
Vaccine Design against Chagas Disease Focused on the Use of Nucleic Acids


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Vaccine Rationale
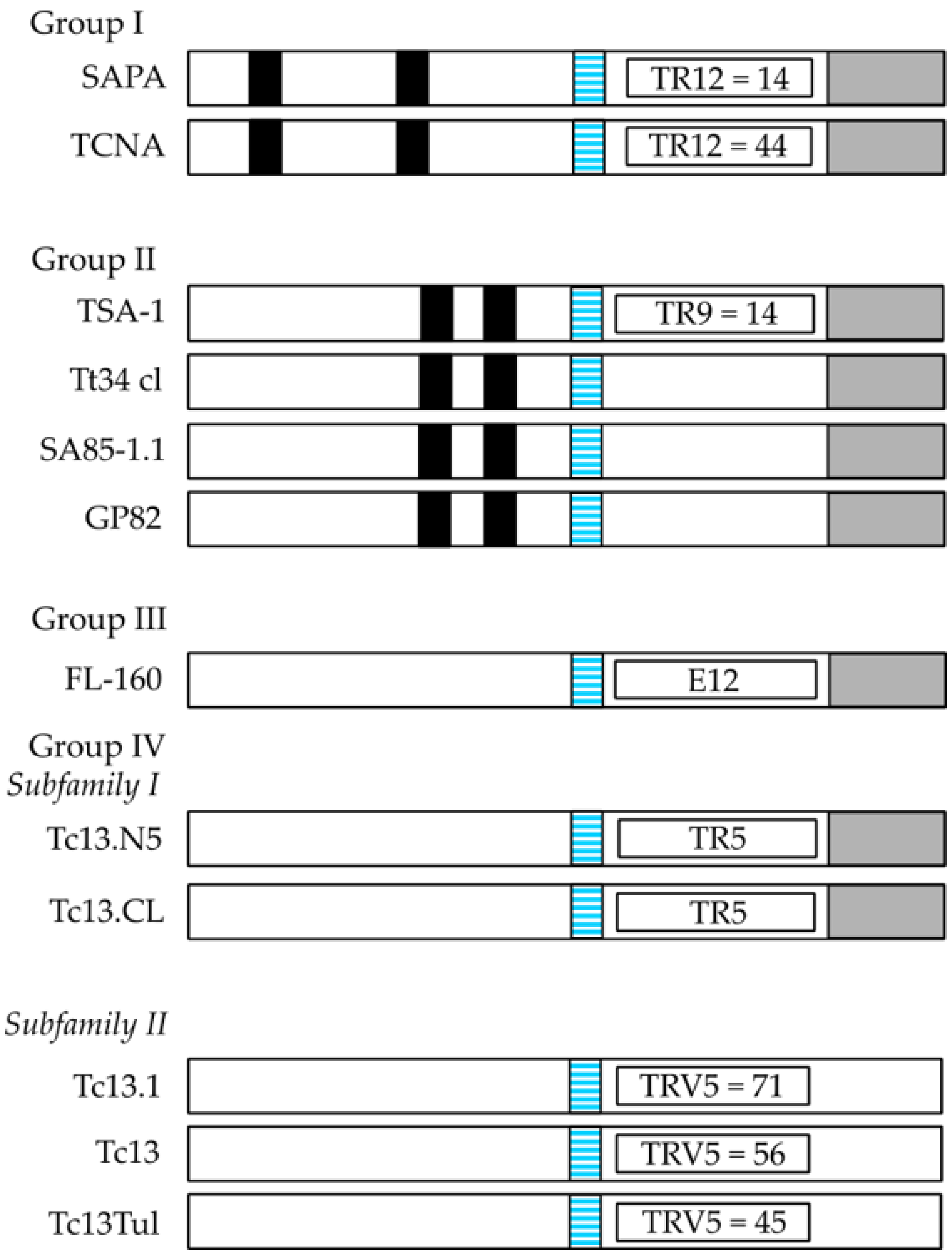
2.1. Trypanosoma Cruzi Surface Antigens
2.2. Most Likely Candidate Antigens for Vaccine Development
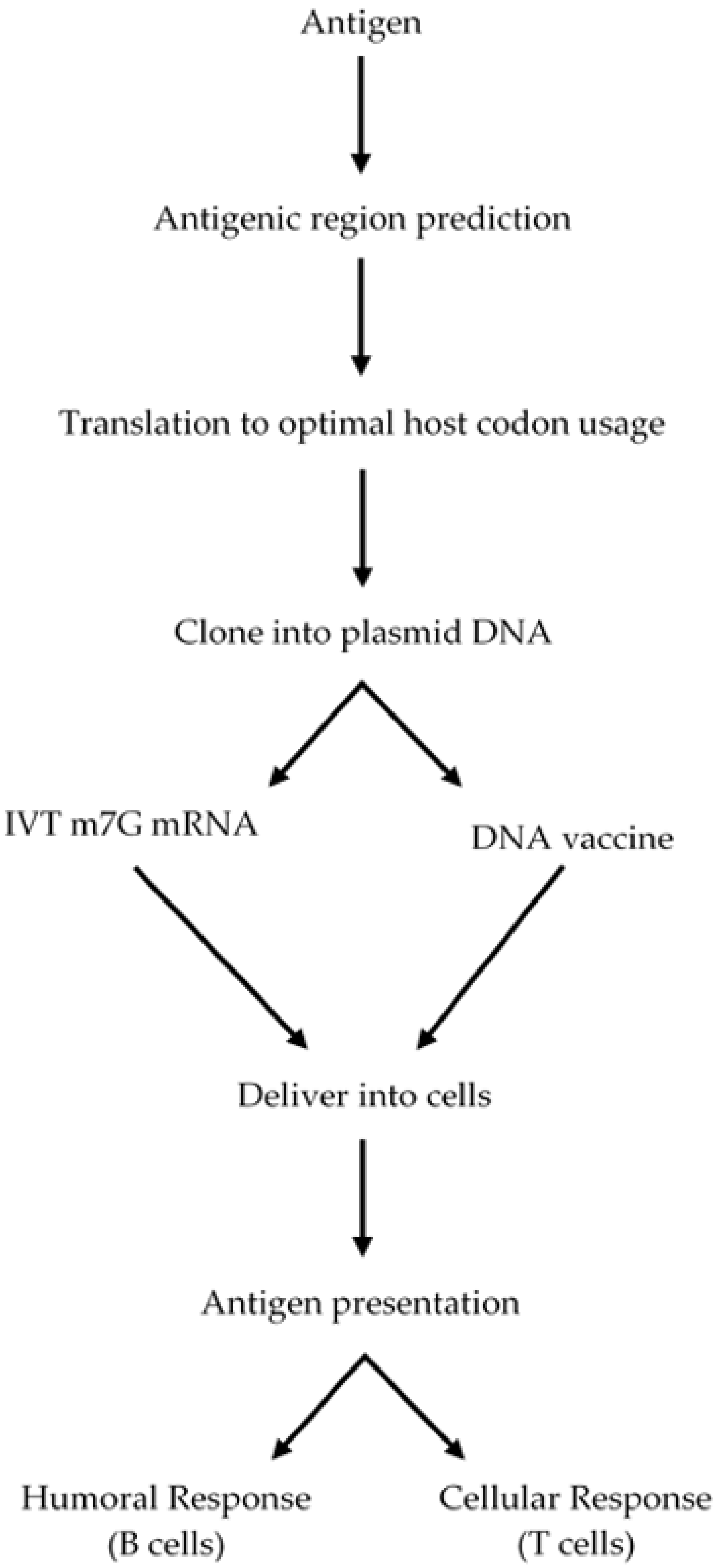
2.3. DNA-Based Vaccines
2.4. RNA-Based Vaccines
2.5. Live Attenuated Vaccines
2.6. T4 Bacteriophage Nanoparticles for Vaccine Delivery
2.7. Co-Adjuvants
2.7.1. Co-Adjuvants for Protein Antigen-Based Vaccines
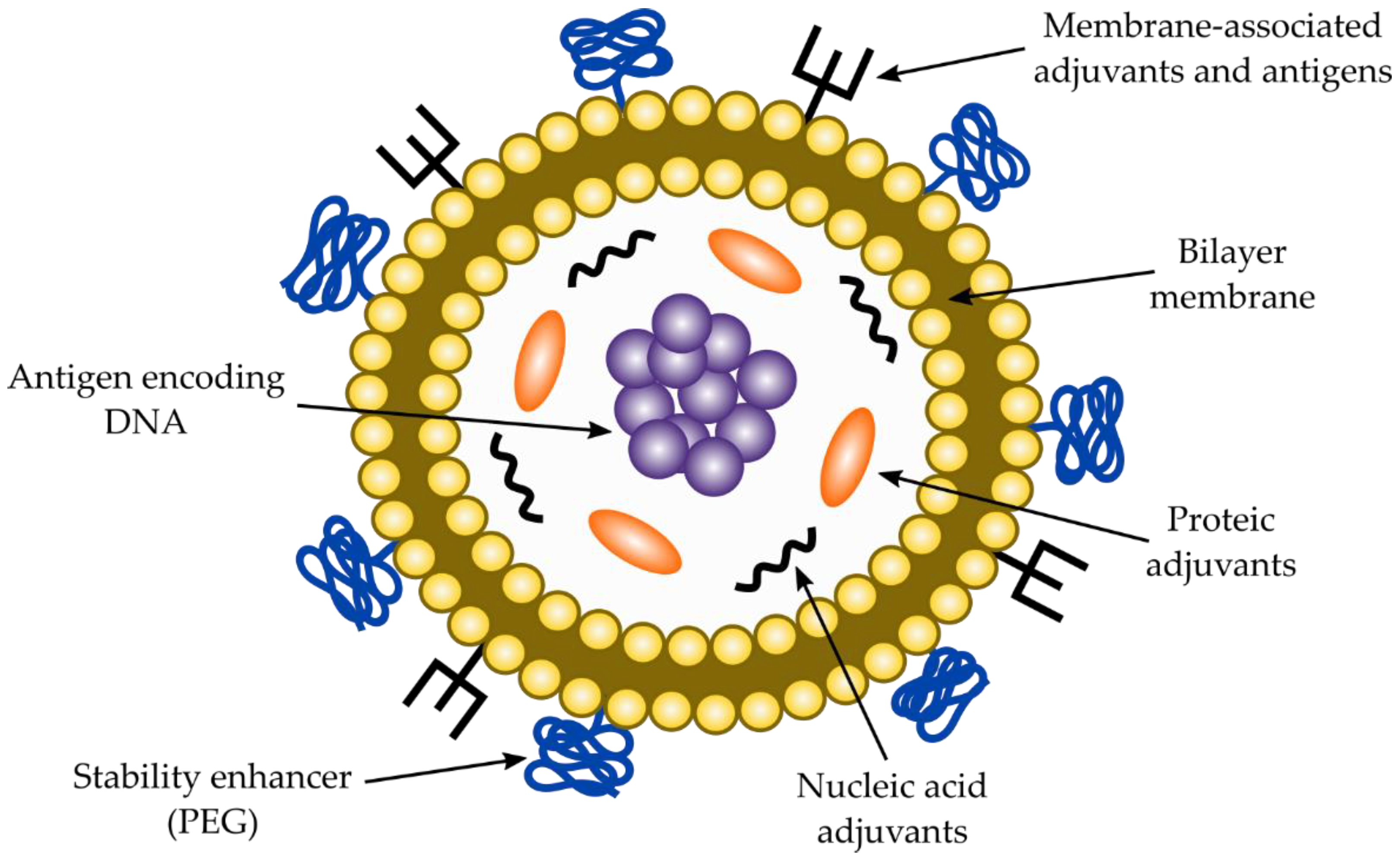
2.7.2. Co-Adjuvants for Nucleic Acid-Based Vaccines
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yeung, C.; Mendoza, I.; Echeverria, L.E.; Baranchuk, A. Chagas’ cardiomyopathy and Lyme carditis: Lessons learned from two infectious diseases affecting the heart. Trends Cardiovasc. Med. 2021, 31, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Antinori, S.; Galimberti, L.; Bianco, R.; Grande, R.; Galli, M.; Corbellino, M. Chagas disease in Europe: A review for the internist in the globalized world. Eur. J. Intern. Med. 2017, 43, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Kourbeli, V.; Chontzopoulou, E.; Moschovou, K.; Pavlos, D.; Mavromoustakos, T.; Papanastasiou, I.P. An overview on target-based drug design against kinetoplastid protozoan infections: Human African trypanosomiasis, Chagas disease and leishmaniases. Molecules 2021, 26, 4629. [Google Scholar] [CrossRef]
- Vermelho, A.B.; Rodrigues, G.C.; Supuran, C.T. Why hasn’t there been more progress in new Chagas disease drug discovery? Expert Opin. Drug Discov. 2020, 15, 145–158. [Google Scholar] [CrossRef]
- Vekemans, J.; Hasso-Agopsowicz, M.; Kang, G.; Hausdorff, W.P.; Fiore, A.; Tayler, E.; Klemm, E.J.; Laxminarayan, R.; Srikantiah, P.; Friede, M.; et al. Leveraging vaccines to reduce antibiotic use and prevent antimicrobial resistance: A World Health Organization action framework. Clin. Infect. Dis. 2021, 73, e1011–e1017. [Google Scholar] [CrossRef]
- Bartsch, S.M.; Avelis, C.M.; Asti, L.; Hertenstein, D.L.; Ndeffo-Mbah, M.; Galvani, A.; Lee, B.Y. The economic value of identifying and treating Chagas disease patients earlier and the impact on Trypanosoma cruzi transmission. PLoS Negl. Trop. Dis. 2018, 12, e0006809. [Google Scholar] [CrossRef]
- Lee, B.Y.; Bartsch, S.M.; Skrip, L.; Hertenstein, D.L.; Avelis, C.M.; Ndeffo-Mbah, M.; Tilchin, C.; Dumonteil, E.O.; Galvani, A. Are the London Declaration’s 2020 goals sufficient to control Chagas disease?: Modeling scenarios for the Yucatan Peninsula. PLoS Negl. Trop. Dis. 2018, 12, e0006337. [Google Scholar] [CrossRef] [Green Version]
- Dumonteil, E.; Bottazzi, M.E.; Zhan, B.; Heffernan, M.J.; Jones, K.; Valenzuela, J.G.; Kamhawi, S.; Ortega, J.; Ponce de Leon Rosales, S.; Lee, B.Y.; et al. Accelerating the development of a therapeutic vaccine for human Chagas disease: Rationale and prospects. Expert Rev. Vaccines 2012, 11, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Bottazzi, M.E. Vaccines against neglected tropical diseases: Promising interventions to rescue the poorest populations in the Americas. Immunotherapy 2014, 6, 117–119. [Google Scholar] [CrossRef]
- Camargo, E.P.; Gazzinelli, R.T.; Morel, C.M.; Precioso, A.R. Why do we still have not a vaccine against Chagas disease? Mem Inst Oswaldo Cruz Rio De Jan. 2021, 116. [Google Scholar] [CrossRef]
- Engman, D.M.; Dragon, E.A.; Donelson, J.E. Human humoral immunity to hsp70 during Trypanosoma cruzi infection. J. Immunol. 1990, 144, 3987–3991. [Google Scholar] [PubMed]
- Campetella, O.; Sánchez, D.; Cazzulo, J.J.; Frasch, A.C. A superfamily of Trypanosoma cruzi surface antigens. Parasitol. Today 1992, 8, 378–381. [Google Scholar] [CrossRef]
- De Marchi, C.R.; Di Noia, J.M.; Frasch, A.C.C.; Neto, V.A.; Almeida, I.C.; Buscaglia, C.A. Evaluation of a recombinant Trypanosoma cruzi mucin-like antigen for serodiagnosis of Chagas’ disease. Clin. Vaccine Immunol. 2011, 18, 1850–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buscaglia, C.A.; Campo, V.A.; Frasch, A.C.C.; Di Noia, J.M. Trypanosoma cruzi surface mucins: Host-dependent coat diversity. Nat. Rev. Microbiol. 2006, 4, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Cánepa, G.E.; Mesías, A.C.; Yu, H.; Chen, X.; Buscaglia, C.A. Structural features affecting trafficking, processing, and secretion of Trypanosoma cruzi mucins. J. Biol. Chem. 2012, 287, 26365–26376. [Google Scholar] [CrossRef] [Green Version]
