DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant

Abstract
:1. Introduction
2. Experimental
2.1. Cloning of Vaccination Expression Constructs and Molecular Mechanism Constructs
2.3. Mice and Vaccinations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | n | Total Immunizations | Route a | ImmunogenpOVA | Adjuvant pFliC(-gly) | Empty Vector pcDNA3.1/Zeo(+) | N3 Lipid |
|---|---|---|---|---|---|---|---|
| 1 | 7 | 2 | g.g. | 0.5 µg | - | 0.5 µg | - |
| 2 | 7 | 2 | g.g. | 0.5 µg | 0.1 µg | 0.4 µg | - |
| 3 | 8 | 2 | g.g. | 0.5 µg | 0.2 µg | 0.3 µg | - |
| 4 | 8 | 2 | g.g. | 0.5 µg | 0.5 µg | - | - |
| 5 | 7 | 2 | i.m. | 10 µg | - | 10 µg | - |
| 6 | 8 | 2 | i.m. | 10 µg | 2 µg | 8 µg | - |
| 7 | 8 | 2 | i.m. | 10 µg | 5 µg | 5 µg | - |
| 8 | 8 | 2 | i.m. | 10 µg | 10 µg | - | - |
| 9 | 8 | 2 | i.na. | 4 µg | - | 4 µg | - |
| 10 | 7 | 2 | i.na. | 4 µg | - | 4 µg | 1% |
| 11 | 8 | 2 | i.na. | 4 µg | 1 µg | 3 µg | 1% |
| 12 | 8 | 2 | i.na. | 4 µg | 2 µg | 2 µg | 1% |
| 13 | 7 | 2 | i.na. | 4 µg | 4 µg | - | 1% |
| Group | n | Total Immunizations | Priming | Boosting | |||
|---|---|---|---|---|---|---|---|
| ImmunogenP a | Adjuvant | EmptyVectorpcDNA3.1/Zeo(+) | ImmunogenB b | Adjuvant | |||
| 1 | 35 | 3 | pgp160(5 µg) pGagp24(5 µg) | None | - | rgp160/rp24gag (1 µg each) | None |
| 2 | 35 | 3 | pgp160(5 µg) pGagp24(5 µg) | N3 (1%) | - | rgp160/rp24gag (1 µg each) | L3B(2%) |
| 3 | 35 | 3 | pgp160(5 µg) pGagp24(5 µg) | pFliC(-gly) (5 µg) | - | rgp160/rp24gag (1 µg each) | L3B(2%) |
| 4 | 35 | 3 | pgp160(5 µg) pGagp24( 5µg) | N3(1%) + pFliC(gly) (5 µg) | - | rgp160/rp24gag (1 µg each) | N3(1%) + pFliC(gly) (5 µg) |
| 5 | 35 | 3 | pgp160(5 µg) pGagp24(5 µg) | None | 5 µg | rgp160/rp24gag (1 µg each) | N3(1%) + pcDNA3.1 (5 µg) |
| 6 | 35 | 3 | pgp160(5 µg) pGagp24(5 µg) | N3(1%) | 5 µg | rgp160/rp24gag (1 µg each) | N3(1%) + pcDNA3.1 (5 µg) |
| Saline | 30 | 3 | Saline | ||||
2.4. Antibody, Mucosal Cytokines and T Cell Analysis
2.5. HIV-1 Neutralization Assay
3. Results and Discussion
