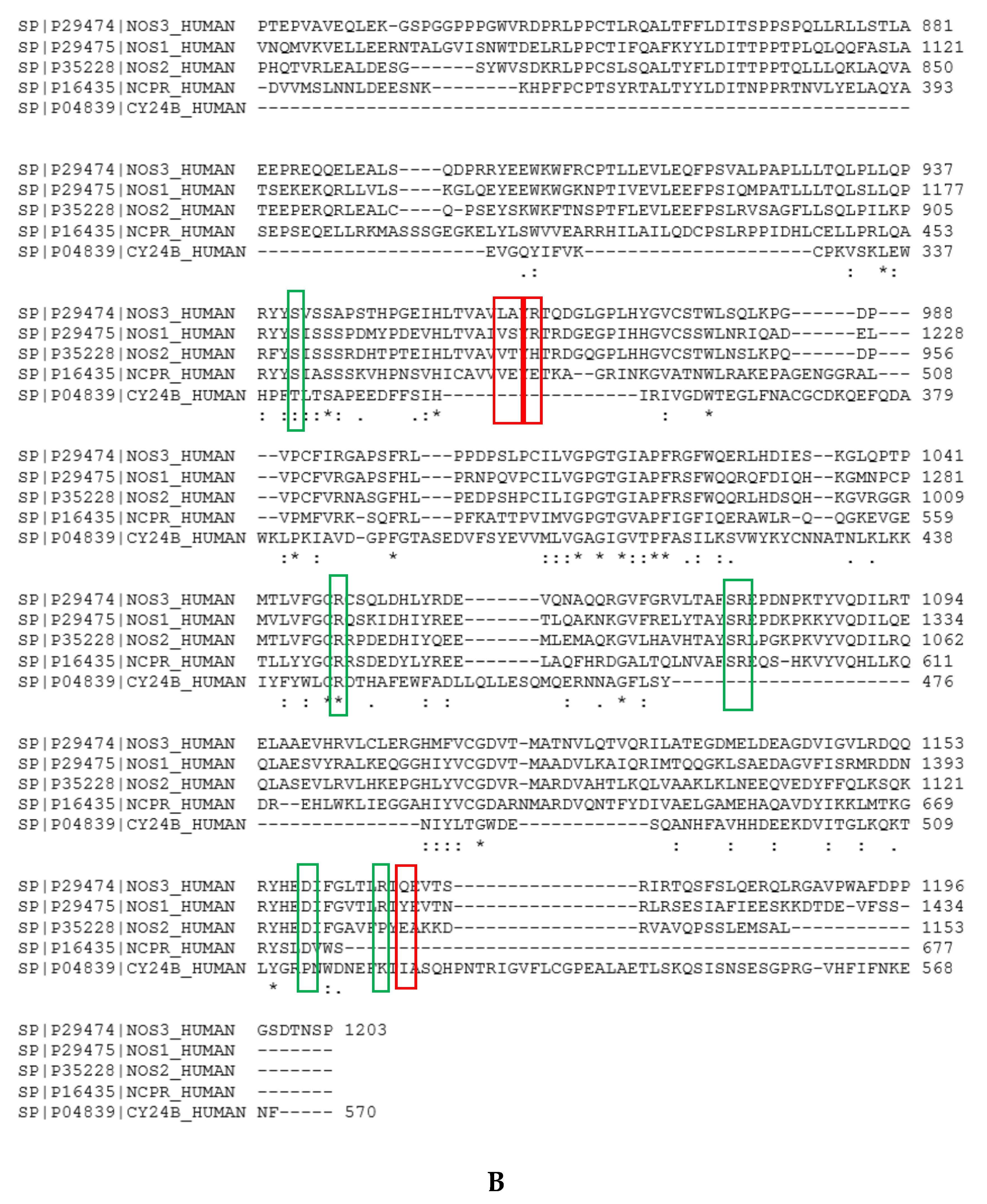
- Specificity for eNOS introduced by the hook 2. Here we target eNOS by exploiting sequence variations between the NADPH site of eNOS vs that of nNOS. Visual inspection of the sequence alignment (Figure A6) shows white or pale blue areas corresponding to variable residues, highlighting an available chemical space for implementing specificity for eNOS vs nNOS at this C-terminal domain. We choose to target A961, R963 and Q1167 of eNOS. By generating a more extended sequence alignment of human eNOS, nNOS, iNOS, P450 reductase, and NOX2, we can see that A961 with its nearby L960 and Q1167 were unique to eNOS and R963 was only shared with nNOS among nNOS, iNOS, P450red, NOX2. This suggested that by properly targeting variable residues, an expected selectivity should be achievable, and we used that information to design NT3_5h. Additionally, R1165, which only shared by eNOS and nNOS, was also recognized by the 3’ carboxylate of NT3_5 and NT3_5h. Indeed, the extended hook2 of NT3_5h contributed to multiple interactions with eNOS C-terminal and, overall, NT3_5h was well buried in its binding site (Figure 5). Its interactions with the variable residues were relatively stable over the trajectories. In contrast, hook1 of NT3_5 targeted a conserved arginine as R776 and its interactions with A961, R963 and Q1177 were relatively unstable (Figure 5B,C). The contribution of hook 2 to the NT3_5h interaction energy was at least 24%, based on its specific interaction energy with the variable residues A961, R963, and Q1167. Synthesis of this compound is ongoing for further experimental validation.
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A








References
- Forstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anavi, S.; Tirosh, O. iNOS as a metabolic enzyme under stress conditions. Free Radic. Biol. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, J.L.; Martin, K.C.; Lawrence, B.P. Novel cellular targets of AhR underlie alterations in neutrophilic inflammation and inducible nitric oxide synthase expression during influenza virus infection. J. Immunol. 2013, 190, 659–668. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Nakamura, T.; Lipton, S.A. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol. Neurobiol. 2010, 41, 55–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, R.B. Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res. 2009, 42, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Balligand, J.L.; Feron, O.; Dessy, C. eNOS activation by physical forces: From short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 2009, 89, 481–534. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Pappas, P.J.; Hobson, R.W., 2nd; Boric, M.P.; Sessa, W.C.; Duran, W.N. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 2006, 574, 275–281. [Google Scholar] [CrossRef]
- Forstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Heeschen, C.; Aicher, A.; Ziebart, T.; Honold, J.; Urbich, C.; Rossig, L.; Koehl, U.; Koyanagi, M.; Mohamed, A.; et al. Ex vivo pretreatment of bone marrow mononuclear cells with endothelial NO synthase enhancer AVE9488 enhances their functional activity for cell therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 14537–14541. [Google Scholar] [CrossRef] [Green Version]
- Morigaki, K.; Mizutani, K.; Kanemura, E.; Tatsu, Y.; Yumoto, N.; Imaishi, H. Photoregulation of cytochrome P450 activity by using caged compound. Anal. Chem. 2012, 84, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L.; Cohen, B.E.; Brubaker, M.; Mesecar, A.D.; Koshland, D.E., Jr. Millisecond Laue structures of an enzyme-product complex using photocaged substrate analogs. Nat. Struct. Biol. 1998, 5, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Santolini, J.; Adak, S.; Curran, C.M.; Stuehr, D.J. A kinetic simulation model that describes catalysis and regulation in nitric-oxide synthase. J. Biol. Chem. 2001, 276, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkmann, N.; Martasek, P.; Roman, L.J.; Xu, X.P.; Page, C.; Swift, M.; Hanein, D.; Masters, B.S. Holoenzyme structures of endothelial nitric oxide synthase—An allosteric role for calmodulin in pivoting the FMN domain for electron transfer. J. Struct. Biol. 2014, 188, 46–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaumont, E.; Lambry, J.C.; Blanchard-Desce, M.; Martasek, P.; Panda, S.P.; van Faassen, E.E.; Brochon, J.C.; Deprez, E.; Slama-Schwok, A. NO formation by neuronal NO-synthase can be controlled by ultrafast electron injection from a nanotrigger. ChemBioChem 2009, 10, 690–701. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, E.; Lambry, J.C.; Gautier, C.; Robin, A.C.; Gmouh, S.; Berka, V.; Tsai, A.L.; Blanchard-Desce, M.; Slama-Schwok, A. Synchronous photoinitiation of endothelial NO synthase activity by a nanotrigger targeted at its NADPH site. J. Am. Chem. Soc. 2007, 129, 2178–2186. [Google Scholar] [CrossRef]
- Beaumont, E.; Lambry, J.C.; Robin, A.C.; Martasek, P.; Blanchard-Desce, M.; Slama-Schwok, A. Two photon-induced electron injection from a nanotrigger in native endothelial NO-synthase. ChemPhysChem 2008, 9, 2325–2331. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Bogliotti, N.; Chennoufi, R.; Henry, E.; Tauc, P.; Salas, E.; Roman, L.J.; Slama-Schwok, A.; Deprez, E.; Xie, J. Convergent synthesis and properties of photoactivable NADPH mimics targeting nitric oxide synthases. Org. Biomol. Chem. 2016, 14, 9519–9532. [Google Scholar] [CrossRef]
