
A Synthetic Biology Approach for Vaccine Candidate Design against Delta Strain of SARS-CoV-2 Revealed Disruption of Favored Codon Pair as a Better Strategy over Using Rare Codons


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequence Retrieval
2.2. Odds Ratio Analysis
2.3. Relative Synonymous Codon Usage (RSCU) Analysis
2.4. Codon Context Analysis
2.5. High Occurring Codon Pairs
2.6. Rare Codon Analysis
2.7. Codon Pair Score
2.8. mRNA Stability Calculation
2.9. Codon Adaptation Index (CAI) Calculation
3. Results
3.1. Compositional Features of SARS-CoV-2 Delta Strain Structural Genes Revealed at Richness
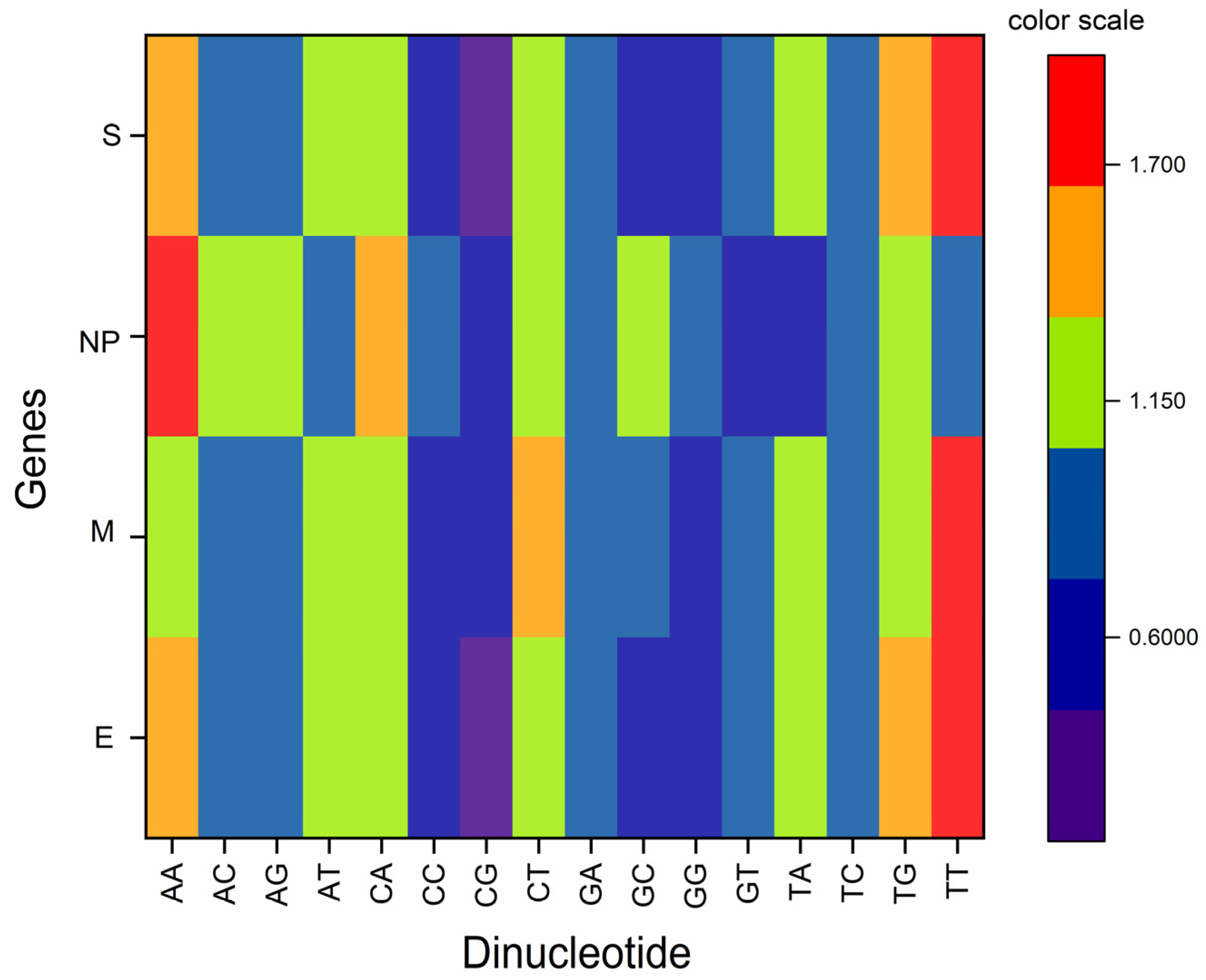
3.2. Odds Ratio Analysis Indicated Both under and Overrepresentation of Some Mirror Dinucleotides
3.3. Dinucleotide Bias at the Junction of Codons
3.4. RSCU Values of Codons from Four Structural Genes Revealed That for All Genes; Preferred Codons Are Not the Same
3.5. Codon Usage Comparison for Other Variants of Concern (VOCs) of SARS-CoV-2 and Representative Sarbecoviruses
3.6. ACT-, AAT-, TTT- and TTG-Initiated Codons Were Preferred in at Least Three out of Four Genes
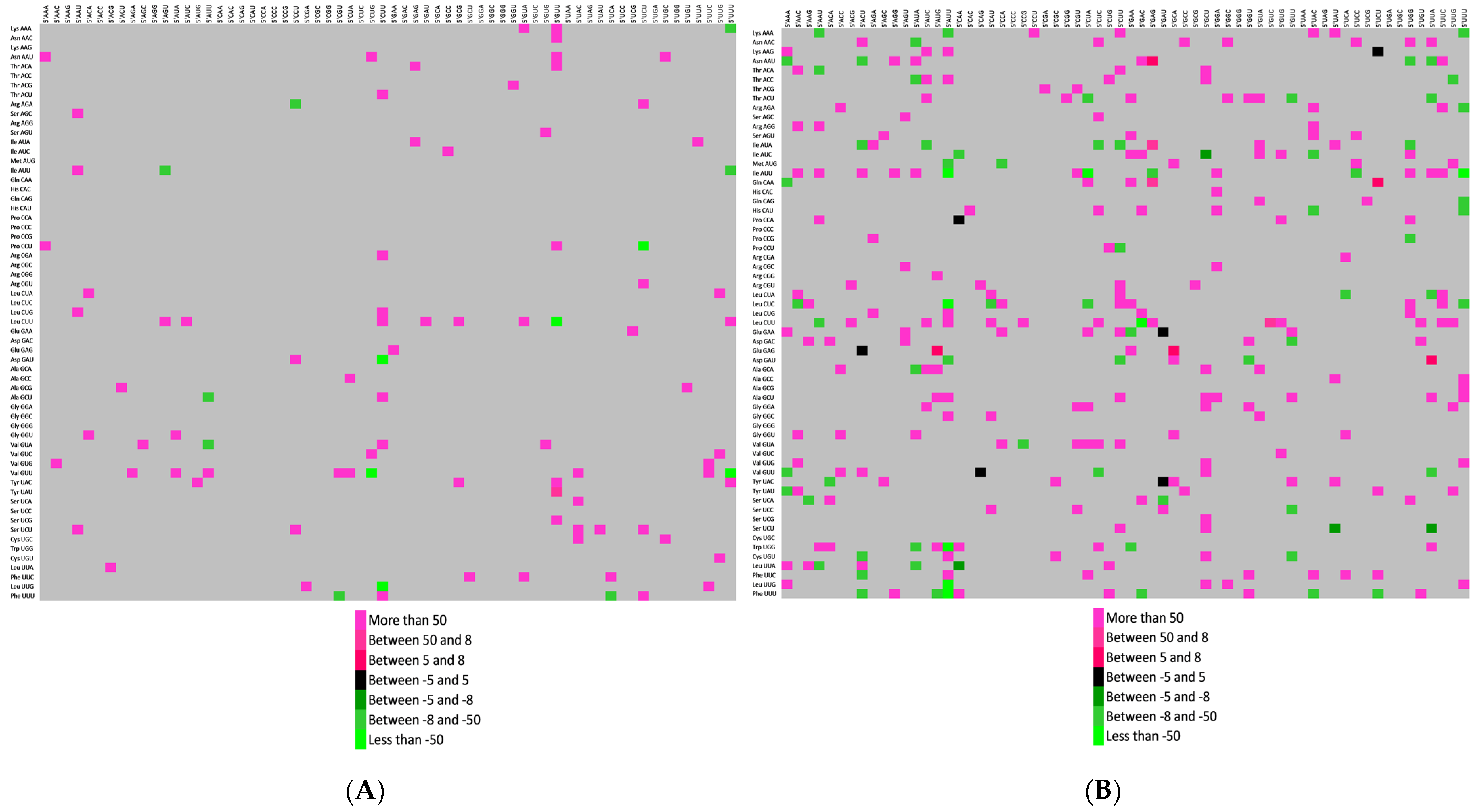
3.7. Preferred Codon Pair Analysis in Sarbecoviruses and Other SARS-CoV-2 VoCs
3.8. Codon Context Revealed Highest Codon Pair Bias in Spike Protein
3.9. Codons CGG (Arg), CCG (Pro) and CAC (His) Were Rare in All the Genes
3.10. Codon Preference of SARS-CoV-2 Gene Delta Sequences Is towards Rare Human Isoacceptor tRNAs
3.11. Vaccine Candidate Designing Using Information Generated in the Study
3.12. CpG Suppression in Different Constructs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Virus | Virus Type/Name Assigned | %GC | CpG | TpA | ∆CpG | ∆TpA | Impact of CpG and TpA Enhancement | Reference |
|---|---|---|---|---|---|---|---|---|
| HIV-1 | WT | * | 02 | -- | -- | -- | High replicative fitness | [100] |
| L | * | 39 | -- | 37 | -- | ~100-fold lower levels than HIV-1 WT | ||
| LCG-HI | * | 43 | -- | 41 | -- | |||
| Influenza A virus | Wild type | 46 | 28 | 43 | -- | -- | High replicative fitness | [102] |