- Pech-Canul, A.d.l.C.; Monteón, V.; Solís-Oviedo, R.L. A brief view of the surface membrane proteins from Trypanosoma cruzi. J. Parasitol. Res. 2017. [Google Scholar] [CrossRef] [Green Version]
- Barreto-Bergter, E.; Vermelho, A.B. Structures of glycolipids found in trypanosomatids: Contribution to parasite functions. Open Parasitol. J. 2010, 4, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Cánepa, G.E.; Degese, M.S.; Budu, A.; Garcia, C.R.S.; Buscaglia, C.A. Involvement of TSSA (trypomastigote small surface antigen) in Trypanosoma cruzi invasion of mammalian cells. Biochem. J. 2012, 444, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Nakayasu, E.S.; Yashunsky, D.V.; Nohara, L.L.; Torrecilhas, A.C.T.; Nikolaev, A.V.; Almeida, I.C. GPIomics: Global analysis of glycosylphosphatidylinositol-anchored molecules of Trypanosoma cruzi. Mol. Syst. Biol. 2009, 5, 261. [Google Scholar] [CrossRef]
- Urban, I.; Santurio, L.B.; Chidichimo, A.; Yu, H.; Chen, X.; Mucci, J.; Agüero, F.; Buscaglia, C.A. Molecular diversity of the Trypanosoma cruzi TcSMUG family of mucin genes and proteins. Biochem. J. 2011, 438, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pablos, L.M.; Osuna, A. Conserved regions as markers of different patterns of expression and distribution of the mucin-associated surface proteins of Trypanosoma cruzi. Infect. Immun. 2012, 80, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenkman, S.; Jiang, M.S.; Hart, G.W.; Nussenzweig, V. A novel cell surface trans-sialidase of Trypanosoma cruzi generates a stage-specific epitope required for invasion of mammalian cells. Cell 1991, 65, 1117–1125. [Google Scholar] [CrossRef]
- El-Sayed, N.M.; Myler, P.J.; Bartholomeu, D.C.; Nilsson, D.; Aggarwal, G.; Tran, A.-N.; Ghedin, E.; Worthey, E.A.; Delcher, A.L.; Blandin, G.; et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science 2005, 309, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Frasch, A.C.C. Functional diversity in the trans-sialidase and mucin families in Trypanosoma cruzi. Parasitol. Today 2000, 16, 282–286. [Google Scholar] [CrossRef]
- Freitas, L.M.; dos Santos, S.L.; Rodrigues-Luiz, G.F.; Mendes, T.A.O.; Rodrigues, T.S.; Gazzinelli, R.T.; Teixeira, S.M.R.; Fujiwara, R.T.; Bartholomeu, D.C. Genomic analyses, gene expression and antigenic profile of the trans-sialidase superfamily of Trypanosoma cruzi reveal an undetected level of complexity. PLoS ONE 2011, 6, e25914. [Google Scholar] [CrossRef] [Green Version]
- Chiurillo, M.A.; Cortez, D.R.; Lima, F.M.; Cortez, C.; Ramírez, J.L.; Martins, A.G.; Serrano, M.G.; Teixeira, M.M.G.; da Silveira, J.F. The diversity and expansion of the trans-sialidase gene family is a common feature in Trypanosoma cruzi clade members. Infect. Genet. Evol. 2016, 37, 266–274. [Google Scholar] [CrossRef]
- Dc-Rubin, S.S.C.; Schenkman, S. Trypanosoma cruzi trans-sialidase as a multifunctional enzyme in Chagas’ disease. Cell. Microbiol. 2012, 14, 1522–1530. [Google Scholar] [CrossRef]
- Freire-de-Lima, L.; Fonseca, L.M.; Oeltmann, T.; Mendoca-Previato, L.; Previato, J.O. The trans-sialidase, the major Trypanosoma cruzi virulence factor: Three decades of studies. Glycobiology 2015, 25, 1142–1149. [Google Scholar] [CrossRef] [Green Version]
- Lantos, A.B.; Carlevaro, G.; Araoz, B.; Diaz, P.R.; de los Milagros Camara, M.; Buscaglia, C.A.; Bossi, M.; Yu, H.; Chen, X.; Bertozzi, C.R.; et al. Sialic acid glycobiology unveils Trypanosoma cruzi trypomastigote membrane physiology. PLoS Pathog. 2016, 12, e1005559. [Google Scholar] [CrossRef] [Green Version]
- Bivona, A.E.; Alberti, A.S.; Cerny, N.; Trinitario, S.N.; Malchiodi, E.L. Chagas disease vaccine design: The search for an efficient Trypanosoma cruzi immune-mediated control. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165658. [Google Scholar] [CrossRef] [PubMed]
- Schenkman, S.; Eichinger, D.; Pereira, M.E.; Nussenzweig, V. Structural and functional properties of Trypanosoma trans-sialidase. Annu. Rev. Microbiol. 1994, 48, 499–523. [Google Scholar] [CrossRef] [PubMed]
- Bayer-Santos, E.; Aguilar-Bonavides, C.; Rodrigues, S.P.; Cordero, E.M.; Marques, A.F.; Varela-Ramirez, A.; Choi, H.; Yoshida, N.; Silveira, J.F.; Almeida, I.C. Proteomic analysis of Trypanosoma cruzi secretome: Characterization of two populations of extracellular vesicles and soluble proteins. J. Proteome Res. 2013, 12, 883–897. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.B.; Lorca, M.; Muñoz, P.; Frasch, A.C. Fetal IgG specificities against Trypanosoma cruzi antigens in infected newborns. Proc. Natl. Acad. Sci. USA 1990, 87, 2846–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prioli, R.P.; Ortega-Barria, E.; Mejia, J.S.; Pereira, M.E. Mapping of a B-cell epitope present in the neuraminidase of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1992, 52, 85–96. [Google Scholar] [CrossRef]