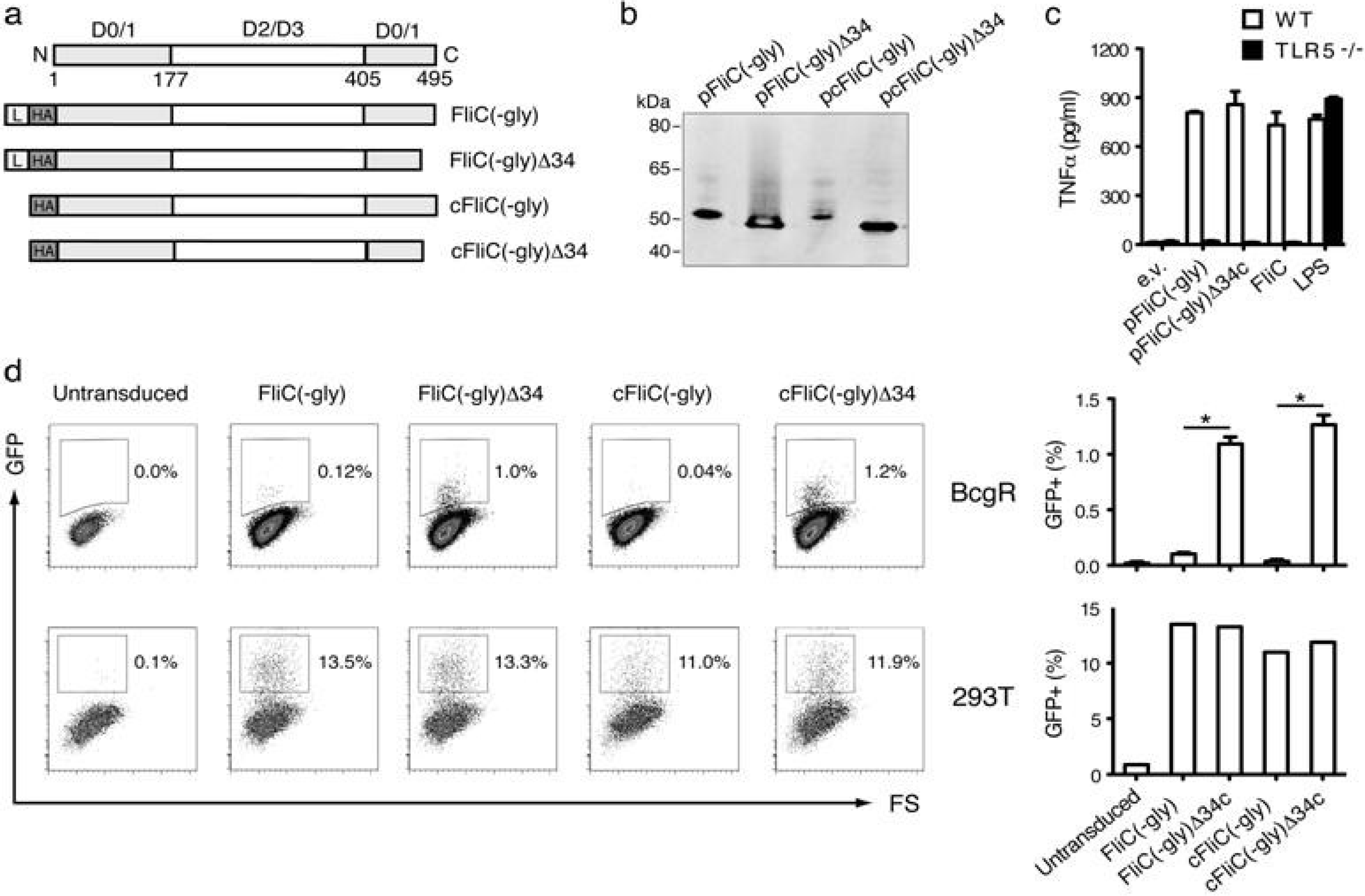
3.1. Construction of Secreted FliC Adjuvant
3.2. pOVA DNA Vaccinations; Timeline, Routes, and Dose
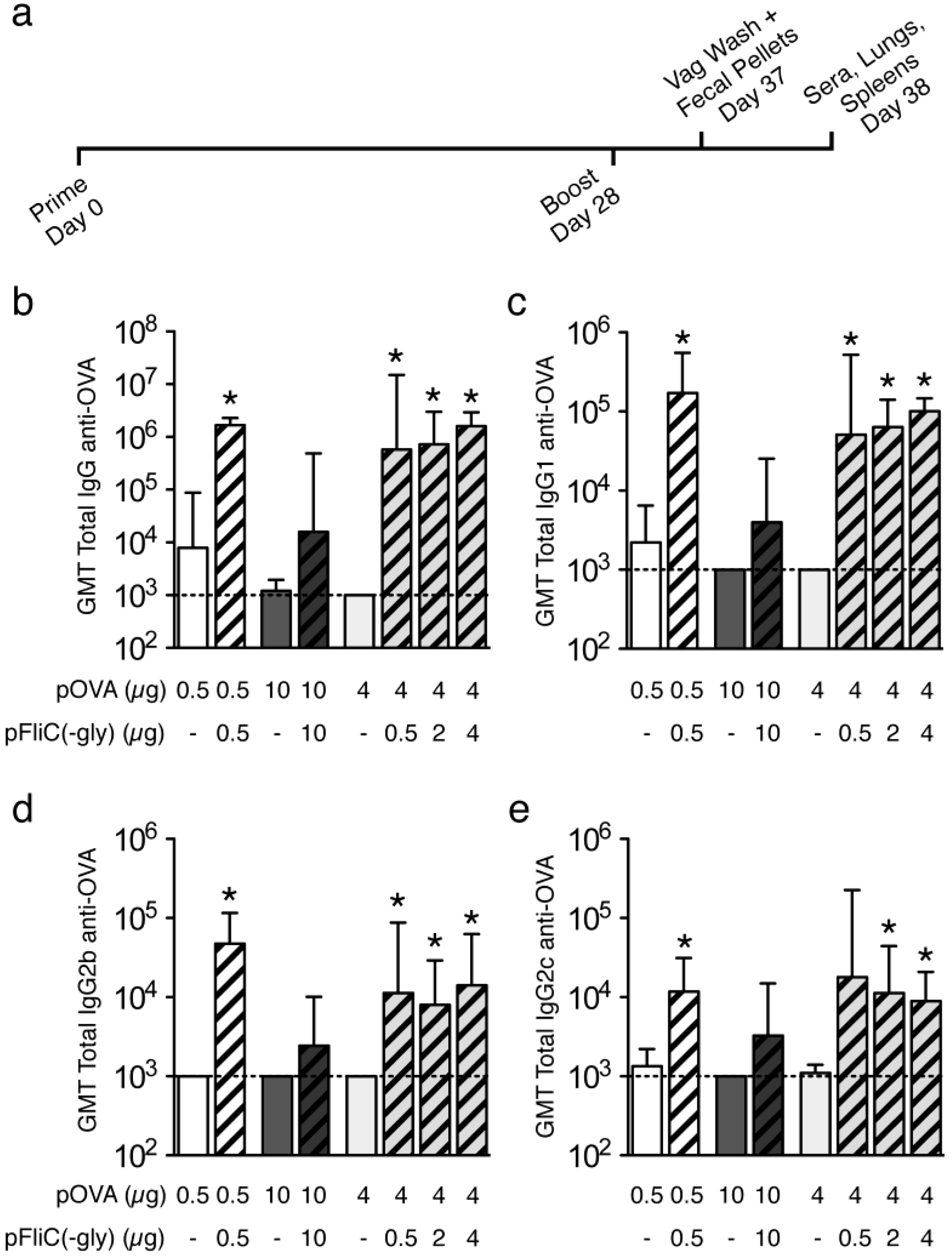
3.3. Antibody Immune Responses to pOVA DNA Vaccination



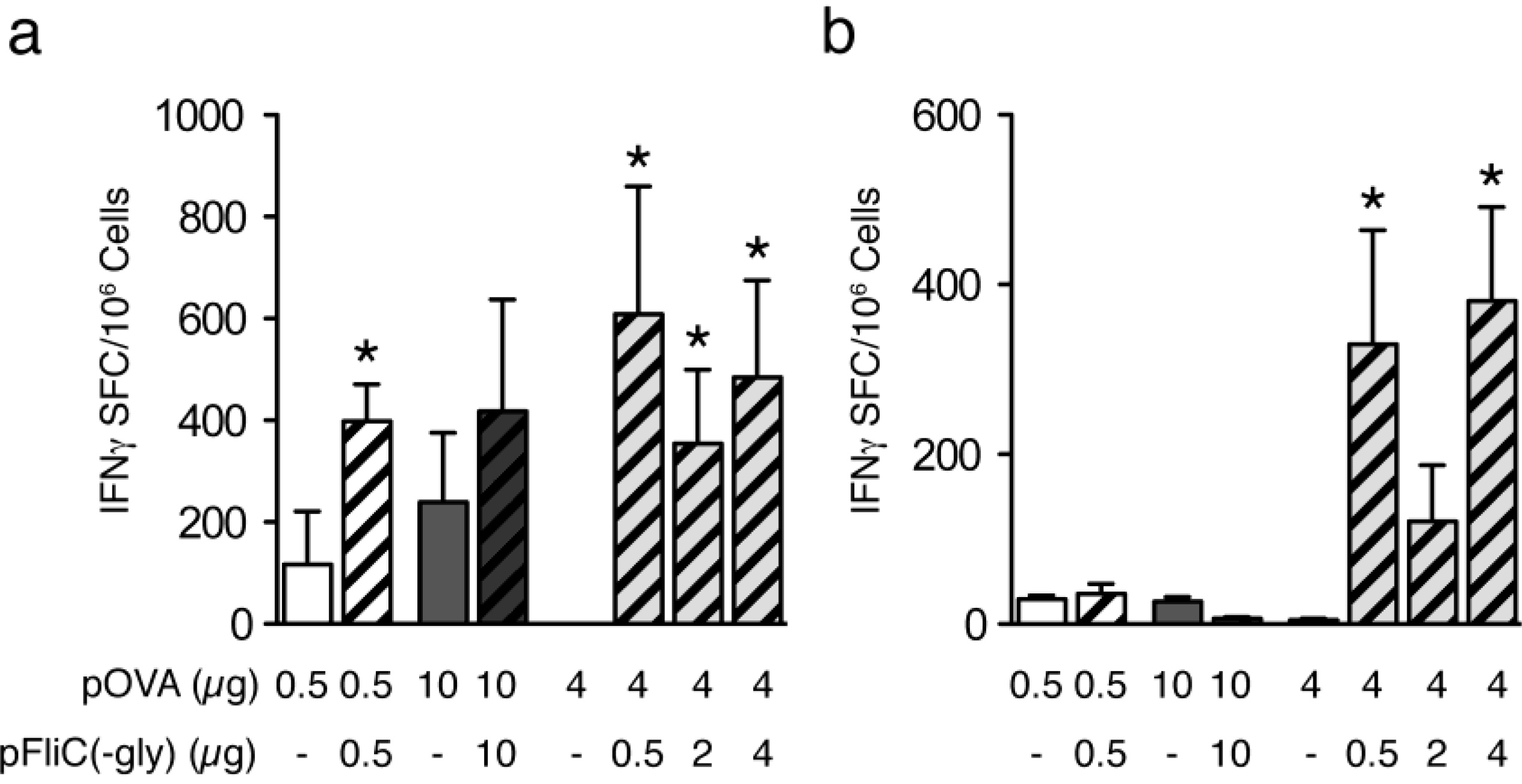
3.4. Cellular Immune Responses to pOVA DNA Vaccination

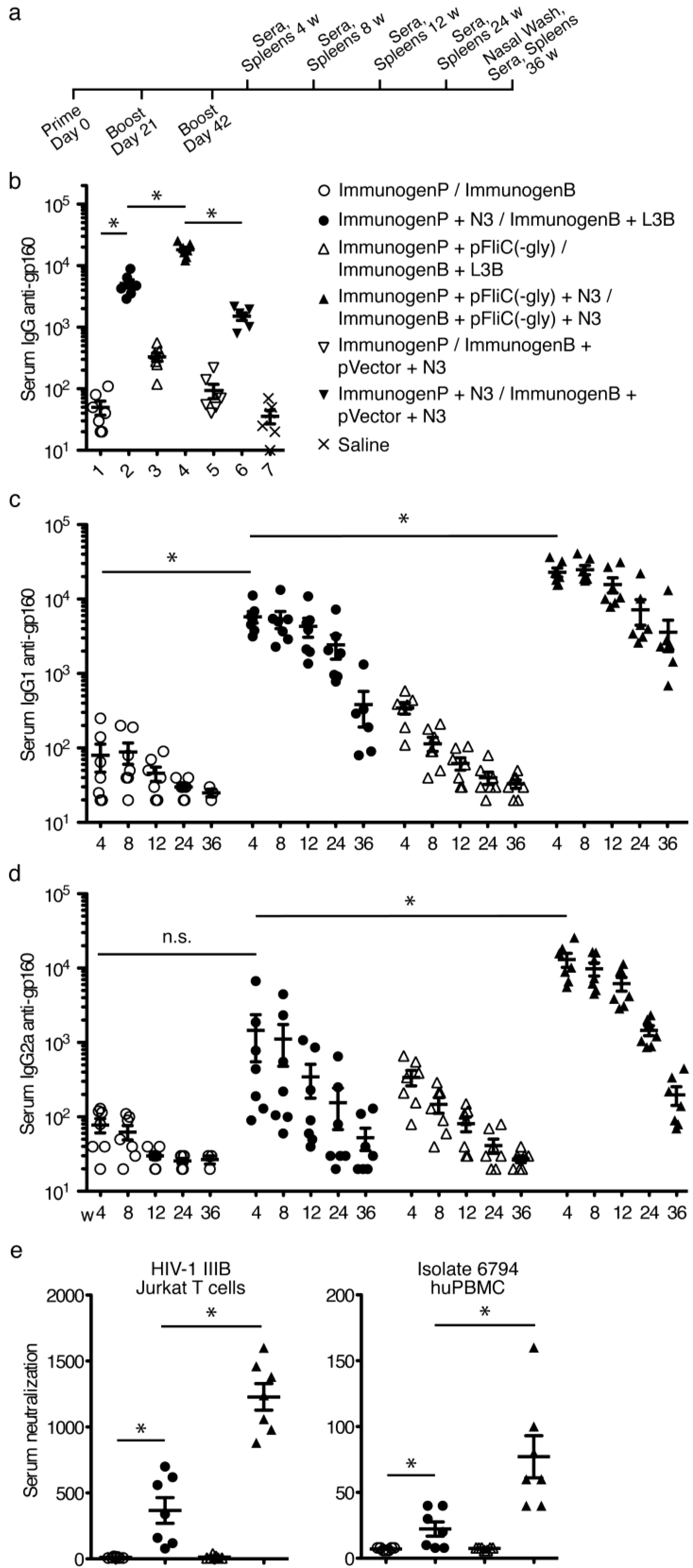
3.5. gp160 DNA and Protein Vaccinations; Timeline and Antibody Responses



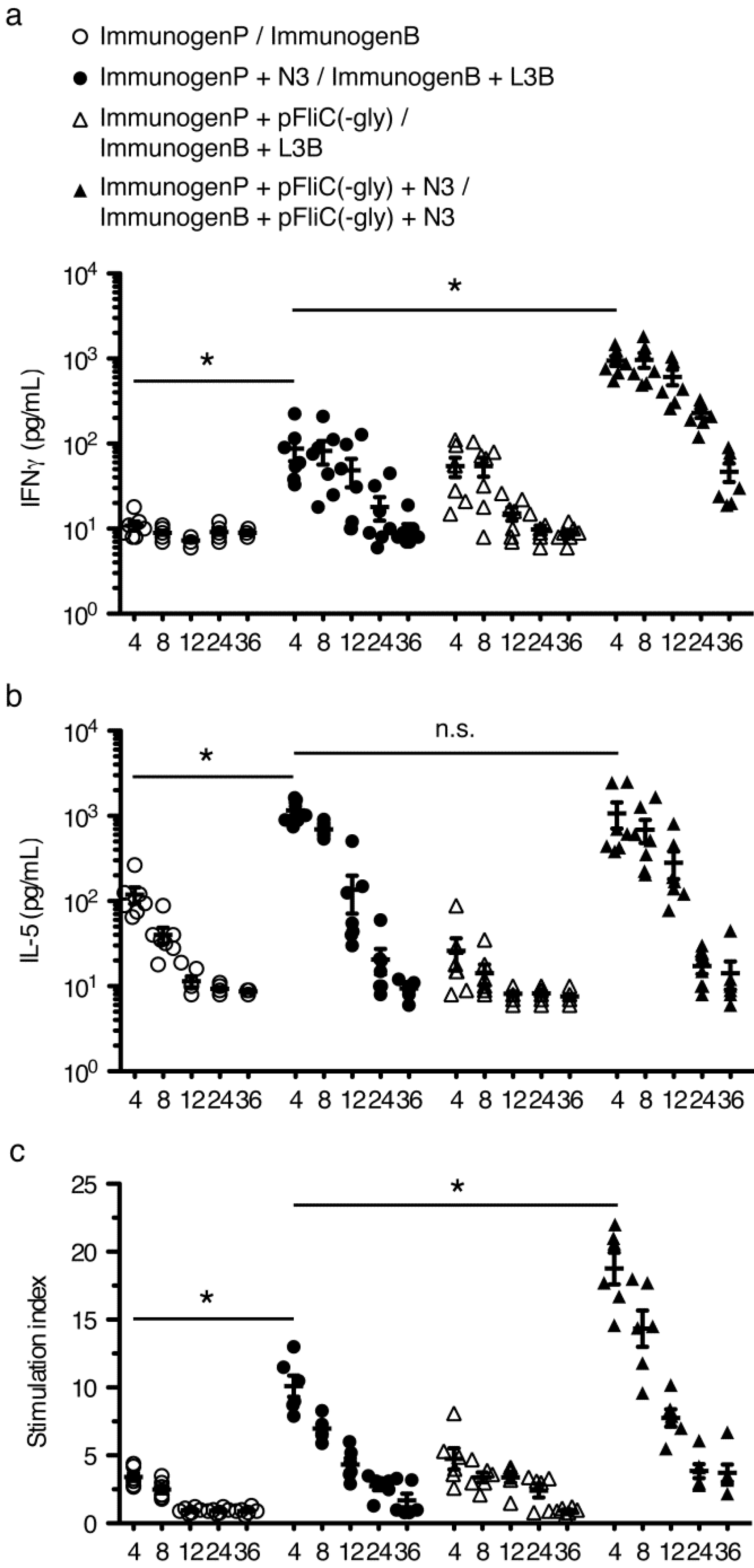
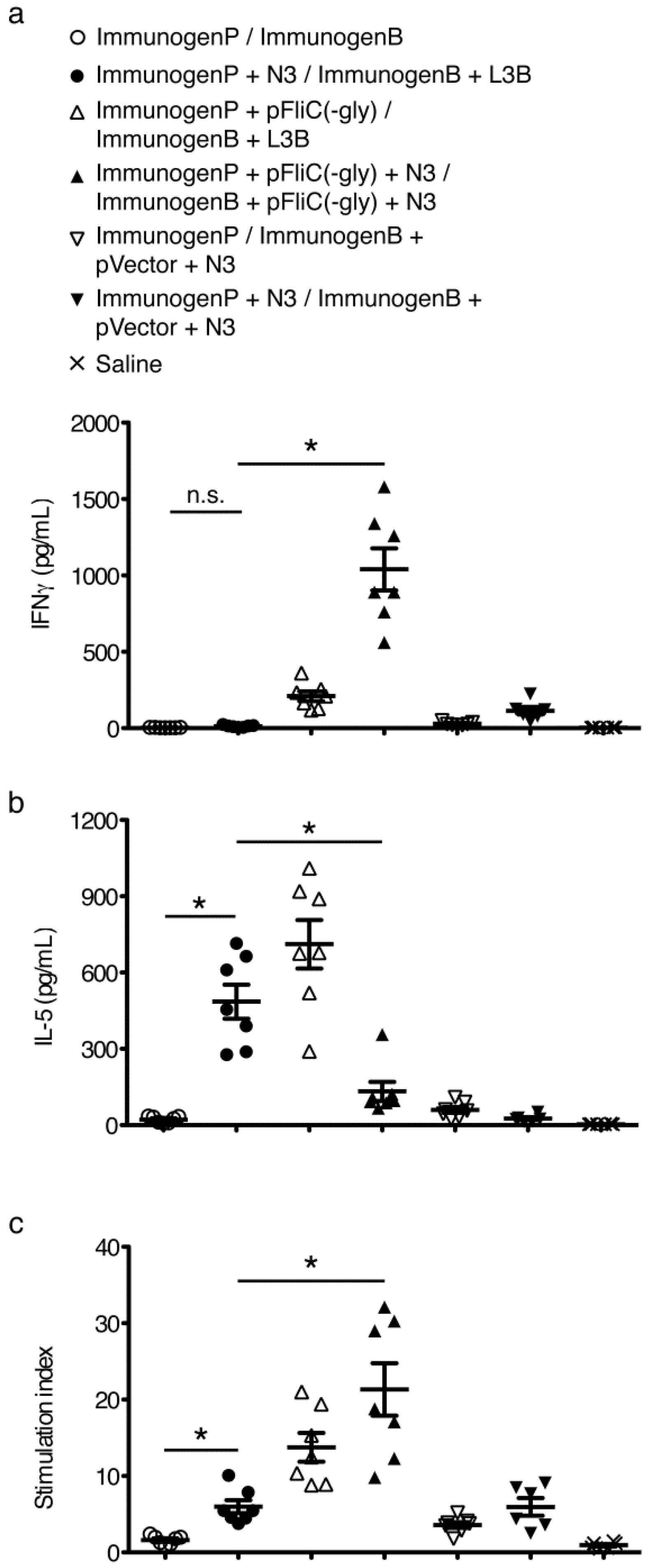
3.6. gp160 and p24gag DNA and Protein Vaccinations; Cellular Responses
3.7. Immune Activation Potential of Adjuvants
3.8. Longevity of Immune Responses



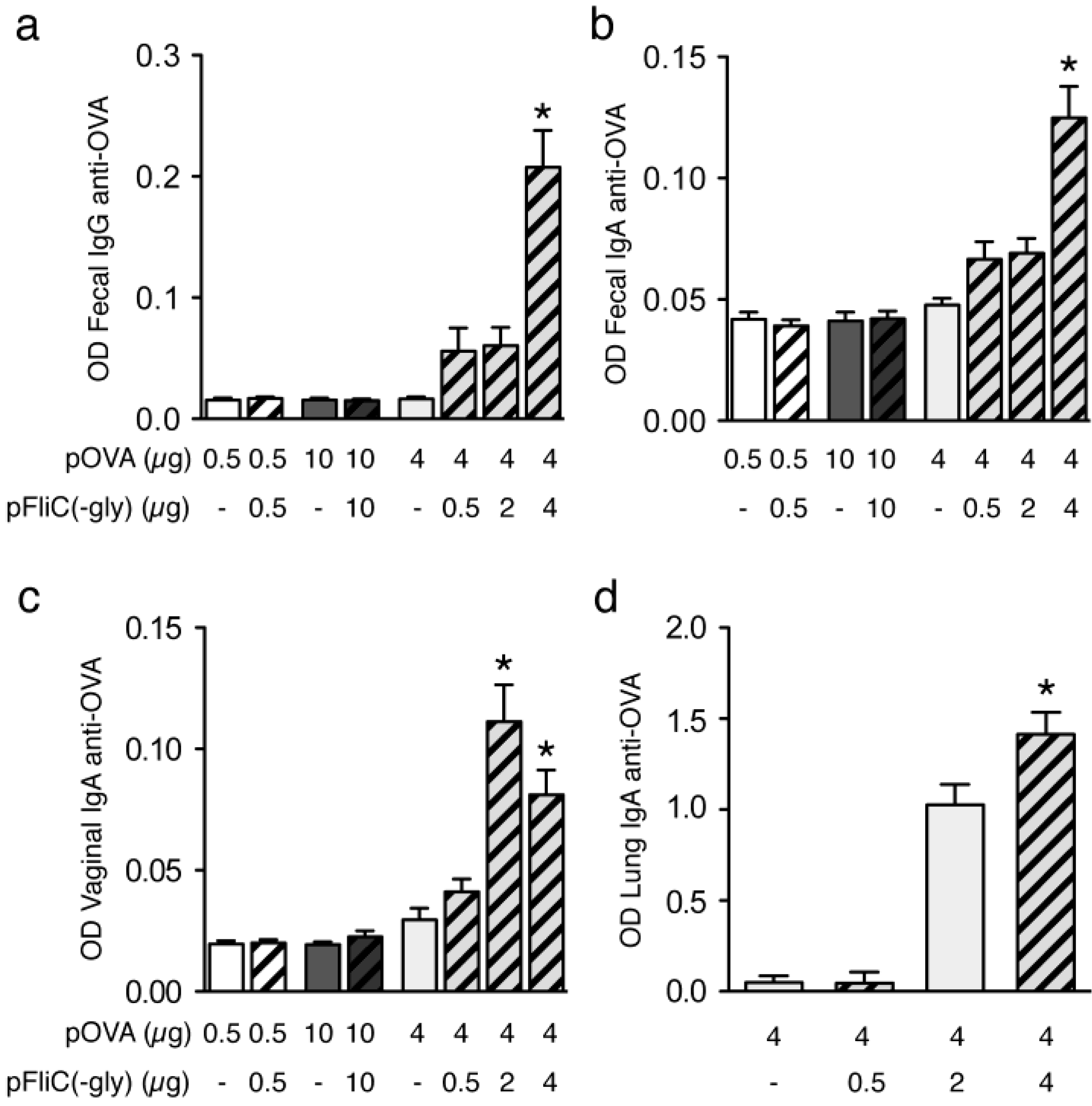
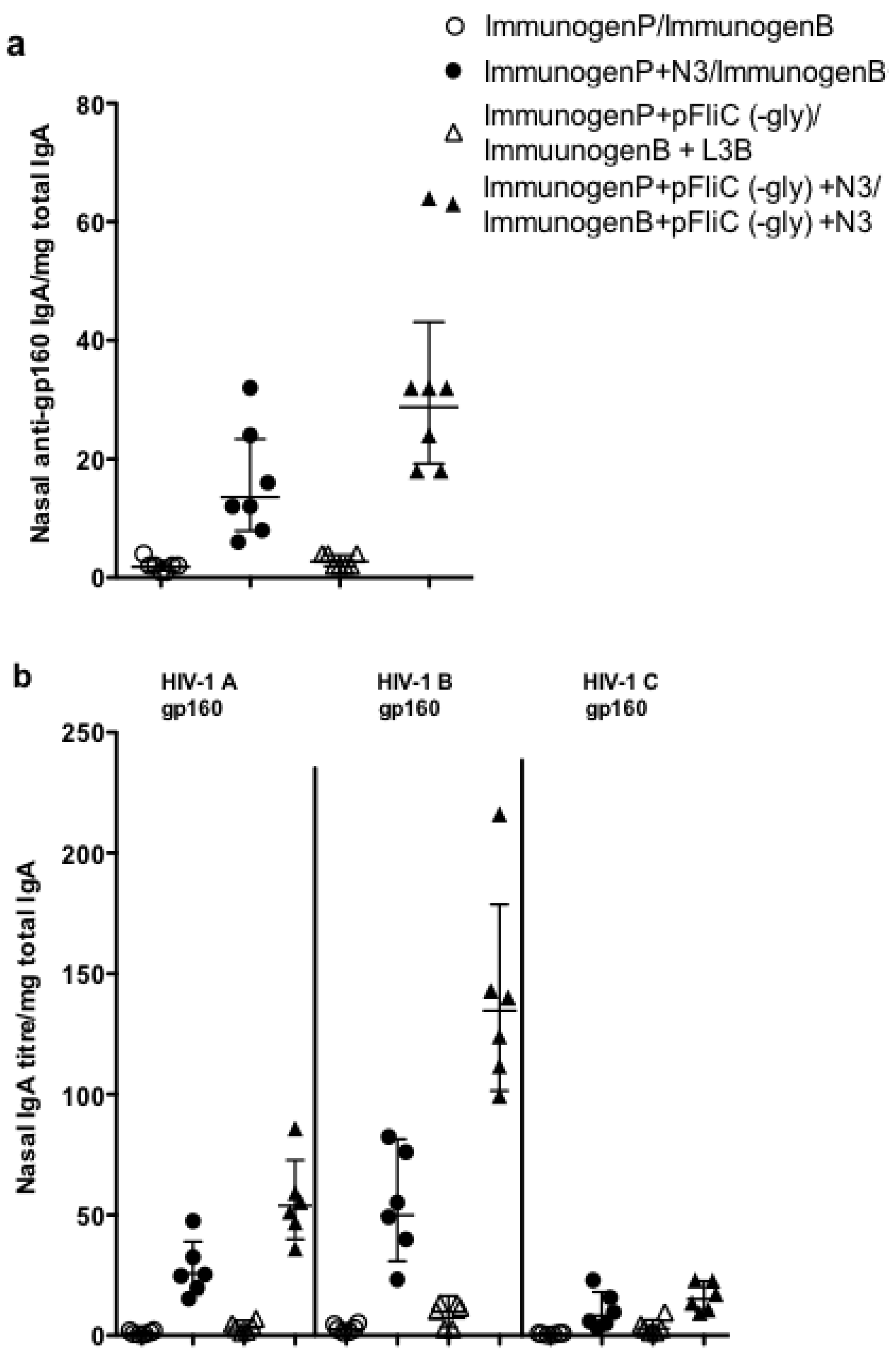
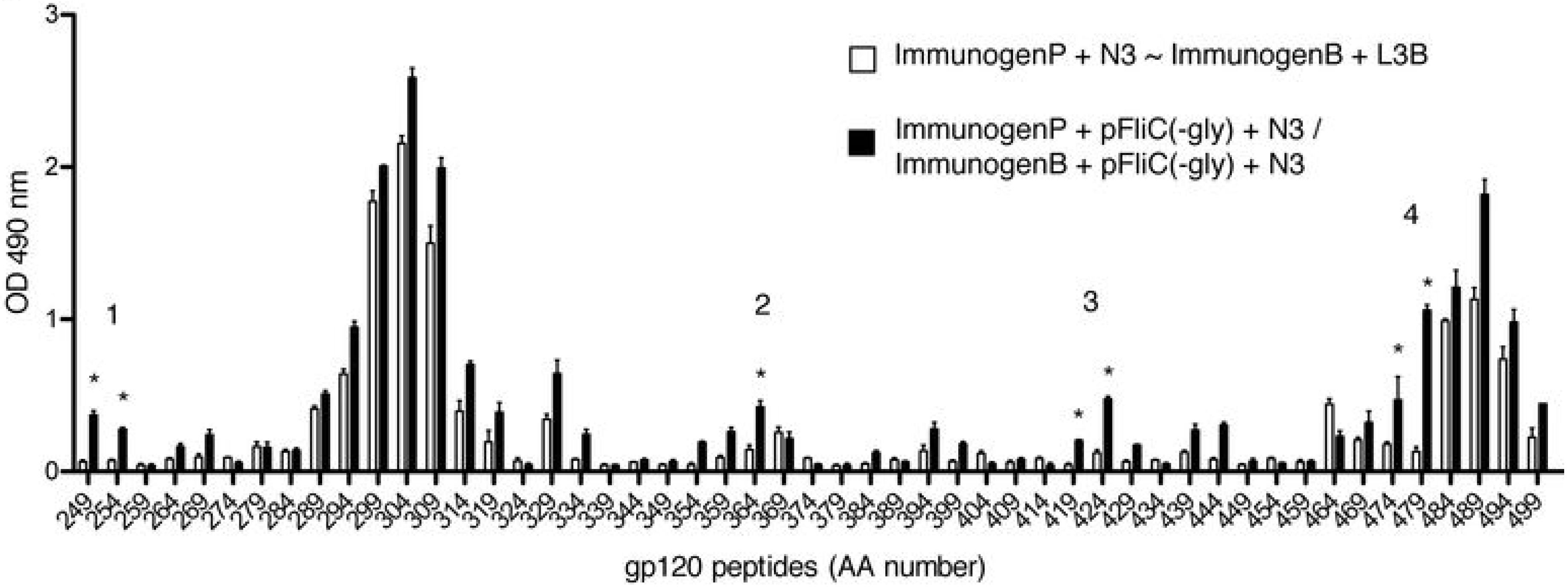
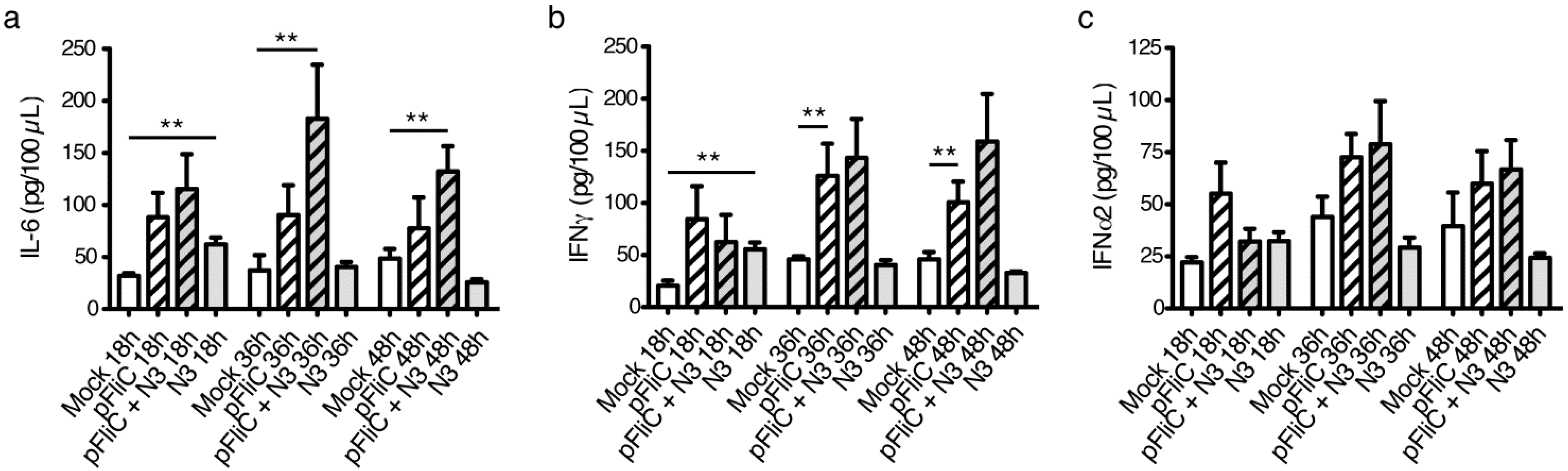
3.9. Impact of Adjuvant on Local Innate Immunity
4. Conclusions
Abbreviations
| TLR | Toll-like Receptor |
| NLRC4 | Nod-like Receptor family CARD domain-containing protein 4 |
| Naip5 | Neuronal apoptosis inhibitory protein 5 |
| HIV-1 | Human Immunodeficiency Virus-1 |
Acknowledgments
Conflicts of Interest
References
- Jones, S.; Evans, K.; McElwaine-Johnn, H.; Sharpe, M.; Oxford, J.; Lambkin-Williams, R.; Mant, T.; Nolan, A.; Zambon, M.; Ellis, J.; et al. DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccine 2009, 27, 2506–2512. [Google Scholar] [CrossRef]
- Girard, M.P.; Bansal, G.P. HIV/AIDS vaccines: A need for new concepts? Int. Rev. Immunol. 2008, 27, 447–471. [Google Scholar] [CrossRef]
- Jechlinger, W. Optimization and delivery of plasmid DNA for vaccination. Expert Rev. Vaccines 2006, 5, 803–825. [Google Scholar] [CrossRef]
- Miao, E.A.; Andersen-Nissen, E.; Warren, S.E.; Aderem, A. TLR5 and Ipaf: Dual sensors of bacterial flagellin in the innate immune system. Semin. Immunopathol. 2007, 29, 275–288. [Google Scholar] [CrossRef]
