
Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures

 ,
,  , , and
, , and
Abstract
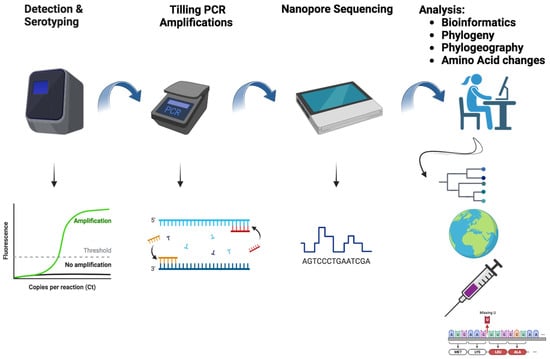
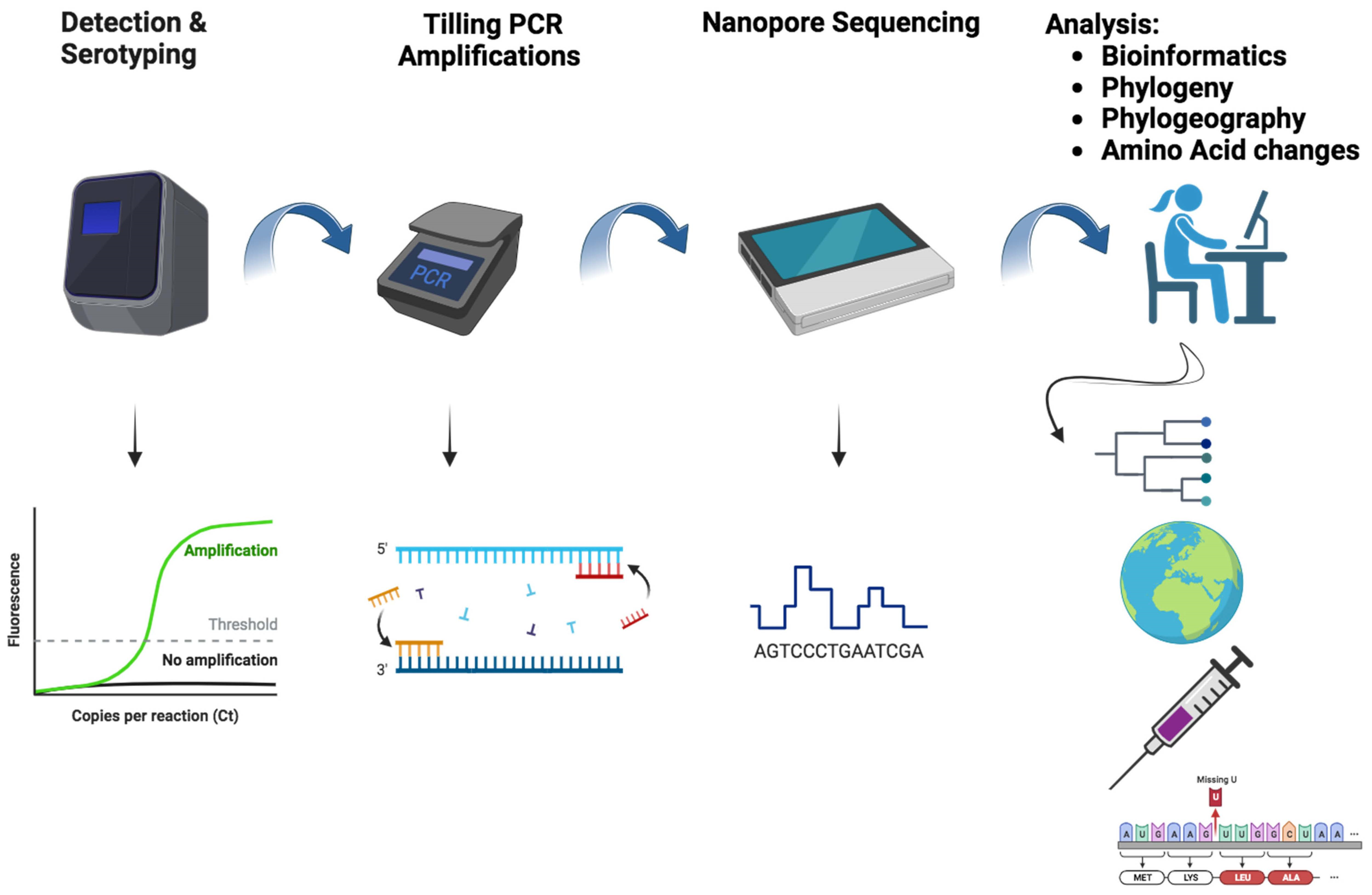
:
1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. RNA Extraction
2.3. Dengue Virus Detection and Serotyping with qRT-PCR
2.4. cDNA Synthesis and Tilling PCR Amplification
2.5. Sequencing and Genome Assembly
2.6. Dataset Construction
2.7. Phylogenetic Analysis
2.8. SNP Calling and Population Genetics Analysis
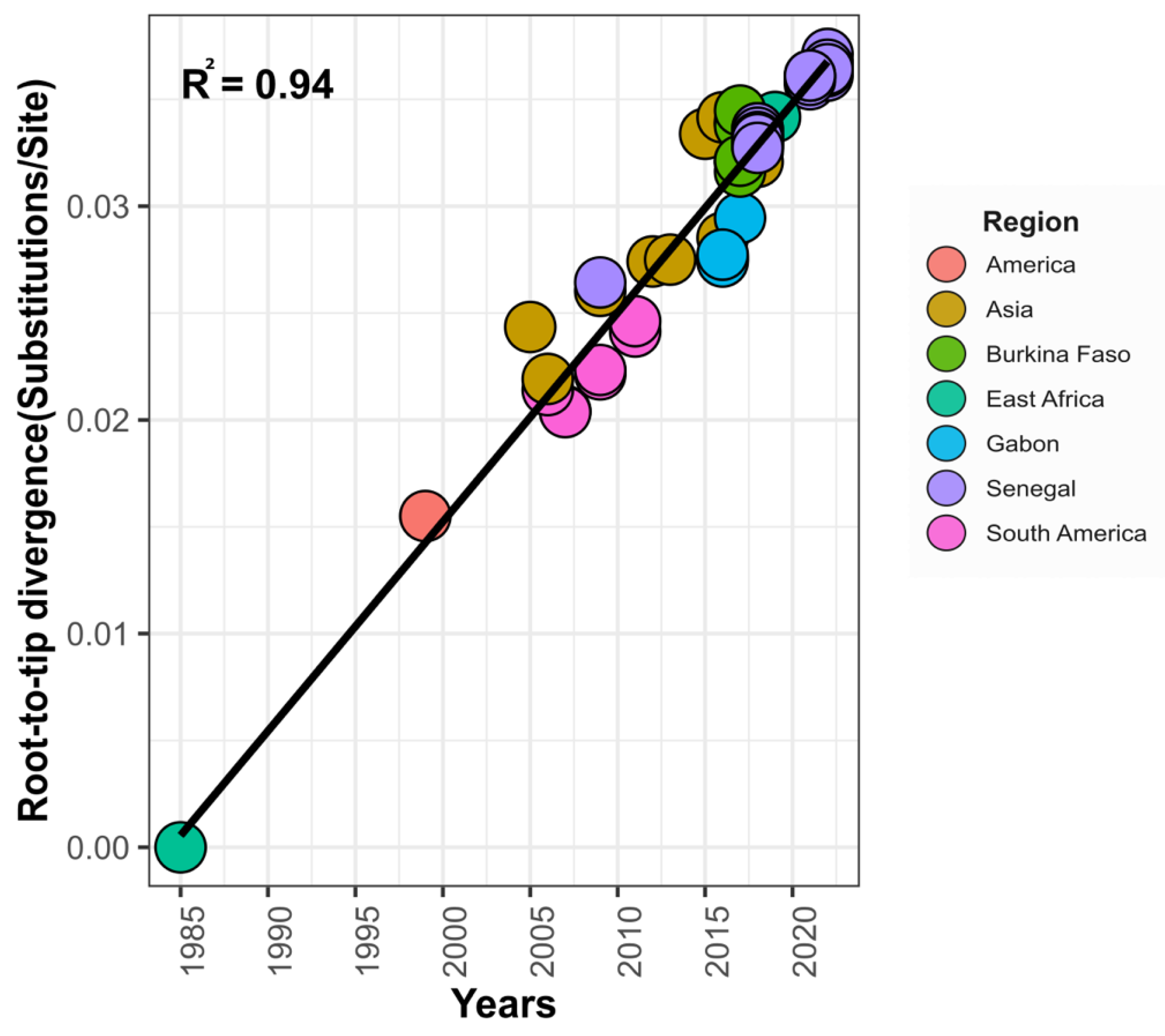
2.9. Bayesian Phylogeographic Analysis
2.10. Amino Acid Changes against Vaccinal Strains and Human DENV−3 mAb Epitopes
3. Results
3.1. Spatial and Temporal Distribution of Sequenced Strains
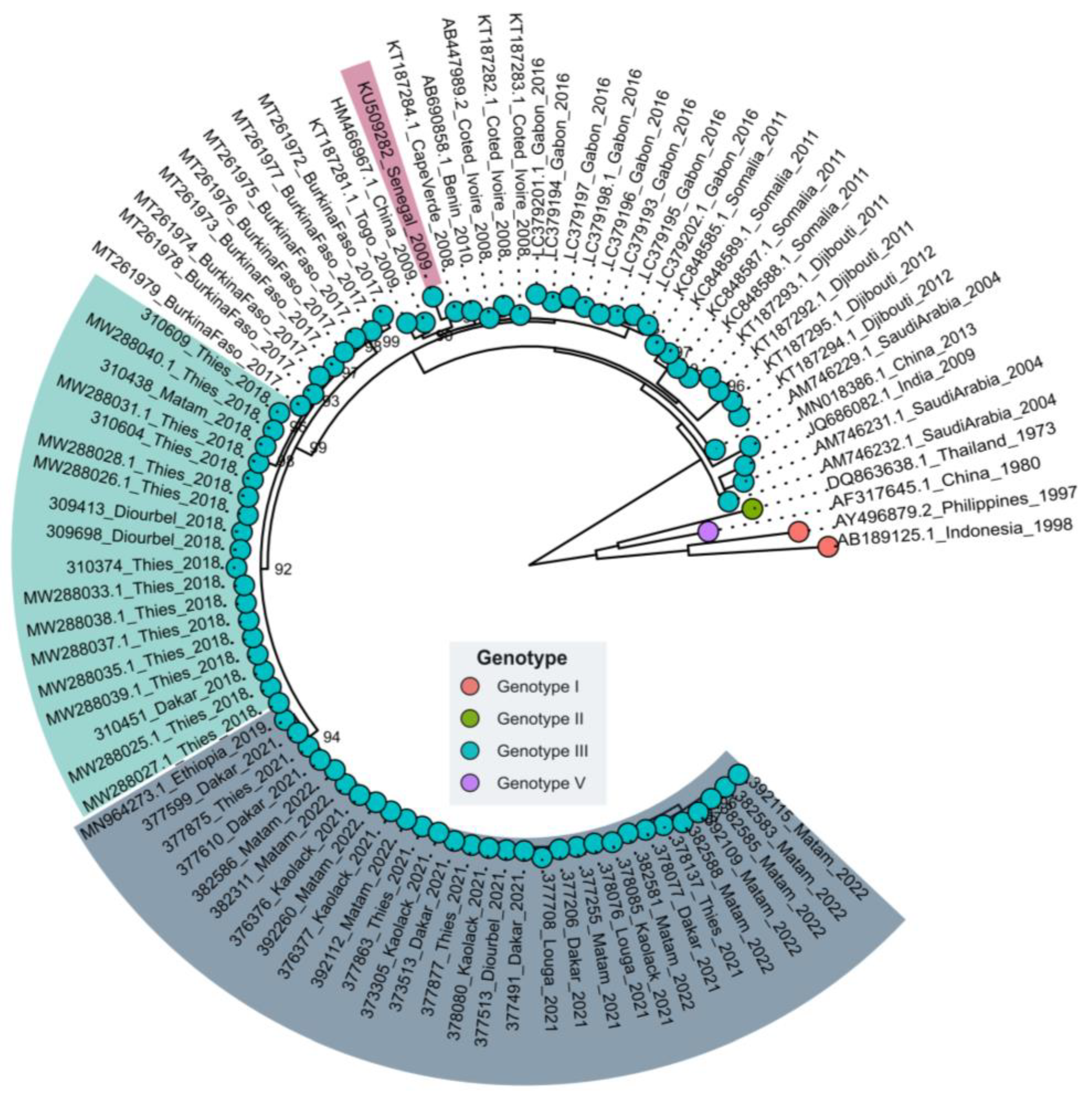
3.2. Phylogenetic Relationships of Senegalese DENV−3
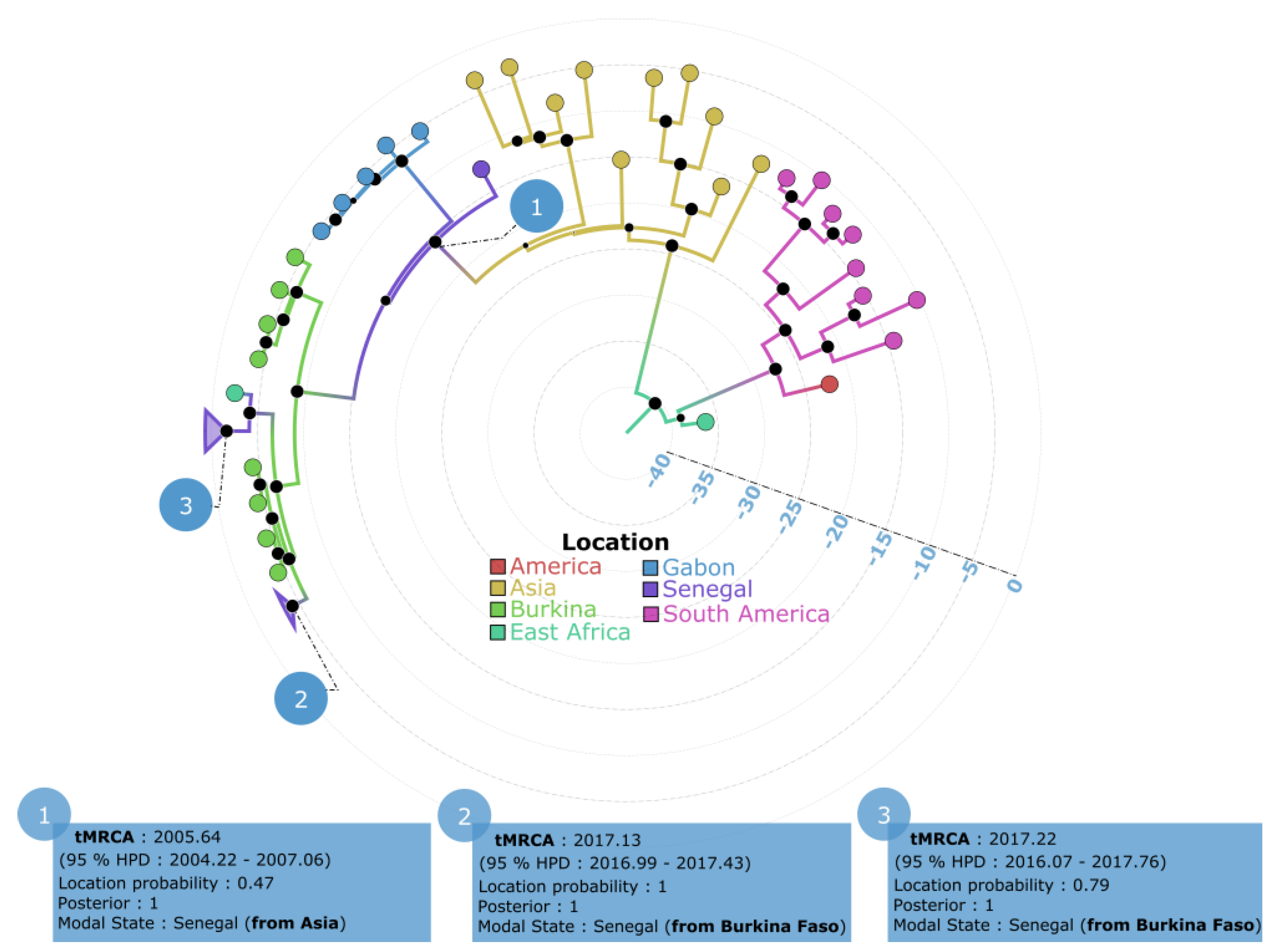
3.3. Phylogeographic Pattern of DENV−3 Circulation in Senegal
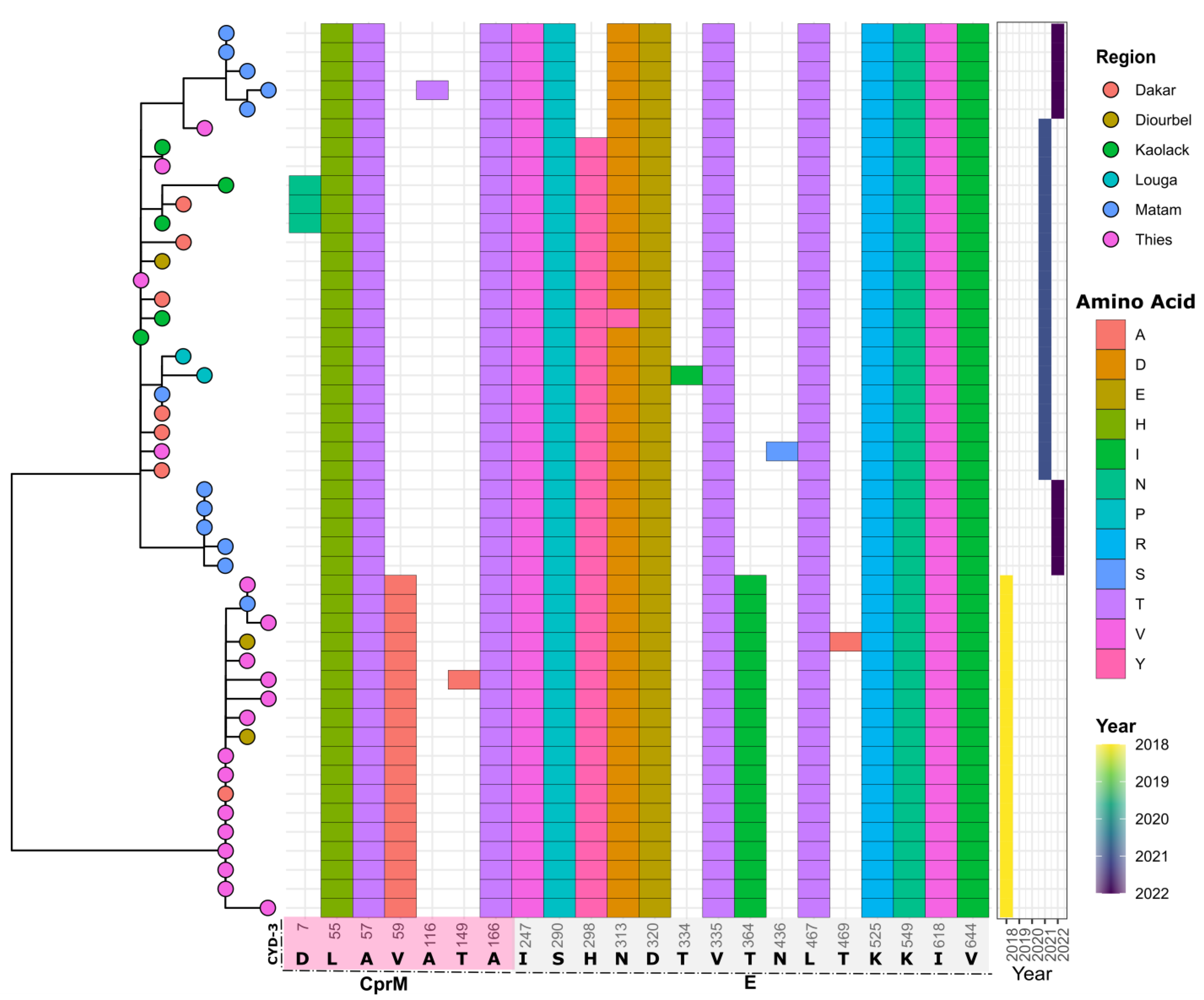
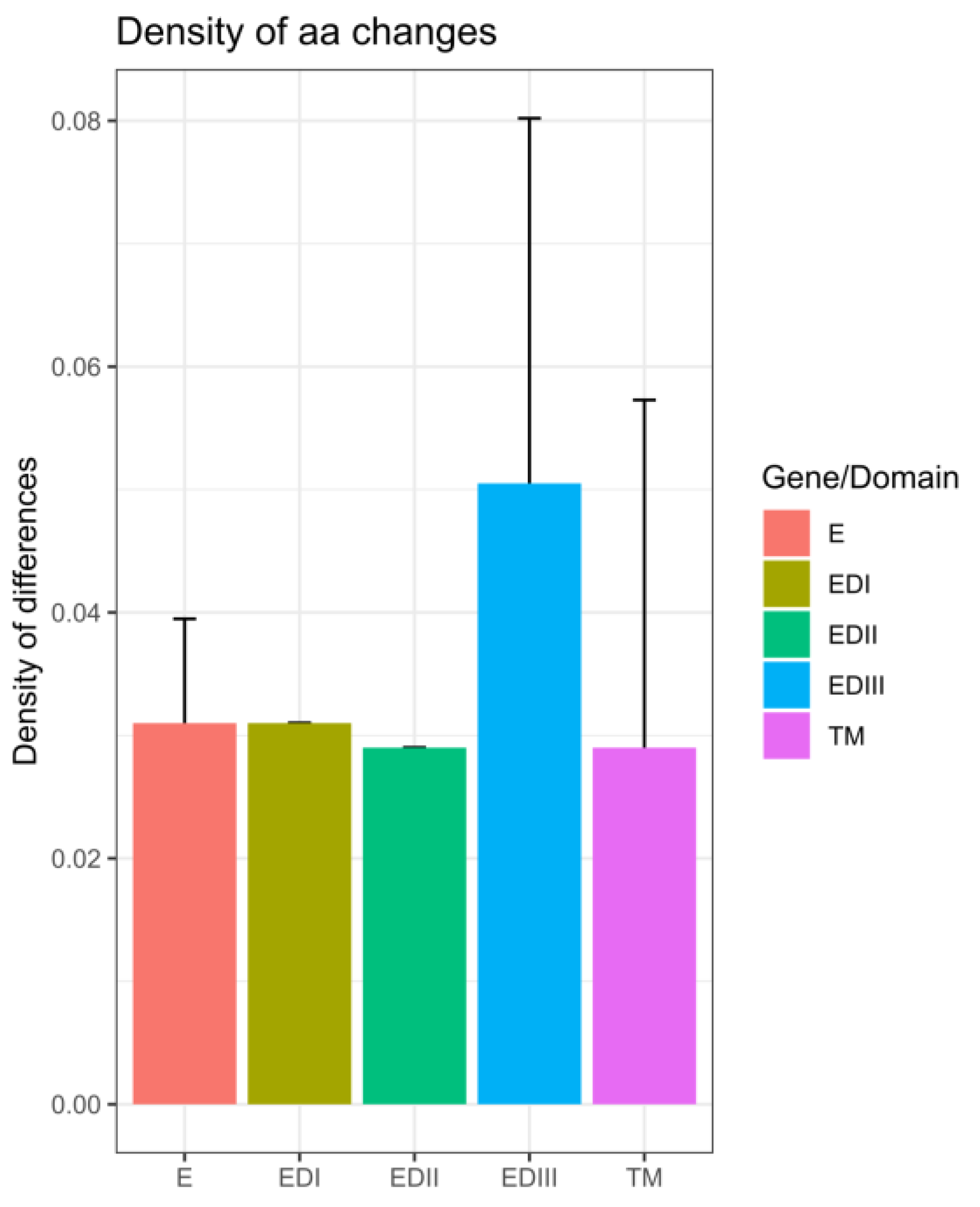
