Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Abnormal Intracellular Signaling and Oxidative Stress Characterizes CLL
2.1. Oxidation Affects the Dynamic Coordination between Kinases and Phosphatases in Cancer Cells
2.2. Imbalance between Kinases and Phosphatases Activities: The Key Mechanism Underlying CLL Development
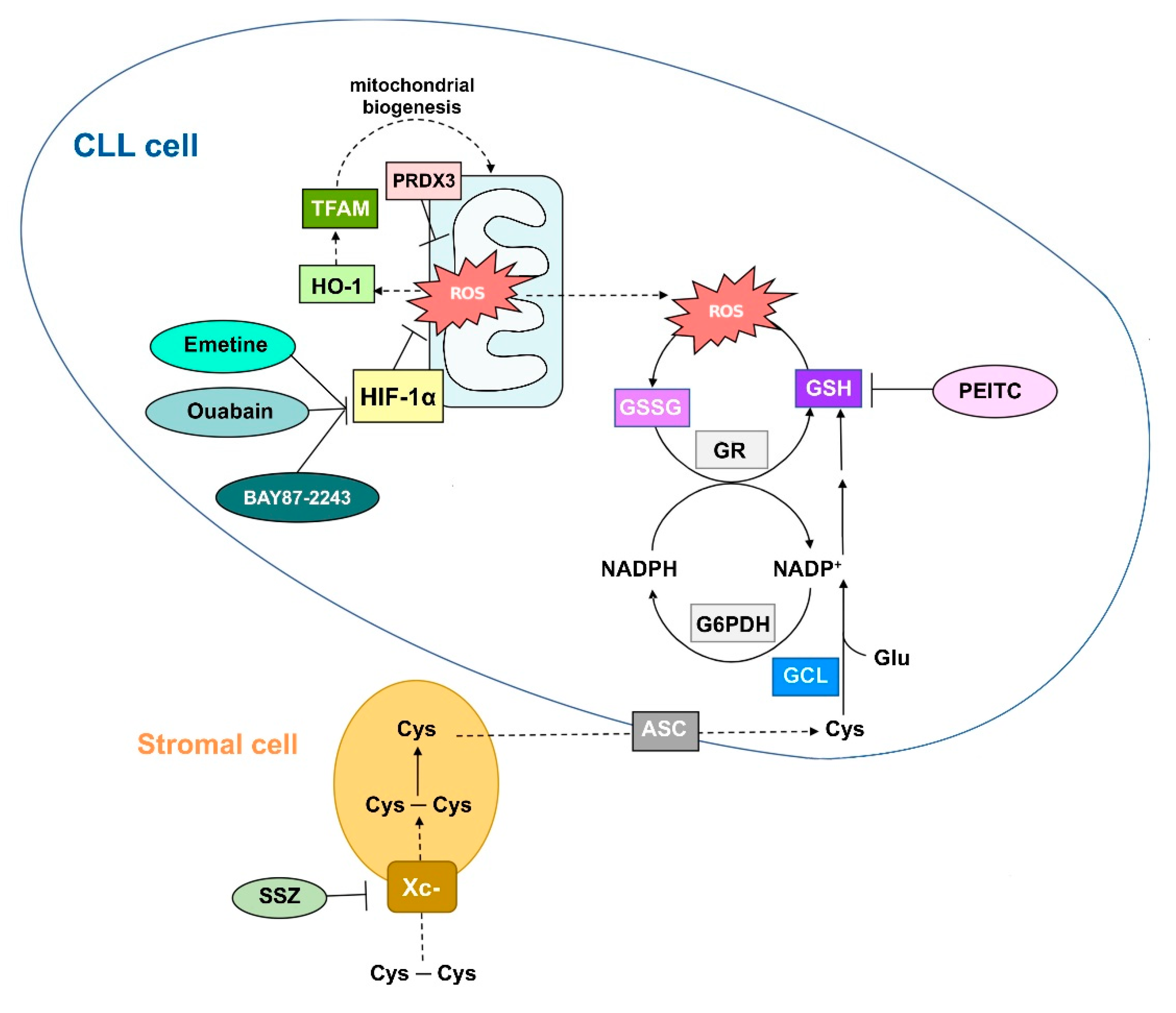
2.2.1. CLL Cells Exhibit High ROS-Buffering Capacity to Preserve Survival
2.2.2. Potentiation of Antioxidant Defense Systems and CLL
3. Erythropoiesis: The Interplay between Oxidation and Signal Transduction Pathways
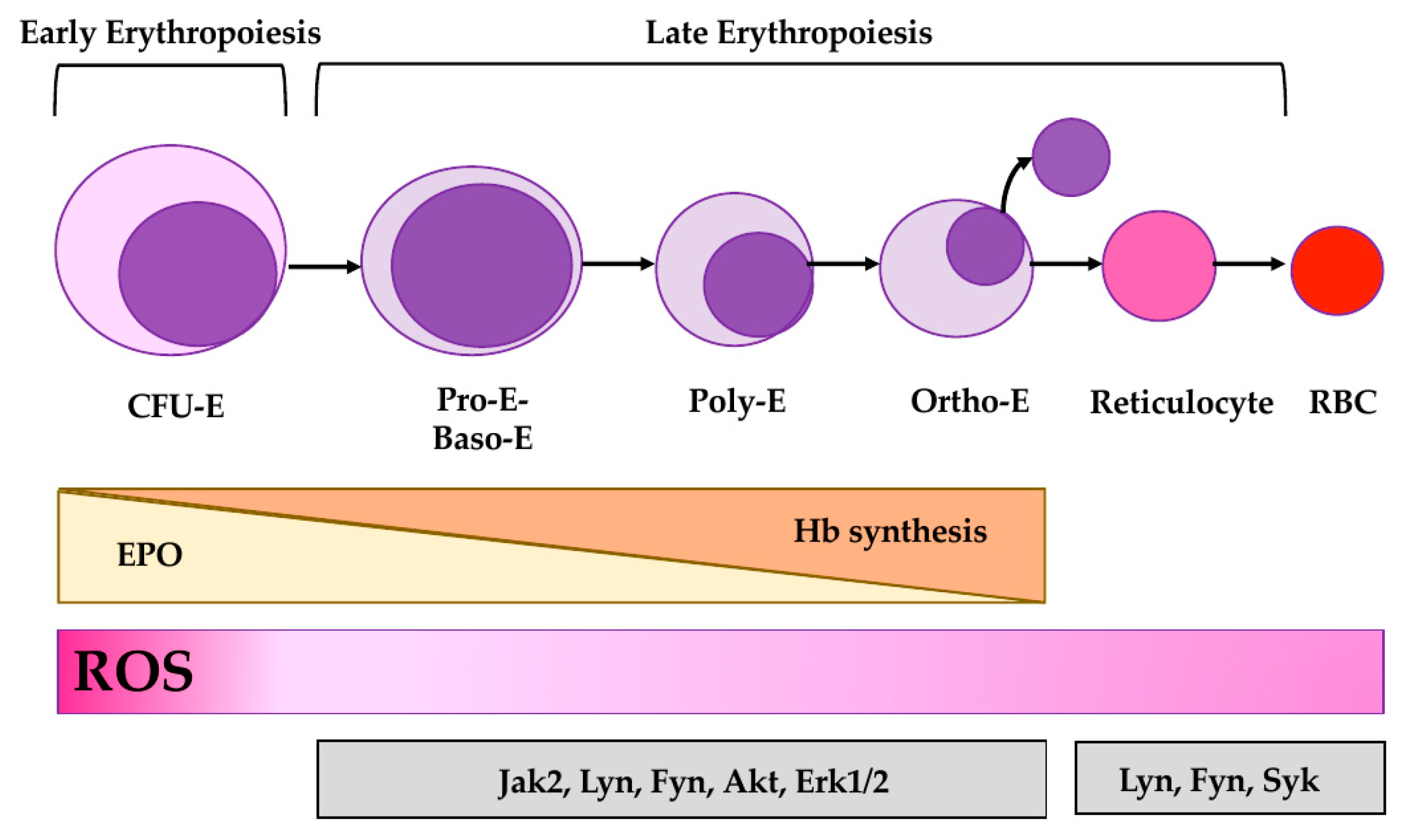
3.1. Redox Interfaces with Cell Signaling in Erythropoiesis
3.2. Antioxidant System(s) and Stress Erythropoiesis
4. Redox Affects Signaling Pathways Involved in Red Cell Homeostasis
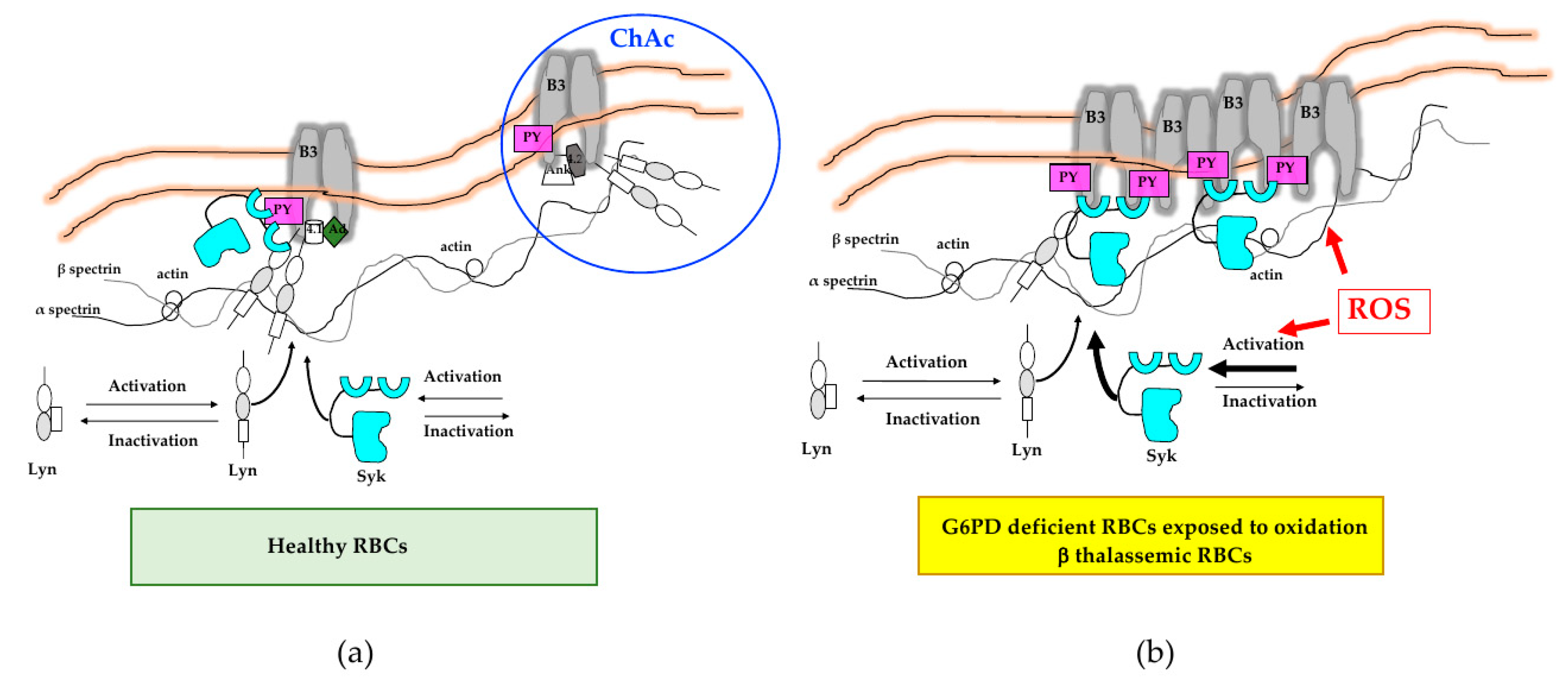
4.1. Oxidation and Signal Transduction Pathways in Red Cell Pathologies
4.2. Functional Interplay between Oxidation and Cell Signaling Towards Regulation of Erythrocyte Volume
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Russell, E.G.; Cotter, T.G. New Insight into the Role of Reactive Oxygen Species (ROS) in Cellular Signal-Transduction Processes. Int. Rev. Cell Mol. Biol. 2015, 319, 221–254. [Google Scholar] [PubMed]
- Woolley, J.F.; Stanicka, J.; Cotter, T.G. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem. Sci. 2013, 38, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef]
- Juarez, J.C.; Manuia, M.; Burnett, M.E.; Betancourt, O.; Boivin, B.; Shaw, D.E.; Tonks, N.K.; Mazar, A.P.; Donate, F. Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 7147–7152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Franceschi, L.; Villa-Moruzzi, E.; Biondani, A.; Siciliano, A.; Brugnara, C.; Alper, S.L.; Lowell, C.A.; Berton, G. Regulation of K-Cl cotransport by protein phosphatase 1alpha in mouse erythrocytes. Pflug. Arch. 2006, 451, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Mallozzi, C.; De Franceschi, L.; Brugnara, C.; Di Stasi, A.M. Protein phosphatase 1alpha is tyrosine-phosphorylated and inactivated by peroxynitrite in erythrocytes through the src family kinase fgr. Free Radic. Biol. Med. 2005, 38, 1625–1636. [Google Scholar] [CrossRef] [PubMed]
- Heppner, D.E.; Dustin, C.M.; Liao, C.; Hristova, M.; Veith, C.; Little, A.C.; Ahlers, B.A.; White, S.L.; Deng, B.; Lam, Y.W.; et al. Direct cysteine sulfenylation drives activation of the Src kinase. Nat. Commun. 2018, 9, 4522. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E. Src kinases are important regulators of mitochondrial functions. Int. J. Biochem. Cell. Biol. 2013, 45, 90–98. [Google Scholar] [CrossRef]
- Giannoni, E.; Buricchi, F.; Raugei, G.; Ramponi, G.; Chiarugi, P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol. Cell. Biol. 2005, 25, 6391–6403. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Fumagalli, L.; Olivieri, O.; Corrocher, R.; Lowell, C.A.; Berton, G. Deficiency of Src family kinases Fgr and Hck results in activation of erythrocyte K/Cl cotransport. J. Clin. Investig. 1997, 99, 220–227. [Google Scholar] [CrossRef]
- Beneduce, E.; Matte, A.; De Falco, L.; Mbiandjeu, S.; Chiabrando, D.; Tolosano, E.; Federti, E.; Petrillo, S.; Mohandas, N.; Siciliano, A.; et al. Fyn kinase is a novel modulator of erythropoietin signaling and stress erythropoiesis. Am. J. Hematol. 2019, 94, 10–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zonta, F.; Pagano, M.A.; Trentin, L.; Tibaldi, E.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Pavan, V.; Ribaudo, G.; et al. Lyn sustains oncogenic signaling in chronic lymphocytic leukemia by strengthening SET-mediated inhibition of PP2A. Blood 2015, 125, 3747–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibaldi, E.; Pagano, M.A.