- Chennoufi, R.; Cabrie, A.; Nguyen, N.H.; Bogliotti, N.; Simon, F.; Cinquin, B.; Tauc, P.; Boucher, J.L.; Slama-Schwok, A.; Xie, J.; et al. Light-induced formation of NO in endothelial cells by photoactivatable NADPH analogues targeting nitric-oxide synthase. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1127–1137. [Google Scholar] [CrossRef]
- Carugo, O.; Argos, P. NADP-dependent enzymes. I: Conserved stereochemistry of cofactor binding. Proteins 1997, 28, 10–28. [Google Scholar] [CrossRef]
- Carugo, O.; Argos, P. NADP-dependent enzymes. II: Evolution of the mono- and dinucleotide binding domains. Proteins 1997, 28, 29–40. [Google Scholar] [CrossRef]
- Garcin, E.D.; Bruns, C.M.; Lloyd, S.J.; Hosfield, D.J.; Tiso, M.; Gachhui, R.; Stuehr, D.J.; Tainer, J.A.; Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 2004, 279, 37918–37927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambry, J.C.; Beaumont, E.; Tarus, B.; Blanchard-Desce, M.; Slama-Schwok, A. Selective probing of a NADPH site controlled light-induced enzymatic catalysis. J. Mol. Recognit. 2010, 23, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Chennoufi, R.; Bogliotti, N.; Deprez, E.; Slama-Schwok, A.; Xie, J. Unpublished work. 2019.
- Moali, C.; Boucher, J.L.; Sari, M.A.; Stuehr, D.J.; Mansuy, D. Substrate specificity of NO synthases: Detailed comparison of L-arginine, homo-L-arginine, their N omega-hydroxy derivatives, and N omega-hydroxynor-L-arginine. Biochemistry 1998, 37, 10453–10460. [Google Scholar] [CrossRef] [PubMed]
- Roman, L.J.; Sheta, E.A.; Martasek, P.; Gross, S.S.; Liu, Q.; Masters, B.S. High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 8428–8432. [Google Scholar] [CrossRef] [Green Version]
- Thierry, E.; Lebourgeois, S.; Simon, F.; Delelis, O.; Deprez, E. Probing Resistance Mutations in Retroviral Integrases by Direct Measurement of Dolutegravir Fluorescence. Sci. Rep. 2017, 7, 14067. [Google Scholar] [CrossRef] [Green Version]
- Carayon, K.; Leh, H.; Henry, E.; Simon, F.; Mouscadet, J.F.; Deprez, E. A cooperative and specific DNA-binding mode of HIV-1 integrase depends on the nature of the metallic cofactor and involves the zinc-containing N-terminal domain. Nucleic Acids Res. 2010, 38, 3692–3708. [Google Scholar] [CrossRef] [Green Version]
- Lazareno, S.; Birdsall, N.J. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br. J. Pharmacol. 1993, 109, 1110–1119. [Google Scholar] [CrossRef]
- Deprez, E.E.A. Unpublished work. 2019.