- Leguizamón, M.S.; Campetella, O.E.; Gonzalez-Cappa, S.M.; Frasch, A.C. Mice infected with Trypanosoma cruzi produce antibodies against the enzymatic domain of trans-sialidase that inhibit its activity. Infect. Immun. 1994, 62, 3441–3446. [Google Scholar] [CrossRef] [Green Version]
- Prioli, R.P.; Rosenberg, I.; Pereira, M.E. High-and low-density lipoproteins enhance infection of Trypanosoma cruzi in vitro. Mol. Biochem. Parasitol. 1990, 38, 191–198. [Google Scholar] [CrossRef]
- Prioli, R.P.; Rosenberg, I.; Pereira, M.E. Specific inhibition of Trypanosoma cruzi neuraminidase by the human plasma glycoprotein “cruzin”. Proc. Natl. Acad. Sci. USA 1987, 84, 3097–3101. [Google Scholar] [CrossRef] [Green Version]
- Prioli, R.P.; Ordovas, J.M.; Rosenberg, I.; Schaefer, E.J.; Pereira, M.E. Similarity of cruzin, an inhibitor of Trypanosoma cruzi neuraminidase, to high-density lipoprotein. Science 1987, 238, 1417–1419. [Google Scholar] [CrossRef]
- Briones, M.R.; Egima, C.M.; Schenkman, S. Trypanosoma cruzi trans-sialidase gene lacking C-terminal repeats and expressed in epimastigote forms. Mol. Biochem. Parasitol. 1995, 70, 9–17. [Google Scholar] [CrossRef]
- Rubin-De-Celis, S.S.C.; Uemura, H.; Yoshida, N.; Schenkman, S. Expression of trypomastigote trans-sialidase in metacyclic forms of Trypanosoma cruzi increases parasite escape from its parasitophorous vacuole. Cell. Microbiol. 2006, 8, 1888–1898. [Google Scholar] [CrossRef] [PubMed]
- Magdesian, M.H.; Giordano, R.; Ulrich, H.; Juliano, M.A.; Juliano, L.; Schumacher, R.I.; Colli, W.; Alves, M.J. Infection by Trypanosoma cruzi: Identification of a parasite ligand and its host cell receptor. J. Biol. Chem. 2001, 276, 19382–19389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, R.R.; Giordano, R.J.; Barbu, E.M.; Torrecilhas, A.C.; Kobayashi, G.S.; Langley, R.R.; Arap, W.; Pasqualina, R.; Colli, W.; Alves, M.J.M. Role of the gp85/trans-sialidases in Trypanosoma cruzi tissue tropism: Preferential binding of a conserved peptide motif to the vasculature in vivo. PLoS Negl. Trop. Dis. 2010, 4, e864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, A.A.R.; de Vasconcelos, V.d.C.S.; Colli, W.; Alves, M.J.M.; Giordano, R.J. Trypanosoma cruzi binds to cytokeratin through conserved peptide motifs found in the laminin-G-like domain of the gp85/trans-sialidase proteins. PLoS Negl. Trop. Dis. 2015, 9, e0004099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, S.; Wleklinski, M.; Aruffo, A.; Farr, A.; Coder, D.; Kahn, M. Trypanosoma cruzi amastigote adhesion to macrophages is facilitated by the mannose receptor. J. Exp. Med. 1995, 182, 1243–1258. [Google Scholar] [CrossRef]
- Staquicini, D.I.; Martins, R.M.; Macedo, S.; Sasso, G.R.S.; Atayde, V.D.; Juliano, M.A.; Yoshida, N. Role of GP82 in the selective binding to gastric mucin during oral infection with Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2010, 4, e613. [Google Scholar] [CrossRef]
- Kipnis, T.L.; David, J.R.; Alper, C.A.; Sher, A.; da Silva, W.D. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc. Natl. Acad. Sci. USA 1981, 78, 602–605. [Google Scholar] [CrossRef] [Green Version]
- Beucher, M.; Norris, K.A. Sequence diversity of the Trypanosoma cruzi complement regulatory protein family. Infect. Immun. 2008, 76, 750–758. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, T.K.; Cotrim, P.C.; da Silveira, J.F.; Stolf, A.M.S.; Umezawa, E.S. Trypanosoma cruzi: Isolation of an immunodominant peptide of TESA (trypomastigote excreted-secreted antigens) by gene cloning. Diagn. Microbiol. Infect. Dis. 2002, 42, 187–192. [Google Scholar] [CrossRef]
- Berrizbeitia, M.; Ndao, M.; Bubis, J.; Gottschalk, M.; Aché, A.; Lacouture, S.; Medina, M.; Ward, B.J. Purified excreted-secreted antigens from Trypanosoma cruzi trypomastigotes as tools for diagnosis of Chagas’ disease. J. Clin. Microbiol. 2006, 44, 291–296. [Google Scholar] [CrossRef] [Green Version]
- García, E.A.; Ziliani, M.; Agüero, F.; Bernabó, G.; Sánchez, D.O.; Tekiel, V. TcTASV: A novel protein family in Trypanosoma cruzi identified from a subtractive trypomastigote cDNA library. PLoS Negl. Trop. Dis. 2010, 4, e841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floridia-Yapur, N.; Monje-Rumi, M.; Ragone, P.; Lauthier, J.J.; Tomasini, N.; D’Amato, A.A.; Diosque, P.; Cimino, R.; Gil, J.F.; Sanchez, D.O.; et al. TcTASV antigens of Trypanosoma cruzi: Utility for diagnosis and high accuracy as biomarkers of treatment efficacy in pediatric patients. Am. J. Trop. Med. Hyg. 2019, 101, 1135. [Google Scholar] [CrossRef] [PubMed]