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Pavan, V.; Bordin, L.; Visentin, A.; et al. Targeted activation of the SHP-1/PP2A signaling axis elicits apoptosis of chronic lymphocytic leukemia cells. Haematologica 2017, 102, 1401–1412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lupo, F.; Tibaldi, E.; Matte, A.; Sharma, A.K.; Brunati, A.M.; Alper, S.L.; Zancanaro, C.; Benati, D.; Siciliano, A.; Bertoldi, M.; et al. A new molecular link between defective autophagy and erythroid abnormalities in chorea-acanthocytosis. Blood 2016, 128, 2976–2987. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Tomelleri, C.; Matte, A.; Brunati, A.M.; Bovee-Geurts, P.H.; Bertoldi, M.; Lasonder, E.; Tibaldi, E.; Danek, A.; Walker, R.H.; et al. Erythrocyte membrane changes of chorea-acanthocytosis are the result of altered Lyn kinase activity. Blood 2011, 118, 5652–5663. [Google Scholar] [CrossRef] [Green Version]
- Merciris, P.; Hardy-Dessources, M.D.; Giraud, F. Deoxygenation of sickle cells stimulates Syk tyrosine kinase and inhibits a membrane tyrosine phosphatase. Blood 2001, 98, 3121–3127. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferru, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Matte, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 Erythrocyte Membrane Protein Acts as Redox Stress Sensor Leading to Its Phosphorylation by p (72) Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- Siciliano, A.; Turrini, F.; Bertoldi, M.; Matte, A.; Pantaleo, A.; Olivieri, O.; De Franceschi, L. Deoxygenation affects tyrosine phosphoproteome of red cell membrane from patients with sickle cell disease. Blood Cells Mol. Dis. 2010, 44, 233–242. [Google Scholar] [CrossRef]
- Pantaleo, A.; De Franceschi, L.; Ferru, E.; Vono, R.; Turrini, F. Current knowledge about the functional roles of phosphorylative changes of membrane proteins in normal and diseased red cells. J. Proteom. 2010, 73, 445–455. [Google Scholar] [CrossRef]
- Iolascon, A.; De Falco, L.; Borgese, F.; Esposito, M.R.; Avvisati, R.A.; Izzo, P.; Piscopo, C.; Guizouarn, H.; Biondani, A.; Pantaleo, A.; et al. A novel erythroid anion exchange variant (Gly796Arg) of hereditary stomatocytosis associated with dyserythropoiesis. Haematologica 2009, 94, 1049–1059. [Google Scholar] [CrossRef]
- Pantaleo, A.; Ferru, E.; Giribaldi, G.; Mannu, F.; Carta, F.; Matte, A.; de Franceschi, L.; Turrini, F. Oxidized and poorly glycosylated band 3 is selectively phosphorylated by Syk kinase to form large membrane clusters in normal and G6PD-deficient red blood cells. Biochem. J. 2009, 418, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 17008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Kipps, T.J. The pathogenesis of chronic lymphocytic leukemia. Annu. Rev. Pathol. 2014, 9, 103–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta. 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Burger, J.A.; Kipps, T.J. Chemokine receptors and stromal cells in the homing and homeostasis of chronic lymphocytic leukemia B cells. Leuk. Lymphoma 2002, 43, 461–466. [Google Scholar] [CrossRef]
- Roy Chowdhury, S.; Banerji, V. Targeting Mitochondrial Bioenergetics as a Therapeutic Strategy for Chronic Lymphocytic Leukemia. Oxid. Med. Cell. Longev. 2018, 2018, 2426712. [Google Scholar] [CrossRef]
- Yosifov, D.Y.; Idler, I.; Bhattacharya, N.; Reichenzeller, M.; Close, V.; Ezerina, D.; Scheffold, A.; Jebaraj, B.M.C.; Kugler, S.; Bloehdorn, J.; et al. Oxidative stress as candidate therapeutic target to overcome microenvironmental protection of CLL. Leukemia 2020, 34, 115–127. [Google Scholar] [CrossRef]