- Applequist, S.E.; Rollman, E.; Wareing, M.D.; Liden, M.; Rozell, B.; Hinkula, J.; Ljunggren, H.G. Activation of innate immunity, inflammation, and potentiation of DNA vaccination through mammalian expression of the TLR5 agonist flagellin. J. Immunol. 2005, 175, 3882–3891. [Google Scholar]
- Song, L.; Nakaar, V.; Kavita, U.; Price, A.; Huleatt, J.; Tang, J.; Jacobs, A.; Liu, G.; Huang, Y.; Desai, P.; et al. Efficacious recombinant influenza vaccines produced by high yield bacterial expression: A solution to global pandemic and seasonal needs. PLoS One 2008, 3, e2257. [Google Scholar] [CrossRef]
- Mizel, S.B.; Graff, A.H.; Sriranganathan, N.; Ervin, S.; Lees, C.J.; Lively, M.O.; Hantgan, R.R.; Thomas, M.J.; Wood, J.; Bell, B. Flagellin-F1-V fusion protein is an effective plague vaccine in mice and two species of nonhuman primates. Clin. Vaccine Immunol. 2009, 16, 21–28. [Google Scholar] [CrossRef]
- Mizel, S.B.; Bates, J.T. Flagellin as an adjuvant: Cellular mechanisms and potential. J. Immunol. 2010, 185, 5677–5682. [Google Scholar] [CrossRef]
- Lycke, N. Recent progress in mucosal vaccine development: Potential and limitations. Nat. Rev. Immunol. 2012, 12, 592–605. [Google Scholar] [CrossRef]
- Chen, W.; Patel, G.B.; Yan, H.; Zhang, J. Recent advances in the development of novel mucosal adjuvants and antigen delivery systems. Hum. Vaccine 2010, 6, 706–714. [Google Scholar] [CrossRef]
- Hinkula, J.; Bratt, G.; Gilljam, G.; Nordlund, S.; Broliden, P.A.; Holmberg, V.; Olausson-Hansson, E.; Albert, J.; Sandstrom, E.; Wahren, B. Immunological and virological interactions in patients receiving passive immunotherapy with HIV-1 neutralizing monoclonal antibodies. J. Acq. Immune Defic. Syndr. 1994, 7, 940–951. [Google Scholar]
- Sutlu, T.; Nystrom, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular anti-viral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef]
- Hinkula, J.; Devito, C.; Zuber, B.; Benthin, R.; Ferreira, D.; Wahren, B.; Schroder, U. A novel DNA adjuvant, N3, enhances mucosal and systemic immune responses induced by HIV-1 DNA and peptide immunizations. Vaccine 2006, 24, 4494–4497. [Google Scholar] [CrossRef]