| CpG high | 46 | 114 | 45 | +86 | +2 | 10–100 fold reduced viral loads in the lungs of mice infected with 200PFU and substantially greater attenuation of pathogenicity | ||
| TpA high | 46 | 29 | 116 | +1 | +73 | 10–100 fold reduced viral loads in lungs of mice infected with 200PFU | ||
| Polio virus Capsid Region | Wild | 47.1 | 28 | 36 | -- | -- | High replicative fitness | [101] |
| ABC7 | 53.3 | 80 | 34 | 52 | −2 | Relative plaque area is 0.651, and relative plaque yield is 0.72 at 37 °C | ||
| ABC8 | 59.3 | 133 | 29 | 105 | −7 | Relative plaque area is 0.549, and relative plaque yield is 0.36 at 37 °C | ||
| Zika | Wild | 49.8 | 60 | 43 | -- | -- | Lethal to mice | [104] |
| Permuted | 49.8 | 60 | 43 | 0 | 0 | Lethal to mice | ||
| E+32CpG | 49.9 | 92 | 42 | 32 | −1 | Replication not reduced | ||
| E+102CpG | 49.9 | 162 | 43 | 102 | 0 | Reduced replication in VERO and RD cells lines | ||
| E/NS1-176CpG | 49.9 | 236 | 43 | 176 | 0 | Reduced replication in VERO and RD cells lines | ||
| Dengue virus type 2 | Wild-type E | * | 20 | 55 | -- | -- | Increased frequencies of CpG and TpA attenuated the virus to degrees proportional to the numbers of additional CpG and UpA dinucleotides incorporated | [105] |
| E recoded | * | 87 | 86 | 67 | 31 | |||
| Wild Type NS3 | * | 32 | 68 | 12 | 13 | |||
| NS3 recoded | * | 99 | 111 | 79 | 56 | |||
| Wild type NS5 | * | 62 | 91 | 42 | 36 | |||
| NS5 recoded | * | 147 | 134 | 127 | 79 | |||
| E7 virus segment 1 | Native (W) | 47.6% | -- | -- | −51 | −62 | High replicative fitness | [40] |
| Permuted (P) | 47.6% | 51 | 62 | -- | -- | |||
| CpG-zero (c) | 44.3% | 0 | 70 | 51 | 8 | A 100-fold increase in relative luminescence as early as 4 h post-transfection in E7 replicon having a luciferase gene that replaces structural genes | ||
| UpA-low (u) | 50.9% | 56 | 19 | 5 | −43 | |||
| Both-low (cu) | 47.5% | 0 | 19 | −51 | −43 | 10-fold enhancements in replication, two-fold greater resistance to IFNβ than WT | ||
| CpG-high (C) | 56.5% | 180 | 52 | 129 | −10 | 100- to 10,000-fold impairments in replication # C/W has 144-fold less replication # U|W has 10 fold greater amplification | ||