- Park, S.Y.; Matte, A.; Jung, Y.; Ryu, J.; Wilson, A.B.; Han, E.Y.A.; Liu, M.; Carbone, C.; Melisi, D.; Nagasawa, T.; et al. Pathologic angiogenesis in the bone marrow of humanized sickle cell mice is reversible by blood transfusion. Blood 2020. [Google Scholar] [CrossRef]
- Crippa, S.; Rossella, V.; Aprile, A.; Silvestri, L.; Rivis, S.; Scaramuzza, S.; Pirroni, S.; Avanzini, M.A.; Basso-Ricci, L.; Hernandez, R.J.; et al. Bone marrow stromal cells from beta-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Invest. 2019, 129, 1566–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rund, D.; Rachmilewitz, E. Beta-thalassemia. N. Engl. J. Med. 2005, 353, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and beta-thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell. Longev. 2013, 2013, 985210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, A.; Federti, E.; Winter, M.; Koerner, A.; Harmeier, A.; Mazer, N.; Tomka, T.; Di Paolo, M.L.; Defalco, L.; Andolfo, I.; et al. Bitopertin, a selective oral GLYT1 inhibitor, improves anemia in a mouse model of beta-thalassemia. JCI Insight 2019, 4, e130111. [Google Scholar] [CrossRef] [PubMed]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef]
- Matte, A.; Cappellini, M.D.; Iolascon, A.; Enrica, F.; De Franceschi, L. Emerging drugs in randomized controlled trials for sickle cell disease: Are we on the brink of a new era in research and treatment? Expert Opin. Investig. Drugs 2020, 29, 23–31. [Google Scholar] [CrossRef]
- Matte, A.; Zorzi, F.; Mazzi, F.; Federti, E.; Olivieri, O.; De Franceschi, L. New Therapeutic Options for the Treatment of Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019002. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Fiorelli, G. Glucose-6-phosphate dehydrogenase deficiency. Lancet 2008, 371, 64–74. [Google Scholar] [CrossRef]
- Luzzatto, L.; Arese, P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N. Engl. J. Med. 2018, 378, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef]
- Yang, J.; Groen, A.; Lemeer, S.; Jans, A.; Slijper, M.; Roe, S.M.; den Hertog, J.; Barford, D. Reversible oxidation of the membrane distal domain of receptor PTPalpha is mediated by a cyclic sulfenamide. Biochemistry 2007, 46, 709–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmeen, A.; Barford, D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signal 2005, 7, 560–577. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Taddei, M.L.; Chiarugi, P. Src redox regulation: Again in the front line. Free Radic. Biol. Med. 2010, 49, 516–527. [Google Scholar] [CrossRef]
- Senga, T.; Miyazaki, K.; Machida, K.; Iwata, H.; Matsuda, S.; Nakashima, I.; Hamaguchi, M. Clustered cysteine residues in the kinase domain of v-Src: Critical role for protein stability, cell transformation and sensitivity to herbimycin A. Oncogene 2000, 19, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Hori, T.; Sato, N.; Sugie, K.; Kawakami, T.; Yodoi, J. Redox regulation of a src family protein tyrosine kinase p56lck in T cells. Oncogene 1993, 8, 3133–3139. [Google Scholar] [PubMed]
- Dustin, C.M.; Heppner, D.E.; Lin, M.J.; van der Vliet, A. Redox regulation of tyrosine kinase signalling: More than meets the eye. J. Biochem. 2020, 167, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Gianni, D.; Bohl, B.; Courtneidge, S.A.; Bokoch, G.M. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol. Biol. Cell. 2008, 19, 2984–2994. [Google Scholar] [CrossRef] [Green Version]
- Herishanu, Y.; Katz, B.Z.; Lipsky, A.; Wiestner, A. Biology of chronic lymphocytic leukemia in different microenvironments: Clinical and therapeutic implications. Hematol. Oncol. Clin. North. Am. 2013, 27, 173–206. [Google Scholar] [CrossRef] [Green Version]