- Petersson, P.; Hedenskog, M.; Alves, D.; Brytting, M.; Schroder, U.; Linde, A.; Lundkvist, A. The Eurocine L3 adjuvants with subunit influenza antigens induce protective immunity in mice after intranasal vaccination. Vaccine 2010, 28, 6491–6497. [Google Scholar] [CrossRef]
- Meyers, G.; Korber, B.; Foley, K.T.; Jeang, J.W.; Mellers, J.W.; Wain-Hobson, S. A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences; Los Alamos, Los Alamos National Laboratory: Washington, DC, USA, 1992. [Google Scholar]
- Brave, A.; Hallengard, D.; Schroder, U.; Blomberg, P.; Wahren, B.; Hinkula, J. Intranasal immunization of young mice with a multigene HIV-1 vaccine in combination with the N3 adjuvant induces mucosal and systemic immune responses. Vaccine 2008, 26, 5075–5078. [Google Scholar] [CrossRef]
- Hinkula, J.; Hagbom, M.; Wahren, B.; Schroder, U. Safety and immunogenicity, after nasal application of HIV-1 DNA gagp37 plasmid vaccine in young mice. Vaccine 2008, 26, 5101–5106. [Google Scholar] [CrossRef]
- Hinkula, J.; Rollman, E.; Lundholm, P.; Benthin, R.; Okuda, K.; Wahren, B. Genetic immunizationwith multiple HIV-1 genes provides protection against HIV-1/MuLV pseudovirus challenge in vivo. Cells Tissues Organs 2004, 177, 169–184. [Google Scholar] [CrossRef]
- Devito, C.; Levi, M.; Broliden, K.; Hinkula, J. Mapping of B-cell epitopes in rabbits immunised with various gag antigens for the production of HIV-1 gag capture ELISA reagents. J. Immunol. Methods 2000, 238, 69–80. [Google Scholar] [CrossRef]
- Lightfield, K.L.; Persson, J.; Brubaker, S.W.; Witte, C.E.; von Moltke, J.; Dunipace, E.A.; Henry, T.; Sun, Y.H.; Cado, D.; Dietrich, W.F.; et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 2008, 9, 1171–1178. [Google Scholar] [CrossRef]
- Nystrom, S.; Antoine, D.J.; Lundback, P.; Lock, J.G.; Nita, A.F.; Hogstrand, K.; Grandien, A.; Erlandsson-Harris, H.; Andersson, U.; Applequist, S.E. TLR activation regulates damage-associated molecular pattern isoforms released during pyroptosis. EMBO J. 2013, 32, 86–99. [Google Scholar]