3.4. In Silico Assessment of Impact of Amino Acid Changes against the Vaccines (CYD-3 and TV003) and Human mAB (mAB 5J7)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harapan, H.; Michie, A.; Sasmono, R.T.; Imrie, A. Dengue: A Minireview. Viruses 2020, 12, 829. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus Genome Organization, Expression, and Replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Fonville, J.M.; Gromowski, G.D.; Arriaga, J.B.; Green, A.; James, S.L.; Smith, D.J. Dengue viruses cluster antigenically but not as discrete serotypes. Science 2015, 349, 1338–1343. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.K.; Carabali, M.; Lee, J.S.; Lee, K.S.; Namkung, S.; Lim, S.K.; Yoon, I.K. Evaluating dengue burden in Africa in passive fever surveillance and seroprevalence studies: Protocol of field studies of the Dengue Vaccine Initiative. BMJ Open 2018, 8, e017673. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Hay, S.I. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, A.; Kuritsky, J.N.; Letson, G.W.; Margolis, H.S. Dengue Virus Infection in Africa. Emerg. Infect. Dis. 2011, 17, 1349–1354. [Google Scholar] [CrossRef]
- Ayolabi, C.I.; Olusola, B.A.; Ibemgbo, S.A.; Okonkwo, G.O. Detection of Dengue viruses among febrile patients in Lagos, Nigeria and phylogenetics of circulating Dengue serotypes in Africa. Infect. Genet. Evol. 2019, 75, 103947. [Google Scholar] [CrossRef]
- Franco, L.; Di Caro, A.; Carletti, F.; Vapalahti, O.; Renaudat, C.; Zeller, H.; Tenorio, A. Recent expansion of dengue virus serotype 3 in West Africa. Eurosurveillance 2010, 15, 19490. [Google Scholar] [CrossRef]
- Abe, H.; Ushijima, Y.; Loembe, M.M.; Bikangui, R.; Nguema-Ondo, G.; Mpingabo, P.I.; Yasuda, J. Re-emergence of dengue virus serotype 3 infections in Gabon in 2016–2017, and evidence for the risk of repeated dengue virus infections. Int. J. Infect. Dis. 2020, 91, 129–136. [Google Scholar] [CrossRef]
- Abreu, C.; Silva-Pinto, A.; Lazzara, D.; Sobrinho-Simões, J.; Guimarães, J.T.; Sarmento, A. Imported dengue from 2013 Angola outbreak: Not just serotype 1 was detected. J. Clin. Virol. 2016, 79, 77–79. [Google Scholar] [CrossRef]
- Dieng, I.; Ndione MH, D.; Fall, C.; Diagne, M.M.; Diop, M.; Gaye, A.; Faye, O. Multifoci and multiserotypes circulation of dengue virus in Senegal between 2017 and 2018. BMC Infect. Dis. 2021, 21, 867. [Google Scholar] [CrossRef] [PubMed]