- Caeiro, L.D.; Alba-Soto, C.D.; Rizzi, M.; Solana, M.E.; Rodriguez, G.; Chidichimo, A.M.; Rodriguez, M.E.; Sánchez, D.O.; Levy, G.V.; Tekiel, V. The protein family TcTASV-C is a novel Trypanosoma cruzi virulence factor secreted in extracellular vesicles by trypomastigotes and highly expressed in bloodstream forms. PLoS Negl. Trop. Dis. 2018, 12, e0006475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atwood, J.A., 3rd; Weatherly, D.B.; Minning, T.A.; Bundy, B.; Cavola, C.; Opperdoes, F.R.; Orlando, R.; Tarleton, R.L. The Trypanosoma cruzi proteome. Science 2005, 309, 473–476. [Google Scholar] [CrossRef]
- Ouaissi, M.A.; Dubremetz, J.F.; Schöneck, R.; Fernandez-Gomez, R.; Gomez-Corvera, R.; Billaut-Mulot, O.; Taibi, A.; Loyens, M.; Tartar, A.; Sergheraert, C. Trypanosoma cruzi: A 52-kDa protein sharing sequence homology with glutathione S-transferase is localized in parasite organelles morphologically resembling reservosomes. Exp. Parasitol. 1995, 81, 453–461. [Google Scholar] [CrossRef]
- Allaoui, A.; François, C.; Zemzoumi, K.; Guilvard, E.; Ouaissi, A. Intracellular growth and metacyclogenesis defects in Trypanosoma cruzi carrying a targeted deletion of a Tc52 protein-encoding allele. Mol. Microbiol. 1999, 32, 1273–1286. [Google Scholar] [CrossRef]
- Schöneck, R.; Plumas-Marty, B.; Taibi, A.; Billaut-Mulot, O.; Loyens, M.; Gras-Masse, H.; Capron, A.; Ouaissi, A. Trypanosoma cruzi cDNA encodes a tandemly repeated domain structure characteristic of small stress proteins and glutathione S-transferases. Biol. Cell 1994, 80, 1–10. [Google Scholar] [CrossRef]
- Matos, M.N.; Cazorla, S.I.; Bivona, A.E.; Morales, C.; Guzmán, C.A.; Malchiodi, E.L. Tc52 amino-terminal-domain DNA carried by attenuated Salmonella enterica serovar Typhimurium induces protection against a Trypanosoma cruzi lethal challenge. Infect. Immun. 2014, 82, 4265–4275. [Google Scholar] [CrossRef] [Green Version]
- Matos, M.N.; Alberti, A.S.; Morales, C.; Cazorla, S.I.; Malchiodi, E.L. A prime-boost immunization with Tc52 N-terminal domain DNA and the recombinant protein expressed in Pichia pastoris protects against Trypanosoma cruzi infection. Vaccine 2016, 34, 3243–3251. [Google Scholar] [CrossRef]
- Matos, M.N.; Cazorla, S.I.; Schulze, K.; Ebensen, T.; Guzmán, C.A.; Malchiodi, E.L. Immunization with Tc52 or its amino terminal domain adjuvanted with c-di-AMP induces Th17+ Th1 specific immune responses and confers protection against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2017, 11, e0005300. [Google Scholar] [CrossRef]
- Santana, J.M.; Grellier, P.; Schrével, J.; Teixeira, A.R. A Trypanosoma cruzi-secreted 80 kDa proteinase with specificity for human collagen types I and IV. Biochem. J. 1997, 325, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, I.M.D.; Grellier, P.; Martins, N.F.; Cadavid-Restrepo, G.; de Souza-Ault, M.R.; Augustyns, K.; Teixeira, A.R.L.; Schrével, J.; Maigret, B.; da Silveira, J.F.; et al. Molecular, functional and structural properties of the prolyl oligopeptidase of Trypanosoma cruzi (POP Tc80), which is required for parasite entry into mammalian cells. Biochem. J. 2005, 388, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, A.E.; Alberti, A.S.; Matos, M.N.; Cerny, N.; Cardoso, A.C.; Morales, C.; González, G.; Cazorla, S.I.; Malchiodi, E.L. Trypanosoma cruzi 80 kDa prolyl oligopeptidase (Tc80) as a novel immunogen for Chagas disease vaccine. PLoS Negl. Trop. Dis. 2018, 12, e0006384. [Google Scholar] [CrossRef] [PubMed]
- Campetella, O.; Henriksson, J.; Aslund, L.; Frasch, A.C.; Pettersson, U.; Cazzulo, J.J. The major cysteine proteinase (cruzipain) from Trypanosoma cruzi is encoded by multiple polymorphic tandemly organized genes located on different chromosomes. Mol. Biochem. Parasitol. 1992, 50, 225–234. [Google Scholar] [CrossRef]
- Beaulieu, C.; Isabel, E.; Fortier, A.; Massé, F.; Mellon, C.; Méthot, N.; Ndao, M.; Nicoll-Griffith, D.; Lee, D.; Park, H.; et al. Identification of potent and reversible cruzipain inhibitors for the treatment of Chagas disease. Bioorganic Med. Chem. Lett. 2010, 20, 7444–7449. [Google Scholar] [CrossRef]
- Duschak, V.G. Major kinds of drug targets in Chagas disease or American Trypanosomiasis. Curr. Drug Targets 2019, 20, 1203–1216. [Google Scholar] [CrossRef]