- Takeshita, F.; Ishii, K.J. Intracellular DNA sensors in immunity. Curr. Opin. Immunol. 2008, 20, 383–388. [Google Scholar]
- Schroder, K.; Muruve, D.A.; Tschopp, J. Innate immunity: Cytoplasmic DNA sensing by the AIM2 inflammasome. Curr. Biol. 2009, 19, R262–R265. [Google Scholar] [CrossRef]
- Uematsu, S.; Fujimoto, K.; Jang, M.H.; Yang, B.G.; Jung, Y.J.; Nishiyama, M.; Sato, S.; Tsujimura, T.; Yamamoto, M.; Yokota, Y.; et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008, 9, 769–776. [Google Scholar] [CrossRef]
- Atif, S.M.; Uematsu, S.; Akira, S.; McSorley, S.J. CD103-CD11b+ dendritic cells regulate the sensitivity of CD4 T-cell responses to bacterial flagellin. Mucosal. Immunol. 2013. [Google Scholar] [CrossRef]
- Wiley, S.R.; Raman, V.S.; Desbien, A.; Bailor, H.R.; Bhardwaj, R.; Shakri, A.R.; Reed, S.G.; Chitnis, C.E.; Carter, D. Targeting TLRs expands the antibody repertoire in response to a malaria vaccine. Sci. Transl. Med. 2011. [Google Scholar] [CrossRef]
- Vajdy, M.; Srivastava, I.; Polo, J.; Donnelly, J.; O’Hagan, D.; Singh, M. Mucosal adjuvants and delivery systems for protein-, DNA- and RNA-based vaccines. Immunol. Cell. Biol. 2004, 82, 617–627. [Google Scholar] [CrossRef]
- Toka, F.N.; Pack, C.D.; Rouse, B.T. Molecular adjuvants for mucosal immunity. Immunol. Rev. 2004, 199, 100–112. [Google Scholar] [CrossRef]
- Han, T.K.; Dao, M.L. Enhancement of salivary IgA response to a DNA vaccine against Streptococcus mutans wall-associated protein A in mice by plasmid-based adjuvants. J. Med. Microbiol. 2007, 56, 675–680. [Google Scholar] [CrossRef]
- Kataoka, K.; McGhee, J.R.; Kobayashi, R.; Fujihashi, K.; Shizukuishi, S. Nasal Flt3 ligand cDNA elicits CD11c+CD8+ dendritic cells for enhanced mucosal immunity. J. Immunol. 2004, 172, 3612–3619. [Google Scholar]
- Melkebeek, V.; Sonck, E.; Verdonck, F.; Goddeeris, B.M.; Cox, E. Optimized FaeG expression and a thermolabile enterotoxin DNA adjuvant enhance priming of an intestinal immune response by an FaeG DNA vaccine in pigs. Clin. Vaccine Immunol. 2007, 14, 28–35. [Google Scholar] [CrossRef]