- Dieng, I.; Fall, C.; Barry, M.A.; Gaye, A.; Dia, N.; Ndione, M.H.D.; Sall, A.A. Re-Emergence of Dengue Serotype 3 in the Context of a Large Religious Gathering Event in Touba, Senegal. Int. J. Environ. Res. Public Health 2022, 19, 16912. [Google Scholar] [CrossRef] [PubMed]
- Gaye, A.; Ndiaye, T.; Sy, M.; Deme, A.B.; Thiaw, A.B.; Sene, A.; Ndiaye, D. Genomic investigation of a dengue virus outbreak in Thiès, Senegal, in 2018. Sci. Rep. 2021, 11, 10321. [Google Scholar] [CrossRef] [PubMed]
- Torres-Flores, J.M.; Reyes-Sandoval, A.; Salazar, M.I. Dengue Vaccines: An Update. BioDrugs 2022, 36, 325–336. [Google Scholar] [CrossRef]
- Usme-Ciro, J.A.; Méndez, J.A.; Laiton, K.D.; Páez, A. The relevance of dengue virus genotypes surveillance at country level before vaccine approval. Hum. Vaccines Immunother. 2014, 10, 2674–2678. [Google Scholar] [CrossRef]
- Letizia, A.G.; Pratt, C.B.; Wiley, M.R.; Fox, A.T.; Mosore, M.; Agbodzi, B.; Sangaré, L. Retrospective Genomic Characterization of a 2017 Dengue Virus Outbreak, Burkina Faso. Emerg. Infect. Dis. 2022, 28, 1198. [Google Scholar] [CrossRef]