- Herndon, T.M.; Chen, S.S.; Saba, N.S.; Valdez, J.; Emson, C.; Gatmaitan, M.; Tian, X.; Hughes, T.E.; Sun, C.; Arthur, D.C.; et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia 2017, 31, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, F.K.; Krysov, S.; Davies, A.J.; Steele, A.J.; Packham, G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood 2011, 118, 4313–4320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, G.; Montserrat, E. Critical molecular pathways in CLL therapy. Mol. Med. 2018, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Garaud, S.; Taher, T.E.; Debant, M.; Burgos, M.; Melayah, S.; Berthou, C.; Parikh, K.; Pers, J.O.; Luque-Paz, D.; Chiocchia, G.; et al. CD5 expression promotes IL-10 production through activation of the MAPK/Erk pathway and upregulation of TRPC1 channels in B lymphocytes. Cell. Mol. Immunol. 2018, 15, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Meijers, R.W.J.; Muggen, A.F.; Leon, L.G.; de Bie, M.; van Dongen, J.J.M.; Hendriks, R.W.; Langerak, A.W. Responsiveness of chronic lymphocytic leukemia cells to B-cell receptor stimulation is associated with low expression of regulatory molecules of the nuclear factor-kappaB pathway. Haematologica 2020, 105, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Trentin, L.; Frasson, M.; Donella-Deana, A.; Frezzato, F.; Pagano, M.A.; Tibaldi, E.; Gattazzo, C.; Zambello, R.; Semenzato, G.; Brunati, A.M. Geldanamycin-induced Lyn dissociation from aberrant Hsp90-stabilized cytosolic complex is an early event in apoptotic mechanisms in B-chronic lymphocytic leukemia. Blood 2008, 112, 4665–4674. [Google Scholar] [CrossRef]
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, S.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Scharenberg, A.M.; Tsukada, S.; Bolen, J.B.; Kinet, J.P.; Kurosaki, T. Paired immunoglobulin-like receptor B (PIR-B) inhibits BCR-induced activation of Syk and Btk by SHP-1. Oncogene 1999, 18, 2291–2297. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Harder, K.W.; Huntington, N.D.; Hibbs, M.L.; Tarlinton, D.M. Lyn tyrosine kinase: Accentuating the positive and the negative. Immunity 2005, 22, 9–18. [Google Scholar]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.J.; Fan, L.; Wang, L.; Xu, J.; Zhang, R.; Tian, T.; Li, J.Y.; Xu, W. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget 2015, 6, 1276–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tibaldi, E.; Brunati, A.M.; Zonta, F.; Frezzato, F.; Gattazzo, C.; Zambello, R.; Gringeri, E.; Semenzato, G.; Pagano, M.A.; Trentin, L. Lyn-mediated SHP-1 recruitment to CD5 contributes to resistance to apoptosis of B-cell chronic lymphocytic leukemia cells. Leukemia 2011, 25, 1768–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagano, M.A.; Tibaldi, E.; Molino, P.; Frezzato, F.; Trimarco, V.; Facco, M.; Zagotto, G.; Ribaudo, G.; Leanza, L.; Peruzzo, R.; et al. Mitochondrial apoptosis is induced by Alkoxy phenyl-1-propanone derivatives through PP2A-mediated dephosphorylation of Bad and Foxo3A in CLL. Leukemia 2019, 33, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Farinelli, G.; Galli, M.; Aiuti, A.; Sitia, R. AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukoc. Biol. 2016, 100, 1071–1079. [Google Scholar] [CrossRef]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.Y.; Tang, M.; Suzuki, M.; Gunasekara, C.; Anbe, Y.; Hiraoka, Y.; Liu, J.; Grasberger, H.; Ohkita, M.; Matsumura, Y.; et al. Essential Role of NADPH Oxidase-Dependent Production of Reactive Oxygen Species in Maintenance of Sustained B Cell Receptor Signaling and B Cell Proliferation. J. Immunol. 2019, 202, 2546–2557. [Google Scholar] [CrossRef]