| UpA-high (U) | 40.9% | 38 | 171 | −12 | 109 | |||
| E7 virus segment 2 | Native (W) | 47.1% | -- | −18 | -- | −48 | High replicative fitness | |
| Permuted (P) | 47.6% | 18 | 48 | 0 | 0 | |||
| CpG-zero (c) | 45.5% | 0 | 48 | −18 | 0 | 6-fold increase in relative luminescence as early as 4 h post-transfection in E7 replicon having a luciferase gene that replaces structural genes | ||
| UpA-low (u) | 50.0% | 21 | 14 | 3 | −34 | |||
| Both-low (cu) | 48.5% | 0 | 38 | −18 | −10 | 10-fold enhancements in replication, two-fold greater resistance to IFNβ than WT | ||
| CpG-high (C) | 56.4% | 135 | 38 | 116 | −10 | 100- to 10,000-fold impairments, two-fold greater susceptibility to IFNβ # W|C has1500-fold less replication # WU like UU | ||
| UpA-high (U) | 39.2% | 15 | 151 | −3 | 103 |
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maassab, H.F.; Bryant, M.L. The development of live attenuated cold-adapted influenza virus vaccine for humans. Rev. Med. Virol. 1999, 9, 237–244. [Google Scholar] [CrossRef]
- Hanley, K.A. The double-edged sword: How evolution can make or break a live-attenuated virus vaccine. Evolution 2011, 4, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Yeh, M.T.; Bujaki, E.; Dolan, P.T.; Smith, M.; Wahid, R.; Konz, J.; Weiner, A.J.; Bandyopadhyay, A.S.; Van Damme, P.; De Coster, I.; et al. Engineering the Live-Attenuated Polio Vaccine to Prevent Reversion to Virulence. Cell Host Microbe 2020, 27, 736–751.e8. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, L.; Chen, Y.; Gao, L.; Wu, G.; Qin, L.; Wang, Y.; Ren, X.; Gao, Y.; Gao, H.; et al. Mutations of residues 249 and 256 in VP2 are involved in the replication and virulence of infectious Bursal disease virus. PLoS ONE 2013, 8, e70982. [Google Scholar] [CrossRef]
- Appel, M.J.G. Reversion to Virulence of Attenuated Canine Distemper Virus in Vivo and in Vitro. J. Gen. Virol. 1978, 41, 385–393. [Google Scholar] [CrossRef]
- Liu, P.; Bai, Y.; Jiang, X.; Zhou, L.; Yuan, S.; Yao, H.; Yang, H.; Sun, Z. High reversion potential of a cell-adapted vaccine candidate against highly pathogenic porcine reproductive and respiratory syndrome. Vet. Microbiol. 2018, 227, 133–142. [Google Scholar] [CrossRef]