- Hill, S.C.; de Vasconcelos, J.N.; Granja, B.G.; Thézé, J.; Jandondo, D.; Neto, Z.; Afonso, J.M. Early Genomic Detection of Cosmopolitan Genotype of Dengue Virus Serotype 2, Angola, 2018. Emerg. Infect. Dis. 2019, 25, 784–787. [Google Scholar] [CrossRef]
- Dieng, I.; Barry, M.A.; Talla, C.; Sow, B.; Faye, O.; Diagne, M.M.; Faye, O. Analysis of a Dengue Virus Outbreak in Rosso, Senegal 2021. Trop. Med. Infect. Dis. 2022, 7, 420. [Google Scholar] [CrossRef]
- Wagner, D.; de With, K.; Huzly, D.; Hufert, F.; Weidmann, M.; Breisinger, S.; Bauer, T.M. Nosocomial Acquisition of Dengue. Emerg. Infect. Dis. 2004, 10, 1872–1873. [Google Scholar] [CrossRef]
- Dieng, I.; Talla, C.; Fauver, J.; Ndiaye, M.; Sagne, S.N.; Barry, M.A.; Faye, O. Reemergence of Sylvatic Dengue Virus in Southern Senegal, 2021. In Infectious Diseases; Aparecida Sperança, M., Ed.; IntechOpen: London, UK, 2023; Available online: https://www.intechopen.com/chapters/86883 (accessed on 8 August 2023).