- Souto-Padron, T.; Campetella, O.E.; Cazzulo, J.J.; de Souza, W. Cysteine proteinase in Trypanosoma cruzi: Immunocytochemical localization and involvement in parasite-host cell interaction. J. Cell Sci. 1990, 96, 485–490. [Google Scholar] [CrossRef]
- Berasain, P.; Carmona, C.; Frangione, B.; Cazzulo, J.J.; Goñi, F. Specific cleavage sites on human IgG subclasses by cruzipain, the major cysteine proteinase from Trypanosoma cruzi. Mol. Biochem. Parasitol. 2003, 130, 23–29. [Google Scholar] [CrossRef]
- Duschak, V.G.; Riarte, A.; Segura, E.L.; Laucella, S.A. Humoral immune response to cruzipain and cardiac dysfunction in chronic Chagas disease. Immunol. Lett. 2001, 78, 135–142. [Google Scholar] [CrossRef]
- Giordanengo, L.; Guiñazú, N.; Stempin, C.; Fretes, R.; Cerban, F.; Gea, S. Cruzipain, a major Trypanosoma cruzi antigen, conditions the host immune response in favor of parasite. Eur. J. Immunol. 2002, 32, 1003–1011. [Google Scholar] [CrossRef]
- Santos, V.C.; Rocha-Oliveira, A.E.; Broilo-Campos, A.C.; Reis-Cunha, J.L.; Bartholomeu, D.C.; Teixeira, S.M.R.; Lima, A.P.C.A.; Ferreira, R.S. The gene repertoire of the main cysteine protease of Trypanosoma cruzi, cruzipain, reveals four sub-types with distinct active sites. Sci. Rep. 2021, 11, 18231. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Arnaiz, M.; Mirkin, G.A.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Redirection of the immune response to the functional catalytic domain of the cystein proteinase cruzipain improves protective immunity against Trypanosoma cruzi infection. J. Infect. Dis. 2010, 202, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duschak, V.G.; Couto, A.S. Cruzipain, the major cysteine protease of Trypanosoma cruzi: A sulfated glycoprotein antigen as relevant candidate for vaccine development and drug target. A review. Curr. Med. Chem. 2009, 16, 3174–3202. [Google Scholar] [CrossRef]
- Cuevas, I.C.; Cazzulo, J.J.; Sánchez, D.O. gp63 homologues in Trypanosoma cruzi: Surface antigens with metalloprotease activity and a possible role in host cell infection. Infect. Immun. 2003, 71, 5739–5749. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, S.M.; Russell, D.G.; Kirchhoff, L.V.; Donelson, J.E. A differentially expressed gene family encoding “amastin,” a surface protein of Trypanosoma cruzi amastigotes. J. Biol. Chem. 1994, 269, 20509. [Google Scholar] [CrossRef]
- Cruz, M.C.; Souza-Melo, N.; da Silva, C.V.; DaRocha, W.D.; Bahia, D.; Araújo, P.R.; Teixeira, S.R.; Mortara, R.A. Trypanosoma cruzi: Role of δ-amastin on extracellular amastigote cell invasion and differentiation. PLoS ONE 2012, 7, e51804. [Google Scholar] [CrossRef] [Green Version]
- Gunter, S.M.; Jones, K.M.; Zhan, B.; Essigmann, H.T.; Murray, K.O.; Garcia, M.N.; Gorchakov, R.; Bottazzi, M.E.; Hotez, P.J.; Brown, E.L. Identification and characterization of the Trypanosoma cruzi B-cell superantigen Tc24. Am. J. Trop. Med. Hyg. 2016, 94, 114. [Google Scholar] [CrossRef] [Green Version]
- Arnal, A.; Villanueva-Lizama, L.; Teh-Poot, C.; Herrera, C.; Dumonteil, E. Extent of polymorphism and selection pressure on the Trypanosoma cruzi vaccine candidate antigen Tc24. Evol. Appl. 2020, 13, 2663–2672. [Google Scholar] [CrossRef]
- Versteeg, L.; Adhikari, R.; Poveda, C.; Villar-Mondragon, M.J.; Jones, K.M.; Hotez, P.J.; Bottazzi, M.E.; Tijhaar, E.; Pollet, J. Location and expression kinetics of Tc24 in different life stages of Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2021, 15, e0009689. [Google Scholar] [CrossRef]
- Gunter, S.M.; Versteeg, L.; Jones, K.M.; Keegan, B.P.; Strych, U.; Bottazzi, M.E.; Hotez, P.J.; Brown, E.L. Covalent vaccination with Trypanosoma cruzi Tc24 induces catalytic antibody production. Parasite Immunol. 2018, 40, e12585. [Google Scholar] [CrossRef]
- Rappuoli, R. Reverse vaccinology. Curr. Opin. Microbiol. 2000, 3, 445–450. [Google Scholar] [CrossRef]
- Moxon, R.; Reche, P.A.; Rappuoli, R. Reverse vaccinology. Front. Immunol. 2019, 10, 2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Sinha, M.; Luxon, B.; Garg, N. Utility of the Trypanosoma cruzi sequence database for identification of potential vaccine candidates by in silico and in vitro screening. Infect. Immun. 2004, 72, 6245–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Garg, N.J. Previously unrecognized vaccine candidates control Trypanosoma cruzi infection and immunopathology in mice. Clin. Vaccine Immunol. 2008, 15, 1158–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Garg, N.J. A two-component DNA-prime/protein-boost vaccination strategy for eliciting long-term, protective T cell immunity against Trypanosoma cruzi. PLoS Pathog. 2015, 11, e1004828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- das Neves, R.F.C.; Adade, C.M.; Fernandes, A.C.S.; Lopes, A.H.; Souto-Padrón, T. Trypanosoma cruzi: Cell surface dynamics in trypomastigotes of different strains. Parasitology 2020, 147, 310–321. [Google Scholar] [CrossRef]