- Collins, R.J.; Verschuer, L.A.; Harmon, B.V.; Prentice, R.L.; Pope, J.H.; Kerr, J.F. Spontaneous programmed death (apoptosis) of B-chronic lymphocytic leukaemia cells following their culture in vitro. Br. J. Haematol. 1989, 71, 343–350. [Google Scholar] [CrossRef]
- Munk Pedersen, I.; Reed, J. Microenvironmental interactions and survival of CLL B-cells. Leuk. Lymphoma 2004, 45, 2365–2372. [Google Scholar] [CrossRef]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1alpha protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Chan, D.A.; Sutphin, P.D.; Yen, S.E.; Giaccia, A.J. Coordinate regulation of the oxygen-dependent degradation domains of hypoxia-inducible factor 1 alpha. Mol. Cell. Biol. 2005, 25, 6415–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordonez, A.; Corral-Escariz, M.; Soro, I.; Lopez-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarczuk, K.; Sasi, B.K.; Gobessi, S.; Innocenti, I.; Pozzato, G.; Laurenti, L.; Efremov, D.G. BCR signaling inhibitors differ in their ability to overcome Mcl-1-mediated resistance of CLL B cells to ABT-199. Blood 2016, 127, 3192–3201. [Google Scholar] [CrossRef]
- Burger, J.A.; O’Brien, S. Evolution of CLL treatment - from chemoimmunotherapy to targeted and individualized therapy. Nat. Rev. Clin. Oncol. 2018, 15, 510–527. [Google Scholar] [CrossRef]
- Schneider, C.; Steinbrecher, D.; Stilgenbauer, S. Targeted therapy in CLL: Changing the treatment paradigm. Oncotarget 2019, 10, 4002–4003. [Google Scholar] [CrossRef]
- Karur, V.G.; Lowell, C.A.; Besmer, P.; Agosti, V.; Wojchowski, D.M. Lyn kinase promotes erythroblast expansion and late-stage development. Blood 2006, 108, 1524–1532. [Google Scholar] [CrossRef]
- Ingley, E.; McCarthy, D.J.; Pore, J.R.; Sarna, M.K.; Adenan, A.S.; Wright, M.J.; Erber, W.; Tilbrook, P.A.; Klinken, S.P. Lyn deficiency reduces GATA-1, EKLF and STAT5, and induces extramedullary stress erythropoiesis. Oncogene 2005, 24, 336–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingley, E. Integrating novel signaling pathways involved in erythropoiesis. IUBMB Life 2012, 64, 402–410. [Google Scholar] [CrossRef]
- Slavova-Azmanova, N.S.; Kucera, N.; Satiaputra, J.; Stone, L.; Magno, A.; Maxwell, M.J.; Quilici, C.; Erber, W.; Klinken, S.P.; Hibbs, M.L.; et al. Gain-of-function Lyn induces anemia: Appropriate Lyn activity is essential for normal erythropoiesis and Epo receptor signaling. Blood 2013, 122, 262–271. [Google Scholar] [CrossRef] [Green Version]
- Tilbrook, P.A.; Palmer, G.A.; Bittorf, T.; McCarthy, D.J.; Wright, M.J.; Sarna, M.K.; Linnekin, D.; Cull, V.S.; Williams, J.H.; Ingley, E.; et al. Maturation of erythroid cells and erythroleukemia development are affected by the kinase activity of Lyn. Cancer Res. 2001, 61, 2453–2458. [Google Scholar]
- Franco, S.S.; De Falco, L.; Ghaffari, S.; Brugnara, C.; Sinclair, D.A.; Matte, A.; Iolascon, A.; Mohandas, N.; Bertoldi, M.; An, X.; et al. Resveratrol accelerates erythroid maturation by activation of FoxO3 and ameliorates anemia in beta-thalassemic mice. Haematologica 2014, 99, 267–275. [Google Scholar] [CrossRef]
- Matte, A.; De Falco, L.; Federti, E.; Cozzi, A.; Iolascon, A.; Levi, S.; Mohandas, N.; Zamo, A.; Bruno, M.; Lebouef, C.; et al. Peroxiredoxin-2: A Novel Regulator of Iron Homeostasis in Ineffective Erythropoiesis. Antioxid. Redox Signal. 2018, 28, 1–14. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, L.; Bertoldi, M.; De Falco, L.; Santos Franco, S.; Ronzoni, L.; Turrini, F.; Colancecco, A.; Camaschella, C.; Cappellini, M.D.; Iolascon, A. Oxidative stress modulates heme synthesis and induces peroxiredoxin-2 as a novel cytoprotective response in beta-thalassemic erythropoiesis. Haematologica 2011, 96, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Camprecios, G.; Rimmele, P.; Liang, R.; Yalcin, S.; Mungamuri, S.K.; Barminko, J.; D’Escamard, V.; Baron, M.H.; Brugnara, C.; et al. FOXO3-mTOR metabolic cooperation in the regulation of erythroid cell maturation and homeostasis. Am. J. Hematol. 2014, 89, 954–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, A.; De Franceschi, L. Oxidation and erythropoiesis. Curr. Opin. Hematol. 2019, 26, 145–151. [Google Scholar] [CrossRef]