- Dieng, I.; Diallo, A.; Ndiaye, M.; Mhamadi, M.; Diagne, M.M.; Sankhe, S.; Faye, O. Full genome analysis of circulating DENV-2 in Senegal reveals a regional diversification into separate clades. J. Med. Virol. 2022, 94, 5593–5600. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. 2007, 1, 117693430500100003. [Google Scholar] [CrossRef]
- R Core Team. R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Faye, O.; Ba, Y.; Faye, O.; Talla, C.; Diallo, D.; Chen, R.; Sall, A.A. Urban Epidemic of Dengue Virus Serotype 3 Infection, Senegal, 2009. Emerg. Infect. Dis. 2014, 20, 456–459. [Google Scholar] [CrossRef]
- Sokhna, C.; Mboup, B.M.; Sow, P.G.; Camara, G.; Dieng, M.; Sylla, M.; Gautret, P. Communicable and non-communicable disease risks at the Grand Magal of Touba: The largest mass gathering in Senegal. Travel Med. Infect. Dis. 2017, 18, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Tarnagda, Z.; Cissé, A.; Bicaba, B.W.; Diagbouga, S.; Sagna, T.; Ilboudo, A.K.; Muscatello, D.J. Dengue Fever in Burkina Faso, 2016. Emerg. Infect. Dis. 2018, 24, 170–172. [Google Scholar] [CrossRef]
- Gubler, D.J. Dengue and Dengue Hemorrhagic Fever. Clin. Microbiol. Rev. 1998, 11, 17. [Google Scholar] [CrossRef] [PubMed]