- Rodrigues da Cunha, G.M.; Azevedo, M.A.; Nogueira, D.S.; de Carvalho Clímaco, M.; Ayala, E.V.; Chunga, J.A.J.; La Valle, R.J.Y.; da Cunha Galvão, L.M.; Chiari, E.; Brito, C.R.N.; et al. α-Gal immunization positively impacts Trypanosoma cruzi colonization of heart tissue in a mouse model. PLoS Negl. Trop. Dis. 2021, 15, e0009613. [Google Scholar] [CrossRef]
- Alberti, A.S.; Bivona, A.E.; Cerny, N.; Schulze, K.; Weißmann, S.; Ebensen, T.; Morales, C.; Padilla, A.M.; Cazorla, S.I.; Tarleton, R.L.; et al. Engineered trivalent immunogen adjuvanted with a STING agonist confers protection against Trypanosoma cruzi infection. NPJ Vaccines 2017, 2, 9. [Google Scholar] [CrossRef]
- Pereira, I.R.; Vilar-Pereira, G.; Marques, V.; da Silva, A.A.; Caetano, B.; Moreira, O.C.; Machado, A.V.; Bruna-Romero, O.; Rodrigues, M.M.; Gazzinelli, R.T. A human type 5 adenovirus-based Trypanosoma cruzi therapeutic vaccine re-programs immune response and reverses chronic cardiomyopathy. PLoS Pathog. 2015, 11, e1004594. [Google Scholar] [CrossRef]
- Lokugamage, N.; Choudhuri, S.; Davies, C.; Chowdhury, I.H.; Garg, N.J. Antigen-based nano-immunotherapy controls parasite persistence, inflammatory and oxidative stress, and cardiac fibrosis, the hallmarks of chronic Chagas cardiomyopathy, in a mouse model of Trypanosoma cruzi infection. Vaccines 2020, 8, 96. [Google Scholar] [CrossRef] [Green Version]
- Bontempi, I.; Leal, K.; Prochetto, E.; Díaz, G.; Cabrera, G.; Bortolotti, H.R.; Borsuk, S.; Dellagostin, O.A.; Marcipar, I. Recombinant Mycobacterium bovis BCG is a promising platform to develop vaccines against Trypansoma cruzi infection. Clin. Exp. Immunol. 2020, 201, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Tenney, T.D.; Curtis-Robles, R.; Snowden, K.F.; Hamer, S.A. Shelter dogs as sentinels for Trypanosoma cruzi transmission across Texas. Emerg. Infect. Dis. 2014, 20, 1323. [Google Scholar] [CrossRef] [PubMed]
- Cazorla, S.I.; Matos, M.N.; Cerny, N.; Ramirez, C.; Alberti, A.S.; Bivona, A.E.; Morales, C.; Guzmán, C.A.; Malchiodi, E.L. Oral multicomponent DNA vaccine delivered by attenuated Salmonella elicited immunoprotection against American trypanosomiasis. J. Infect. Dis. 2015, 211, 698–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, M.A.; Versteeg, L.; Wang, Q.; Pollet, J.; Zhan, B.; Gusovsky, F.; Bottazzi, M.E.; Hotez, P.J.; Jones, K.M. A therapeutic vaccine prototype induces protective immunity and reduces cardiac fibrosis in a mouse model of chronic Trypanosoma cruzi infection. PLoS Negl. Trop. Dis. 2019, 13, e0007413. [Google Scholar] [CrossRef]
- Villanueva-Lizama, L.E.; Cruz-Chan, J.V.; Aguilar-Cetina, A.D.C.; Herrera-Sanchez, L.F.; Rodriguez-Perez, J.M.; Rosado-Vallado, M.E.; Ramirez-Sierra, M.J.; Ortega-Lopez, J.; Jones, K.; Hotez, P.; et al. Trypanosoma cruzi vaccine candidate antigens Tc24 and TSA-1 recall memory immune response associated with HLA-A and-B supertypes in Chagasic chronic patients from Mexico. PLoS Negl. Trop. Dis. 2018, 12, e0006240. [Google Scholar] [CrossRef]
- Liu, M.A. DNA vaccines: A review. J. Intern. Med. 2003, 253, 402–410. [Google Scholar] [CrossRef] [Green Version]
- Franck, C.O.; Fanslau, L.; Popov, A.B.; Tyagi, P.; Fruk, L. Biopolymer-based Carriers for DNA Vaccine Design. Angew. Chem. 2021, 133, 13333–13351. [Google Scholar] [CrossRef]
- Shafaati, M.; Saidijam, M.; Soleimani, M.; Hazrati, F.; Mirzaei, R.; Amirheidari, B.; Tanzadehpanah, H.; Karampoor, S.; Kazemi, S.; Yavari, B.; et al. A brief review on DNA vaccines in the era of COVID-19. Future Virol. 2022, 17, 49–66. [Google Scholar] [CrossRef]
- Krinner, S.; Heitzer, A.; Asbach, B.; Wagner, R. Interplay of promoter usage and intragenic CpG content: Impact on GFP reporter gene expression. Hum. Gene Ther. 2015, 26, 826–840. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Cheng, S.; Kadonaga, J.T. Rational design of a super core promoter that enhances gene expression. Nat. Methods 2006, 3, 917–922. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- Balmayor, E.R. Synthetic mRNA–emerging new class of drug for tissue regeneration. Curr. Opin. Biotechnol. 2022, 74, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Ross, J. Control of messenger RNA stability in higher eukaryotes. Trends Genet. 1996, 12, 171–175. [Google Scholar] [CrossRef]