- Bresnahan, C.G.; Reinhardt, C.R.; Bartholow, T.G.; Rumpel, J.P.; North, M.; Bhattacharyya, S. Effect of stacking interactions on the thermodynamics and kinetics of lumiflavin: A study with improved density functionals and density functional tight-binding protocol. J. Phys. Chem. A 2015, 119, 172–182. [Google Scholar] [CrossRef]
- Baraldi, I.; Momicchioli, F.; Ponterinin, G.; Tatikolov, A.S.; Vanossi, D. Photoisimerization of simple merocyanines: A theoretical and experimental comparison with polyenes and symetrical cyanines. Phys. Chem. Chem. Phys. 2003, 5, 979–987. [Google Scholar] [CrossRef]
- El-Gezawy, H.; Rettig, W.; Lapouyade, R. Model studies of spectral and photophysical characteristics of donor-acceptor polyenes. Chem. Phys. Lett. 2005, 401, 140–148. [Google Scholar] [CrossRef]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, K.; Vlachojannis, G.J.; Koyanagi, M.; Boeckel, J.N.; Urbich, C.; Farcas, R.; Bonig, H.; Marquez, V.E.; Zeiher, A.M.; Dimmeler, S. Epigenetic regulation of endothelial lineage committed genes in pro-angiogenic hematopoietic and endothelial progenitor cells. Circ. Res. 2011, 109, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Apparent Kd eNOS (µM) | Hook1 Stabilization in eNOS | NO Formed upon Laser Excitation | Apparent Kd/Kd nNOS (µM) | Flavin Reduction hv | Hook1 Stabilization in nNOS |
|---|---|---|---|---|---|---|
| NT1 | 7 ± 3 | NA | + | ND | + | NA |
| [16] | [15,16] | |||||
| NT2_5 | 16 ± 3 a | NA | + | 19.5 + 1.1 a [19] | + e | NA |
| [19] | [19] | 1.3 ± 0.2 b | ||||
| 4.0 ± 0.7 c | ||||||
| NT3_5 | 10.7 ± 0.7 a | ×1.5 | + f | 11.0 ± 1.5 a | + e | × 1.8 |
| 0.65 ± 0.15 b | ||||||
| 1.7 ± 0.5 c | ||||||
| 0.18 ± 0.04 d | ||||||
| Mean (b,d): 0.36 ± 11 | 4–15 | |||||
| NT2_7 | 24.1 ± 1.9 a | NA | − | 30 ± 4 a | ND | NA |
| [19] | [19] | [19] | ||||
| NT3_7 | 15.7 ± 0.6 a | ×1.5 | − f | 11.5 ± 1,5 a | ND | ×2.6 |
| 0.90 ± 0.12 b | ||||||
| NT2_3 | 14.7 ± 1.1 a | NA | − | 21.5 ± 0,7 a | ND | NA |
| [19] | [19] | [19] | ||||
| NT 3_3 | 5 ± 1 & 21 ± 0.2 a | ×2.9 | − f | 5 ± 1 & 28 ± 2 a | − e | ×4.3 |
| (bi-dose response) | 0.4 ± 0.1b | |||||
| 0.78 ± 0.15 c | ||||||
| 0.10 ± 0.03 d | ||||||
| Mean (b,d): 0.25 ± 0.08 |
| NT Moities | NT1 | NT3_3 | NT3_5 | NT3_7 | NT3_5h |
|---|---|---|---|---|---|
| Phosphate or carboxylate | R1165, R1049, R1079 | 2’: R1165, S1078, R1079 3’: R963, L1164, R1165 | 3’: R1165, S1078, R1049 | 2’: R1079 | 3’: R1165, S1078 R1049, P1014 G1015 |
| Hook, Ethyl ou Methyl | A958 | R776, R963 Amide: Triazole | R776, R1165, V959, T1166, Q1167, Amide: T1163 | R776, R1079 | R776, T956 A961, R963 Q1167 |