- Brunette, G.W.; Nemhauser, J.B. Travel-Related Infectious Diseases. In CDC Yellow Book 2020; Oxford University Press: Oxford, UK, 2019; pp. 169–394. Available online: https://academic.oup.com/book/25289/chapter/191511526 (accessed on 18 July 2023).
- Messer, W.B.; Gubler, D.J.; Harris, E.; Sivananthan, K.; De Silva, A.M. Emergence and Global Spread of a Dengue Serotype 3, Subtype III Virus. Emerg. Infect. Dis. 2003, 9, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Khetarpal, N.; Khanna, I. Dengue Fever: Causes, Complications, and Vaccine Strategies. J. Immunol. Res. 2016, 2016, 6803098. [Google Scholar] [CrossRef]
- Izmirly, A.M.; Alturki, S.O.; Alturki, S.O.; Connors, J.; Haddad, E.K. Challenges in Dengue Vaccines Development: Pre-existing Infections and Cross-Reactivity. Front. Immunol. 2020, 11, 1055. [Google Scholar] [CrossRef]
- Gaillat, J. Vaccin Grippe: L’efficacité en Question. 2019. Available online: https://www.sf2h.net/k-stock/data/uploads/2018/09/06062019_SESSION_PARALLELE_SP09_1640_Salle_Marie_Curie_1_Niveau_1_Jacques_GAILLAT.pdf (accessed on 18 July 2023).