- Van Gils, M.J.; Sanders, R.W. Broadly neutralizing antibodies against HIV-1: Templates for a vaccine. Virology 2013, 435, 46–56. [Google Scholar] [CrossRef]
- Girard, M.P.; Osmanov, S.K.; Kieny, M.P. A review of vaccine research and development: The human immunodeficiency virus (HIV). Vaccine 2006, 24, 4062–4081. [Google Scholar] [CrossRef]
- McGuire, A.T.; Hoot, S.; Dreyer, A.M.; Lippy, A.; Stuart, A.; Cohen, K.W.; Jardina, J.; Menis, S.; Scheid, J.F.; West, A.P.; et al. Engineering HIV envelope protein to activate germline B cell receptors of broadly neutralizing anti-CD4 binding site antibodies. J. Exp. Med. 2013, 210, 656–663. [Google Scholar]
- Narayan, K.M.; Agrawal, N.; Du, S.X.; Murakana, J.E.; Bauer, K.; Leaman, D.P.; Phung, P.; Limoli, K.; Chen, H.; Boenig, R.I.; et al. Prime-boost immunization of rabbits with HIV-1 gp120 elicits potent neutralizing activity against a primary viral isolate. PLoS One 2013, 8, e52732. [Google Scholar]
- Naito, S.; Maeyama, J.-I.; Mizukami, T.; Takahashi, M.; Hamaguchi, I.; Yamaguchi, K. Transcutaneous immunization by merely prolonging the duration of antigen presence in the skin of mice induces a potent antigen-specific immune response even in the absence of an adjuvant. Vaccine 2007, 25, 8762–8770. [Google Scholar] [CrossRef]
- Scharton-Kersten, T.; Glenn, G.M.; Vassell, R.; Yu, J.; Walwender, D.; Alving, C.R. Principles of transcutaneous immunization using cholera toxin as an adjuvant. Vaccine 1999, 17, S37–S43. [Google Scholar] [CrossRef]
- Kasturi, S.P.; Skountzou, I.; Albrecht, R.A.; Koutsonanos, D.; Hua, T.; Nakaya, H.I.; Ravindran, R.; Stewart, S.; Alam, M.; Kwissa, M.; et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 2011, 470, 543–547. [Google Scholar] [CrossRef]
- Napoliatani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef]
- Shen, S.-S.; Yang, Y.-W. Antigen delivery for cross-priming via emulsion vaccine adjuvants. Vaccine 2012, 30, 1560–1571. [Google Scholar] [CrossRef]