- Matte, A.; De Falco, L.; Iolascon, A.; Mohandas, N.; An, X.; Siciliano, A.; Leboeuf, C.; Janin, A.; Bruno, M.; Choi, S.Y.; et al. The Interplay Between Peroxiredoxin-2 and Nuclear Factor-Erythroid 2 Is Important in Limiting Oxidative Mediated Dysfunction in beta-Thalassemic Erythropoiesis. Antioxid. Redox Signal. 2015, 23, 1284–1297. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.S.; Lopez, M.F.; Fleming, M.D.; Rivera, A.; Martin, F.M.; Welsh, M.L.; Boyd, A.; Doctrow, S.R.; Burakoff, S.J. SOD2-deficiency anemia: Protein oxidation and altered protein expression reveal targets of damage, stress response, and antioxidant responsiveness. Blood 2004, 104, 2565–2573. [Google Scholar] [CrossRef]
- Pullen, N.A.; Barnstein, B.O.; Falanga, Y.T.; Wang, Z.; Suzuki, R.; Tamang, T.D.; Khurana, M.C.; Harry, E.A.; Draber, P.; Bunting, K.D.; et al. Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J. Biol. Chem. 2012, 287, 2045–2054. [Google Scholar] [CrossRef] [Green Version]
- Tsygankov, A.Y.; Spana, C.; Rowley, R.B.; Penhallow, R.C.; Burkhardt, A.L.; Bolen, J.B. Activation-dependent tyrosine phosphorylation of Fyn-associated proteins in T lymphocytes. J. Biol. Chem. 1994, 269, 7792–7800. [Google Scholar]
- Laurenzana, I.; Caivano, A.; Trino, S.; De Luca, L.; La Rocca, F.; Simeon, V.; Tintori, C.; D’Alessio, F.; Teramo, A.; Zambello, R.; et al. A Pyrazolo[3,4-d]pyrimidine compound inhibits Fyn phosphorylation and induces apoptosis in natural killer cell leukemia. Oncotarget 2016, 7, 65171–65184. [Google Scholar] [CrossRef]
- Casu, C.; Presti, V.L.; Oikonomidou, P.R.; Melchiori, L.; Abdulmalik, O.; Ramos, P.; Rivella, S. Short-term administration of JAK2 inhibitors reduces splenomegaly in mouse models of beta-thalassemia intermedia and major. Haematologica 2018, 103, e46–e49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wu, K.; Xiao, X.; Liao, J.; Hu, Q.; Chen, H.; Liu, J.; An, X. Autophagy as a regulatory component of erythropoiesis. Int. J. Mol. Sci. 2015, 16, 4083–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.; Ghadami, E.; Dadkhah, T.; Akhavan-Niaki, H. PI3k/AKT signaling pathway: Erythropoiesis and beyond. J. Cell. Physiol. 2019, 234, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Shi, X.; Sheng, K.; Han, G.; Li, W.; Zhao, Q.; Jiang, B.; Feng, J.; Li, J.; Gu, Y. PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol. Med. Rep. 2019, 19, 783–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Macias-Garcia, A.; Velazquez, J.; Paltrinieri, E.; Kaufman, R.J.; Chen, J.J. HRI coordinates translation by eIF2alphaP and mTORC1 to mitigate ineffective erythropoiesis in mice during iron deficiency. Blood 2018, 131, 450–461. [Google Scholar] [CrossRef]
- Guo, F.; Zhang, S.; Grogg, M.; Cancelas, J.A.; Varney, M.E.; Starczynowski, D.T.; Du, W.; Yang, J.Q.; Liu, W.; Thomas, G.; et al. Mouse gene targeting reveals an essential role of mTOR in hematopoietic stem cell engraftment and hematopoiesis. Haematologica 2013, 98, 1353–1358. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, Y.; Marden, J.J.; Banfi, B.; Engelhardt, J.F. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem. J. 2008, 411, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Mallozzi, C.; Di Stasi, A.M.; Minetti, M. Activation of src tyrosine kinases by peroxynitrite. FEBS Lett. 1999, 456, 201–206. [Google Scholar] [CrossRef] [Green Version]