- Grudzien, E.W.A.; Stepinski, J.; Jankowska-Anyszka, M.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel cap analogs for in vitro synthesis of mRNAs with high translational efficiency. RNA 2004, 10, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Urbina, F.; Morales-Pison, S.; Maldonado, E. Enzymatic protein biopolymers as a tool to synthetize eukaryotic messenger ribonucleic acid (mRNA) with uses in vaccination, immunotherapy and nanotechnology. Polymers 2020, 12, 1633. [Google Scholar] [CrossRef]
- Sánchez-Valdéz, F.J.; Brandán, C.P.; Ferreira, A.; Basombrío, M.A. Gene-deleted live-attenuated Trypanosoma cruzi parasites as vaccines to protect against Chagas disease. Expert Rev. Vaccines 2015, 14, 681–697. [Google Scholar] [CrossRef]
- Solana, J.C.; Moreno, J.; Iborra, S.; Soto, M.; Requena, J.M. Live attenuated vaccines, a favorable strategy to provide long-term immunity against protozoan diseases. Trends Parasitol. 2021, 38, 316–334. [Google Scholar] [CrossRef]
- Rodríguez, E.V.A.; Furlan, C.L.A.; Vernengo, F.F.; Montes, C.L.; Gruppi, A. Understanding CD8+ T cell immunity to Trypanosoma cruzi and how to improve it. Trends Parasitol. 2019, 35, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Basombrío, M.A.; Besuschio, S. Trypanosoma cruzi culture used as vaccine to prevent chronic Chagas’ disease in mice. Infect. Immun. 1982, 36, 351–356. [Google Scholar] [CrossRef] [Green Version]
- Basombrío, M.A.; Segura, M.A.; Mora, M.C.; Gomez, L. Field trial of vaccination against American trypanosomiasis (Chagas’ disease) in dogs. Am. J. Trop. Med. Hyg. 1993, 49, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Barrio, A.B.; Van Voorhis, W.C.; Basombrío, M.A. Trypanosoma cruzi: Attenuation of virulence and protective immunogenicity after monoallelic disruption of the cub gene. Exp. Parasitol. 2007, 117, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.P.; Barrio, A.B.; Cardozo, R.M.; Duffy, T.; Schijman, A.G.; Basombrío, M.A. Impairment of infectivity and immunoprotective effect of a LYT1 null mutant of Trypanosoma cruzi. Infect. Immun. 2008, 76, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Tao, P.; Mahalingam, M.; Marasa, B.S.; Zhang, Z.; Chopra, A.K.; Rao, V.B. In vitro and in vivo delivery of genes and proteins using the bacteriophage T4 DNA packaging machine. Proc. Natl. Acad. Sci. USA 2013, 110, 5846–5851. [Google Scholar] [CrossRef] [Green Version]
- Tao, P.; Zhu, J.; Mahalingam, M.; Batra, H.; Rao, V.B. Bacteriophage T4 nanoparticles for vaccine delivery against infectious diseases. Adv. Drug Deliv. Rev. 2019, 145, 57–72. [Google Scholar] [CrossRef]
- Yap, M.L.; Rossmann, M.G. Structure and function of bacteriophage T4. Future Microbiol. 2014, 9, 1319–1327. [Google Scholar] [CrossRef] [Green Version]
- Bode, C.; Zhao, G.; Steinhagen, F.; Kinjo, T.; Klinman, D.M. CpG DNA as a vaccine adjuvant. Expert Rev. Vaccines 2011, 10, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Chu, R.S.; Targoni, O.S.; Krieg, A.M.; Lehmann, P.V.; Harding, C.V. CpG oligodeoxynucleotides act as adjuvants that switch on T helper 1 (Th1) immunity. J. Exp. Med. 1997, 186, 1623–1631. [Google Scholar] [CrossRef] [Green Version]
- Roman, M.; Martin-Orozco, E.; Goodman, J.S.; Nguyen, M.-D.; Sato, Y.; Ronaghy, A.; Kornbluth, R.S.; Richman, D.D.; Carson, D.A.; Raz, E. Immunostimulatory DNA sequences function as T helper-1-promoting adjuvants. Nat. Med. 1997, 3, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Krieg, A.M.; Yi, A.K.; Matson, S.; Waldschmidt, T.J.; Bishop, G.A.; Teasdale, R.; Koretzky, G.A.; Klinman, D.M. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 1995, 374, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Frank, F.M.; Petray, P.B.; Cazorla, S.I.; Muñoz, M.C.; Corral, R.S.; Malchiodi, E.L. Use of a purified Trypanosoma cruzi antigen and CpG oligodeoxynucleotides for immunoprotection against a lethal challenge with trypomastigotes. Vaccine 2003, 22, 77–86. [Google Scholar] [CrossRef]