- Mueller, S.; Coleman, J.R.; Papamichail, D.; Ward, C.B.; Nimnual, A.; Futcher, B.; Skiena, S.; Wimmer, E. Live attenuated influenza virus vaccines by computer-aided rational design. Nat. Biotechnol. 2010, 28, 723–726. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.-Y.; Zhao, Z.; Opriessnig, T.; Subramaniam, S.; Zhou, L.; Cao, D.; Cao, Q.; Yang, H.; Meng, X.-J. Computer-aided codon-pairs deoptimization of the major envelope GP5 gene attenuates porcine reproductive and respiratory syndrome virus. Virology 2014, 450–451, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, M.M.; Nogales, A.; Chiem, K.; Blasco, R.; Martínez-Sobrido, L. Vaccinia Virus Attenuation by Codon Deoptimization of the A24R Gene for Vaccine Development. Microbiol. Spectr. 2022, 10, e0027222. [Google Scholar] [CrossRef]
- Nogales, A.; Baker, S.F.; Ortiz-Riaño, E.; Dewhurst, S.; Topham, D.J.; Martínez-Sobrido, L. Influenza A virus attenuation by codon deoptimization of the NS gene for vaccine development. J. Virol. 2014, 88, 10525–10540. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.Y.H.; Nogales, A.; de la Torre, J.C.; Martínez-Sobrido, L. Development of live-attenuated arenavirus vaccines based on codon deoptimization of the viral glycoprotein. Virology 2017, 501, 35–46. [Google Scholar] [CrossRef]
- Cai, Y.; Iwasaki, M.; Motooka, D.; Liu, D.X.; Yu, S.; Cooper, K.; Hart, R.; Adams, R.; Burdette, T.; Postnikova, E.N.; et al. A Lassa Virus Live-Attenuated Vaccine Candidate Based on Rearrangement of the Intergenic Region. Mbio 2020, 11, e00186-20. [Google Scholar] [CrossRef] [Green Version]
- Van Leuven, J.T.; Ederer, M.M.; Burleigh, K.; Scott, L.; Hughes, R.A.; Codrea, V.; Ellington, A.D.; Wichman, H.A.; Miller, C.R. ΦX174 Attenuation by Whole-Genome Codon Deoptimization. Genome Biol. Evol. 2021, 13, evaa214. [Google Scholar] [CrossRef]
- Meng, J.; Lee, S.; Hotard, A.L.; Moore, M.L. Refining the balance of attenuation and immunogenicity of respiratory syncytial virus by targeted codon deoptimization of virulence genes. Mbio 2014, 5, e01704–e01714. [Google Scholar] [CrossRef] [Green Version]