- Wannatung, T.; Lithanatudom, P.; Leecharoenkiat, A.; Svasti, S.; Fucharoen, S.; Smith, D.R. Increased erythropoiesis of beta-thalassaemia/Hb E proerythroblasts is mediated by high basal levels of ERK1/2 activation. Br. J. Haematol. 2009, 146, 557–568. [Google Scholar] [CrossRef]
- Rhodes, M.M.; Kopsombut, P.; Bondurant, M.C.; Price, J.O.; Koury, M.J. Bcl-x(L) prevents apoptosis of late-stage erythroblasts but does not mediate the antiapoptotic effect of erythropoietin. Blood 2005, 106, 1857–1863. [Google Scholar] [CrossRef] [Green Version]
- Lannutti, B.J.; Shim, M.H.; Blake, N.; Reems, J.A.; Drachman, J.G. Identification and activation of Src family kinases in primary megakaryocytes. Exp. Hematol. 2003, 31, 1268–1274. [Google Scholar] [CrossRef]
- Zennadi, R. MEK1/2 as a Therapeutic Target in Sickle Cell Disease. Int. J. Blood Res. Disord. 2019, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Zennadi, R.; Whalen, E.J.; Soderblom, E.J.; Alexander, S.C.; Thompson, J.W.; Dubois, L.G.; Moseley, M.A.; Telen, M.J. Erythrocyte plasma membrane-bound ERK1/2 activation promotes ICAM-4-mediated sickle red cell adhesion to endothelium. Blood 2012, 119, 1217–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altamura, S.; Vegi, N.M.; Hoppe, P.S.; Schroeder, T.; Aichler, M.; Walch, A.; Okreglicka, K.; Hultner, L.; Schneider, M.; Ladinig, C.; et al. Glutathione peroxidase 4 and vitamin E control reticulocyte maturation, stress erythropoiesis and iron homeostasis. Haematologica 2019, 105, 937–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matte, A.; Pantaleo, A.; Ferru, E.; Turrini, F.; Bertoldi, M.; Lupo, F.; Siciliano, A.; Ho Zoon, C.; De Franceschi, L. The novel role of peroxiredoxin-2 in red cell membrane protein homeostasis and senescence. Free Radic. Biol. Med. 2014, 76, 80–88. [Google Scholar] [CrossRef]
- de Franceschi, L.; Turrini, F.; Honczarenko, M.; Ayi, K.; Rivera, A.; Fleming, M.D.; Law, T.; Mannu, F.; Kuypers, F.A.; Bast, A.; et al. In vivo reduction of erythrocyte oxidant stress in a murine model of beta-thalassemia. Haematologica 2004, 89, 1287–1298. [Google Scholar]
- De Franceschi, L.; Biondani, A.; Carta, F.; Turrini, F.; Laudanna, C.; Deana, R.; Brunati, A.M.; Turretta, L.; Iolascon, A.; Perrotta, S.; et al. PTPepsilon has a critical role in signaling transduction pathways and phosphoprotein network topology in red cells. Proteomics 2008, 8, 4695–4708. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Bosman, G.J.; Mohandas, N. Abnormal red cell features associated with hereditary neurodegenerative disorders: The neuroacanthocytosis syndromes. Curr. Opin. Hematol. 2014, 21, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef] [Green Version]
- Bordin, L.; Fiore, C.; Bragadin, M.; Brunati, A.M.; Clari, G. Regulation of membrane band 3 Tyr-phosphorylation by proteolysis of p72(Syk) and possible involvement in senescence process. Acta Biochim. Biophys. Sin. (Shanghai) 2009, 41, 846–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordin, L.; Ion-Popa, F.; Brunati, A.M.; Clari, G.; Low, P.S. Effector-induced Syk-mediated phosphorylation in human erythrocytes. Biochim. Biophys. Acta 2005, 1745, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Brunati, A.M.; Bordin, L.; Clari, G.; James, P.; Quadroni, M.; Baritono, E.; Pinna, L.A.; Donella-Deana, A. Sequential phosphorylation of protein band 3 by Syk and Lyn tyrosine kinases in intact human erythrocytes: Identification of primary and secondary phosphorylation sites. Blood 2000, 96, 1550–1557. [Google Scholar] [CrossRef] [PubMed]