- Kim, H.; Webster, R.G.; Webby, R.J. Influenza Virus: Dealing with a Drifting and Shifting Pathogen. Viral Immunol. 2018, 31, 174–183. [Google Scholar] [CrossRef]
- Jagtap, S.; Pattabiraman, C.; Sankaradoss, A.; Krishna, S.; Roy, R. Evolutionary dynamics of dengue virus in India. PLoS Pathog. 2023, 19, e1010862. [Google Scholar] [CrossRef]
- Muné, M.; Rodríguez, R.; Ramírez, R.; Soto, Y.; Sierra, B.; Rodríguez Roche, R.; Guzmán, M.G. Carboxy-terminally truncated Dengue 4 virus envelope glycoprotein expressed in Pichia pastoris induced neutralizing antibodies and resistance to Dengue 4 virus challenge in mice. Arch. Virol. 2003, 148, 2267–2273. [Google Scholar] [CrossRef]
- Young, E.; Carnahan, R.H.; Andrade, D.V.; Kose, N.; Nargi, R.S.; Fritch, E.J.; Baric, R.S. Identification of Dengue Virus Serotype 3 Specific Antigenic Sites Targeted by Neutralizing Human Antibodies. Cell Host Microbe 2020, 27, 710–724.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.P.; Yu, M.; Jiang, T.; Deng, Y.Q.; Qin, C.F.; Han, J.F.; Qin, E.D. Identification of a recombinant dengue virus type 1 with 3 recombination regions in natural populations in Guangdong province, China. Arch. Virol. 2008, 153, 1175–1179. [Google Scholar] [CrossRef] [PubMed]
- Leng, C.H.; Liu, S.J.; Tsai, J.P.; Li, Y.S.; Chen, M.Y.; Liu, H.H.; Chen, H.W. A novel dengue vaccine candidate that induces cross-neutralizing antibodies and memory immunity. Microbes Infect. 2009, 11, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.S.; Nuckols, J.T.; Horne, K.M.; Vanlandingham, D.; Lobigs, M.; Higgs, S. Mutagenesis analysis of T380R mutation in the envelope protein of yellow fever virus. Virol. J. 2014, 11, 60. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source of Variation | Sum of Squares | Variance Components | Percentage Variation |
|---|---|---|---|
| Among populations | 130.12 | 4.02 | 80.25 |
| Within populations | 43.46 | 0.99 | 19.74 |
| Total | 173.79 | 5.01 |
| Demographic Growth Model | Relaxed Molecular Clock | |
|---|---|---|
| PS | SS | |
| Skyride | −27,752.10 | −27,752.40 |
| Skygrid | −27,732.34 | −27,732.50 |
| Skyline | −27,737.07 | −27,736.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dieng, I.; Balde, D.; Talla, C.; Camara, D.; Barry, M.A.; Sagne, S.N.; Gueye, K.; Dia, C.A.K.M.; Sambe, B.S.; Fall, G.; et al. Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures. Vaccines 2023, 11, 1537. https://doi.org/10.3390/vaccines11101537
Dieng I, Balde D, Talla C, Camara D, Barry MA, Sagne SN, Gueye K, Dia CAKM, Sambe BS, Fall G, et al. Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures. Vaccines. 2023; 11(10):1537. https://doi.org/10.3390/vaccines11101537
Chicago/Turabian StyleDieng, Idrissa, Diamilatou Balde, Cheikh Talla, Diogop Camara, Mamadou Aliou Barry, Samba Niang Sagne, Khadim Gueye, Cheikh Abdou Khadre Mbacké Dia, Babacar Souleymane Sambe, Gamou Fall, and et al. 2023. "Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures" Vaccines 11, no. 10: 1537. https://doi.org/10.3390/vaccines11101537
APA StyleDieng, I., Balde, D., Talla, C., Camara, D., Barry, M. A., Sagne, S. N., Gueye, K., Dia, C. A. K. M., Sambe, B. S., Fall, G., Sall, A. A., Faye, O., Loucoubar, C., & Faye, O. (2023). Molecular Evolution of Dengue Virus 3 in Senegal between 2009 and 2022: Dispersal Patterns and Implications for Prevention and Therapeutic Countermeasures. Vaccines, 11(10), 1537. https://doi.org/10.3390/vaccines11101537