- Kotsopoulou, E.; Kim, V.N.; Kingsman, A.J.; Kingsman, S.M.; Mitrophanous, K.A. A Rev-independent human immunodeficiency virus type 1 (HIV-1)-based vector that exploits a codon-optimized HIV-1 gag-pol gene. J. Virol. 2000, 74, 4839–4852. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, E.; Martí-Solano, M.; Fillat, C. Codon optimization of the adenoviral fiber negatively impacts structural protein expression and viral fitness. Sci. Rep. 2016, 6, 27546. [Google Scholar] [CrossRef] [Green Version]
- Broadbent, A.J.; Santos, C.P.; Anafu, A.; Wimmer, E.; Mueller, S.; Subbarao, K. Evaluation of the attenuation, immunogenicity, and efficacy of a live virus vaccine generated by codon-pair bias de-optimization of the 2009 pandemic H1N1 influenza virus, in ferrets. Vaccine 2016, 34, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Kunec, D.; Osterrieder, N. Codon Pair Bias Is a Direct Consequence of Dinucleotide Bias. Cell Rep. 2016, 14, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Tulloch, F.; Atkinson, N.J.; Evans, D.J.; Ryan, M.D.; Simmonds, P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 2014, 3, e04531. [Google Scholar] [CrossRef] [Green Version]
- Goonawardane, N.; Nguyen, D.; Simmonds, P. Association of Zinc Finger Antiviral Protein Binding to Viral Genomic RNA with Attenuation of Replication of Echovirus 7. mSphere 2021, 6, e01138-20. [Google Scholar] [CrossRef]
- Groenke, N.; Trimpert, J.; Merz, S.; Conradie, A.M.; Wyler, E.; Zhang, H.; Hazapis, O.-G.; Rausch, S.; Landthaler, M.; Osterrieder, N.; et al. Mechanism of Virus Attenuation by Codon Pair Deoptimization. Cell Rep. 2020, 31, 107586. [Google Scholar] [CrossRef]
- Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 12 February 2023).
- SARS-CoV-2 Delta Variant Now Dominant in Much of the European Region and Efforts Must be Reinforced to Prevent Transmission, Warn WHO/Europe and ECDC. Available online: https://www.ecdc.europa.eu/en/news-events/sars-cov-2-delta-variant-now-dominant-european-region (accessed on 28 November 2022).