- Marichal, T.; Ohata, K.; Bedoret, D.; Mesnil, C.; Sabatel, C.; Kobiyama, K.; Lekeux, B.; Coban, C.; Ishii, K.J.; Bureau, F.; et al. DNA released from dying host cells mediate aluminium adjuvant activity. Nat. Med. 2011, 17, 996–1003. [Google Scholar] [CrossRef]
- Yang, Y.-W.; Wei, A.-C.; Shen, S.-S. The immunogenicity-enhancing effect of emulsion vaccine adjuvant independent of the dispersion type and antigen release rate—A revisit of the role of the hydrophile-lipophile balance (HLB) valus. Vaccine 2005, 23, 2665–2675. [Google Scholar] [CrossRef]
- O’Hagan, D.T.; Ott, G.S.; van Nest, G.; Rappuoli, R.; del Guidice, G. The history of M59 adjuvant: A phoenix that rose from the ashes. Exp. Rev. Vaccines 2013, 12, 13–30. [Google Scholar] [CrossRef]
- Awasti, A.; Kuchroo, V.K. Th17 cells: From precursors to players in inflammation and infection. Int. Immunol. 2009, 20, 489–498. [Google Scholar]
- Makidon, P.E.; Belyakov, I.M.; Blanco, L.P.; Janczak, K.W.; Landers, J.; Bielinska, A.U.; Groom, J.V., 2nd; Naker, J.R., Jr. Nanoemulsion mucosal adjuvant uniquely activates cytokine production by nasal epithelium and induces dendritic cell trafficing. Eur. J. Immunol. 2012, 42, 2073–2086. [Google Scholar] [CrossRef]
- Albert, M.L.; Sauter, B.; Bhardwaj, N. Dendritic cells aquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 1998, 392, 86–89. [Google Scholar] [CrossRef]
- Valensi, J.P.; Carlson, J.R.; van Nest, G.A. Systemic cytokine profiles in BALB/c mice immunized with trivalent influenza vaccine containing MF59 oil emulsion and other advanced adjuvants. J. Immunol. 1994, 153, 4029–4039. [Google Scholar]
- Van Duin, D.; Medzhitov, R.; Shaw, A.C. Triggering TLR signaling in vaccination. Trends Immunol. 2006, 27, 49–55. [Google Scholar] [CrossRef]
- Honko, A.N.; Mizel, S.B. Mucosal administration of flagellin induces innate immunity in the mouse lung. Infect. Immun. 2004, 72, 6676–6679. [Google Scholar] [CrossRef]