- Ferru, E.; Pantaleo, A.; Carta, F.; Mannu, F.; Khadjavi, A.; Gallo, V.; Ronzoni, L.; Graziadei, G.; Cappellini, M.D.; Turrini, F. Thalassemic erythrocytes release microparticles loaded with hemichromes by redox activation of p72Syk kinase. Haematologica 2014, 99, 570–578. [Google Scholar] [CrossRef] [PubMed]
- Shimo, H.; Arjunan, S.N.; Machiyama, H.; Nishino, T.; Suematsu, M.; Fujita, H.; Tomita, M.; Takahashi, K. Particle Simulation of Oxidation Induced Band 3 Clustering in Human Erythrocytes. PLoS Comput. Biol. 2015, 11, e1004210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantaleo, A.; Kesely, K.R.; Pau, M.C.; Tsamesidis, I.; Schwarzer, E.; Skorokhod, O.A.; Chien, H.D.; Ponzi, M.; Bertuccini, L.; Low, P.S.; et al. Syk inhibitors interfere with erythrocyte membrane modification during P falciparum growth and suppress parasite egress. Blood 2017, 130, 1031–1040. [Google Scholar] [CrossRef] [Green Version]
- Kesely, K.R.; Pantaleo, A.; Turrini, F.M.; Olupot-Olupot, P.; Low, P.S. Inhibition of an Erythrocyte Tyrosine Kinase with Imatinib Prevents Plasmodium falciparum Egress and Terminates Parasitemia. PLoS ONE 2016, 11, e0164895. [Google Scholar] [CrossRef]
- Brugnara, C.; De Franceschi, L.; Beuzard, Y. Erythrocyte-active agents and treatment of sickle cell disease. Semin. Hematol. 2001, 38, 324–332. [Google Scholar] [CrossRef]
- Merciris, P.; Claussen, W.J.; Joiner, C.H.; Giraud, F. Regulation of K-Cl cotransport by Syk and Src protein tyrosine kinases in deoxygenated sickle cells. Pflug. Arch. 2003, 446, 232–238. [Google Scholar] [CrossRef]
- Brugnara, C.; Bunn, H.F.; Tosteson, D.C. Ion content and transport and the regulation of volume in sickle cells. Ann. N. Y. Acad. Sci. 1989, 565, 96–103. [Google Scholar] [CrossRef]
- Shmukler, B.E.; Rivera, A.; Bhargava, P.; Nishimura, K.; Kim, E.H.; Hsu, A.; Wohlgemuth, J.G.; Morton, J.; Snyder, L.M.; De Franceschi, L.; et al. Genetic disruption of KCC cotransporters in a mouse model of thalassemia intermedia. Blood Cells Mol. Dis. 2020, 81, 102389. [Google Scholar] [CrossRef] [PubMed]
- McNaughton-Smith, G.A.; Burns, J.F.; Stocker, J.W.; Rigdon, G.C.; Creech, C.; Arrington, S.; Shelton, T.; de Franceschi, L. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 2008, 51, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Casula, S.; Shmukler, B.E.; Wilhelm, S.; Stuart-Tilley, A.K.; Su, W.; Chernova, M.N.; Brugnara, C.; Alper, S.L. A dominant negative mutant of the KCC1 K-Cl cotransporter: Both N- and C-terminal cytoplasmic domains are required for K-Cl cotransport activity. J. Biol. Chem. 2001, 276, 41870–41878. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tibaldi, E.; Federti, E.; Matte, A.; Iatcenko, I.; Wilson, A.B.; Riccardi, V.; Pagano, M.A.; De Franceschi, L. Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders. Antioxidants 2020, 9, 353. https://doi.org/10.3390/antiox9040353
Tibaldi E, Federti E, Matte A, Iatcenko I, Wilson AB, Riccardi V, Pagano MA, De Franceschi L. Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders. Antioxidants. 2020; 9(4):353. https://doi.org/10.3390/antiox9040353
Chicago/Turabian StyleTibaldi, Elena, Enrica Federti, Alessandro Matte, Iana Iatcenko, Anand B. Wilson, Veronica Riccardi, Mario Angelo Pagano, and Lucia De Franceschi. 2020. "Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders" Antioxidants 9, no. 4: 353. https://doi.org/10.3390/antiox9040353
APA StyleTibaldi, E., Federti, E., Matte, A., Iatcenko, I., Wilson, A. B., Riccardi, V., Pagano, M. A., & De Franceschi, L. (2020). Oxidation Impacts the Intracellular Signaling Machinery in Hematological Disorders. Antioxidants, 9(4), 353. https://doi.org/10.3390/antiox9040353