- Schnapp, A.R.; Eickhoff, C.S.; Sizemore, D.; Curtiss, R., 3rd; Hoft, D.F. Cruzipain induces both mucosal and systemic protection against Trypanosoma cruzi in mice. Infect. Immun. 2002, 70, 5065–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araújo, A.F.S.; de Alencar, B.C.G.; Vasconcelos, J.R.C.; Hiyane, M.I.; Marinho, C.R.F.; Penido, M.L.O.; Boscardin, S.B.; Hoft, D.F.; Gazzinelli, R.T.; Rodrigues, M.M. CD8+-T-cell-dependent control of Trypanosoma cruzi infection in a highly susceptible mouse strain after immunization with recombinant proteins based on amastigote surface protein 2. Infect. Immun. 2005, 73, 6017–6025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoft, D.F.; Eickhoff, C.S.; Giddings, O.K.; Vasconcelos, J.R.C.; Rodrigues, M.M. Trans-sialidase recombinant protein mixed with CpG motif-containing oligodeoxynucleotide induces protective mucosal and systemic Trypanosoma cruzi immunity involving CD8+ CTL and B cell-mediated cross-priming. J. Immunol. 2007, 179, 6889–6900. [Google Scholar] [CrossRef] [Green Version]
- Giddings, O.K.; Eickhoff, C.S.; Sullivan, N.L.; Hoft, D.F. Intranasal vaccinations with the trans-sialidase antigen plus CpG Adjuvant induce mucosal immunity protective against conjunctival Trypanosoma cruzi challenges. Infect. Immun. 2010, 78, 1333–1338. [Google Scholar] [CrossRef] [Green Version]
- Ouaissi, A.; Guilvard, E.; Delneste, Y.; Caron, G.; Magistrelli, G.; Herbault, N.; Thieblemont, N.; Jeannin, P. The Trypanosoma cruzi Tc52-released protein induces human dendritic cell maturation, signals via Toll-like receptor 2, and confers protection against lethal infection. J. Immunol. 2002, 168, 6366–6374. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, S.I.; Frank, F.M.; Becker, P.D.; Corral, R.S.; Guzmán, C.A.; Malchiodi, E.L. Prime-boost immunization with cruzipain co-administered with MALP-2 triggers a protective immune response able to decrease parasite burden and tissue injury in an experimental Trypanosoma cruzi infection model. Vaccine 2008, 26, 1999–2009. [Google Scholar] [CrossRef] [Green Version]
- Zygmunt, B.M.; Weissmann, S.F.; Guzman, C.A. NKT cell stimulation with α-galactosylceramide results in a block of Th17 differentiation after intranasal immunization in mice. PLoS ONE 2012, 7, e30382. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, S.; Uchida, S. Multifunctional Immunoadjuvants for Use in Minimalist Nucleic Acid Vaccines. Pharmaceutics 2021, 13, 644. [Google Scholar] [CrossRef] [PubMed]
- Assis, B.R.D.; da Silva, C.D.; Santiago, M.G.; Ferreira, L.A.M.; Goulart, G.A.C. Nanotechnology in adjuvants and vaccine development: What should we know? Nanomedicine 2021, 16, 2565–2568. [Google Scholar] [CrossRef] [PubMed]
- Alfagih, I.M.; Aldosari, B.; AlQuadeib, B.; Almurshedi, A.; Alfagih, M.M. Nanoparticles as adjuvants and nanodelivery systems for mRNA-based vaccines. Pharmaceutics 2021, 13, 45. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Abreu, H.; Casale, C.; Dianzani, U.; Chiocchetti, A. Nano-microparticle platforms in developing next-generation vaccines. Vaccines 2021, 9, 606. [Google Scholar] [CrossRef] [PubMed]
- Tenchov, R.; Bird, R.; Curtze, A.E.; Zhou, Q. Lipid Nanoparticles—From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano 2021, 15, 16982–17015. [Google Scholar] [CrossRef]
- Li, Y.; Tenchov, R.; Smoot, J.; Liu, C.; Watkins, S.; Zhou, Q. A comprehensive review of the global efforts on COVID-19 vaccine development. ACS Cent. Sci. 2021, 7, 512–533. [Google Scholar] [CrossRef]
- Arce-Fonseca, M.; Rios-Castro, M.; Carrillo-Sánchez, S.d.C.; Martínez-Cruz, M.; Rodríguez-Morales, O. Prophylactic and therapeutic DNA vaccines against Chagas disease. Parasites Vectors 2015, 8, 121. [Google Scholar] [CrossRef] [Green Version]
- Lokugamage, N.; Chowdhury, I.H.; Biediger, R.J.; Market, R.V.; Khounlo, S.; Warier, N.D.; Hwang, S.-A.; Actor, J.K.; Woodside, D.G.; Marathi, U.; et al. Use of a small molecule integrin activator as a systemically administered vaccine adjuvant in controlling Chagas disease. NPJ Vaccines 2021, 6, 114. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maldonado, E.; Morales-Pison, S.; Urbina, F.; Solari, A. Vaccine Design against Chagas Disease Focused on the Use of Nucleic Acids. Vaccines 2022, 10, 587. https://doi.org/10.3390/vaccines10040587
Maldonado E, Morales-Pison S, Urbina F, Solari A. Vaccine Design against Chagas Disease Focused on the Use of Nucleic Acids. Vaccines. 2022; 10(4):587. https://doi.org/10.3390/vaccines10040587
Chicago/Turabian StyleMaldonado, Edio, Sebastian Morales-Pison, Fabiola Urbina, and Aldo Solari. 2022. "Vaccine Design against Chagas Disease Focused on the Use of Nucleic Acids" Vaccines 10, no. 4: 587. https://doi.org/10.3390/vaccines10040587
APA StyleMaldonado, E., Morales-Pison, S., Urbina, F., & Solari, A. (2022). Vaccine Design against Chagas Disease Focused on the Use of Nucleic Acids. Vaccines, 10(4), 587. https://doi.org/10.3390/vaccines10040587