- Pritts, T.; Hungness, E.; Wang, Q.; Robb, B.; Hershko, D.; Hasselgren, P.O. Mucosal and enterocyte IL-6 production during sepsis and endotoxemia-role of transcription factors and regulation of stress response. Am. J. Surg. 2002, 183, 372–383. [Google Scholar] [CrossRef]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavelli, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defence. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Songhet, P.; Dumoutier, L.; Ferrero, I.; Janot, L.; Erard, F.; Bertout, J.; Leger, H.; et al. TLR5 signaling stimulates the innate production of IL-17 and IL-22 by CD3(neg)CD127+ immune cells in spleen and mucosa. J. Immunol. 2010, 185, 1177–1185. [Google Scholar] [CrossRef]
- Krieg, A.M. CpG still rocks update on an accidental drug. Nucleic Acid Ther. 2012, 22, 77–89. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nyström, S.; Bråve, A.; Falkeborn, T.; Devito, C.; Rissiek, B.; Johansson, D.X.; Schröder, U.; Uematsu, S.; Akira, S.; Hinkula, J.; et al. DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant. Vaccines 2013, 1, 415-443. https://doi.org/10.3390/vaccines1040415
Nyström S, Bråve A, Falkeborn T, Devito C, Rissiek B, Johansson DX, Schröder U, Uematsu S, Akira S, Hinkula J, et al. DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant. Vaccines. 2013; 1(4):415-443. https://doi.org/10.3390/vaccines1040415
Chicago/Turabian StyleNyström, Sanna, Andreas Bråve, Tina Falkeborn, Claudia Devito, Björn Rissiek, Daniel X. Johansson, Ulf Schröder, Satoshi Uematsu, Shizuo Akira, Jorma Hinkula, and et al. 2013. "DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant" Vaccines 1, no. 4: 415-443. https://doi.org/10.3390/vaccines1040415
APA StyleNyström, S., Bråve, A., Falkeborn, T., Devito, C., Rissiek, B., Johansson, D. X., Schröder, U., Uematsu, S., Akira, S., Hinkula, J., & Applequist, S. E. (2013). DNA-Encoded Flagellin Activates Toll-Like Receptor 5 (TLR5), Nod-like Receptor Family CARD Domain-Containing Protein 4 (NRLC4), and Acts as an Epidermal, Systemic, and Mucosal-Adjuvant. Vaccines, 1(4), 415-443. https://doi.org/10.3390/vaccines1040415