| Adénine | L1164, R1165 C1048, Y1087 | P1014, M1119, L1164, R1165 | A958, P1014, T1016, G1017, Triazole | T1087, M1119 | R776, T956, A958 P1014, A1019, Triazole |
| Chromophore | Aniline: FAD, D1158, F1160, C1114, | Aniline: FAD, D1158, F1160, C1114 | Aniline: FAD, D1158, F1160 A958, T1163, C1114 | Aniline: FAD, D1158, C1114 | FAD L960, C1114 D1158 (sometimes) |
| Chromophore | Planar | not planar | not planar | Planar, | Planar |
| Planarity/stacking to FAD | Stacking of the terminal aniline to FAD | Stacking of the terminal aniline to FAD | Stacking of the terminal aniline to FAD | bad stacking with FAD | Stacking of the terminal aniline to FAD |
| Distance (Ǻ) Ser941-FAD | 4.7 ± 0.8 | 5.1 ± 0.4 | 5.1 ± 0.2 | 4.9 ± 0.5 | 5.5 ± 0.5 |
| H-bond to FAD | S941, T956, V957, A958 | A941, T956, V957, T956, T1016 | T956, A958, T1016, G1017 | S941, T956, V957, A958, T1016 | S941, T956, V957, A958 |
| Compound | Total Interaction Energy (Kcal/mol) | Total Interaction Energy (KJ/mol) | Coulomb KJ/mol | LJ KJ/mol | Hook Total KJ/mol (% stabilization) | Hook Targeting eNOS Residues KJ/mol (%stabilization) |
|---|---|---|---|---|---|---|
| NT1 | −155 ± 4 | −647 ± 18 | −343 ± 11 | −304 ± 7 | −19 ± 2 | |
| NT3_3 | −194 ± 3 | −812 ± 12 | −462 ± 3 | −350 ± 9 | −169 ±7 (21%) | |
| NT3_5 | −150 ± 2 | −627 ± 18 | −277 ± 7 | −350 ± 11 | −185±15 (29.5%) | −40 ± 19 (6.4%) |
| NT3_7 | −136 ± 2 | −568 ± 10 | −248 ± 7 | −320 ± 3 | −164 ± 18 (28.8%) | |
| NT3_5h | −163 ± 4 | −681 ± 15 | −279 ± 12 | −402 ± 3 | −225 ± 12 (33%) | −165 ± 35 (24.4%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dilly, S.; Roman, L.J.; Bogliotti, N.; Xie, J.; Deprez, E.; Slama-Schwok, A. Design of Light-Sensitive Triggers for Endothelial NO-Synthase Activation. Antioxidants 2020, 9, 89. https://doi.org/10.3390/antiox9020089
Dilly S, Roman LJ, Bogliotti N, Xie J, Deprez E, Slama-Schwok A. Design of Light-Sensitive Triggers for Endothelial NO-Synthase Activation. Antioxidants. 2020; 9(2):89. https://doi.org/10.3390/antiox9020089
Chicago/Turabian StyleDilly, Sébastien, Linda J. Roman, Nicolas Bogliotti, Juan Xie, Eric Deprez, and Anny Slama-Schwok. 2020. "Design of Light-Sensitive Triggers for Endothelial NO-Synthase Activation" Antioxidants 9, no. 2: 89. https://doi.org/10.3390/antiox9020089
APA StyleDilly, S., Roman, L. J., Bogliotti, N., Xie, J., Deprez, E., & Slama-Schwok, A. (2020). Design of Light-Sensitive Triggers for Endothelial NO-Synthase Activation. Antioxidants, 9(2), 89. https://doi.org/10.3390/antiox9020089