- A ‘Mix and Match’ Approach to SARS-CoV-2 Vaccination|Nature Medicine. Available online: https://www.nature.com/articles/s41591-021-01463-x (accessed on 12 February 2023).
- Bergwerk, M.; Gonen, T.; Lustig, Y.; Amit, S.; Lipsitch, M.; Cohen, C.; Mandelboim, M.; Levin, E.G.; Rubin, C.; Indenbaum, V.; et al. COVID-19 Breakthrough Infections in Vaccinated Health Care Workers. N. Engl. J. Med. 2021, 385, 1474–1484. [Google Scholar] [CrossRef]
- Murono, E.P.; Washburn, A.L.; Goforth, D.P.; Wu, N. Biphasic effect of basic fibroblast growth factor on 125I-human chorionic gonadotropin binding to cultured immature Leydig cells. Mol. Cell. Endocrinol. 1993, 92, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Tenforde, M.W.; Chappell, J.D.; Gaglani, M.; Ginde, A.A.; McNeal, T.; Ghamande, S.; Douin, D.J.; Talbot, H.K.; Casey, J.D.; et al. Clinical severity of, and effectiveness of mRNA vaccines against, covid-19 from omicron, delta, and alpha SARS-CoV-2 variants in the United States: Prospective observational study. BMJ 2022, 376, e069761. [Google Scholar] [CrossRef]
- Bahl, A.; Mielke, N.; Johnson, S.; Desai, A.; Qu, L. Severe COVID-19 outcomes in pediatrics: An observational cohort analysis comparing Alpha, Delta, and Omicron variants. Lancet Reg. Health-Am. 2023, 18, 100405. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of COVID-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. National Study Group for COVID-19 Vaccination Effectiveness of the BNT162b2 Covid-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Skowronski, D.M.; De Serres, G. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2021, 384, 1576–1577. [Google Scholar] [CrossRef]
- Eyre, D.W.; Taylor, D.; Purver, M.; Chapman, D.; Fowler, T.; Pouwels, K.B.; Walker, A.S.; Peto, T.E. The impact of SARS-CoV-2 vaccination on Alpha & Delta variant transmission. N Engl J Med 2022, 386, 744–756. [Google Scholar] [CrossRef]
- Reis, B.Y.; Barda, N.; Leshchinsky, M.; Kepten, E.; Hernán, M.A.; Lipsitch, M.; Dagan, N.; Balicer, R.D. Effectiveness of BNT162b2 Vaccine against Delta Variant in Adolescents. N. Engl. J. Med. 2021, 385, 2101–2103. [Google Scholar] [CrossRef]
- Luo, C.H.; Morris, C.P.; Sachithanandham, J.; Amadi, A.; Gaston, D.; Li, M.; Swanson, N.J.; Schwartz, M.; Klein, E.Y.; Pekosz, A.; et al. Infection with the SARS-CoV-2 Delta Variant is Associated with Higher Infectious Virus Loads Compared to the Alpha Variant in both Unvaccinated and Vaccinated Individuals. MedRxiv 2021. [Google Scholar] [CrossRef]
- Llanes, A.; Restrepo, C.M.; Caballero, Z.; Rajeev, S.; Kennedy, M.A.; Lleonart, R. Betacoronavirus Genomes: How Genomic Information has been Used to Deal with Past Outbreaks and the COVID-19 Pandemic. Int. J. Mol. Sci. 2020, 21, 4546. [Google Scholar] [CrossRef] [PubMed]
- Pintó, R.M.; Burns, C.C.; Moratorio, G. Editorial: Codon Usage and Dinucleotide Composition of Virus Genomes: From the Virus-Host Interaction to the Development of Vaccines. Front. Microbiol. 2021, 12, 791750. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.M.; Kenigsberg, E.; Tanay, A. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell 2011, 145, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.A.; Soll, S.J.; Emery, A.; Blanco-Melo, D.; Swanstrom, R.; Bieniasz, P.D. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. 2018, 14, e1006824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, N.J.; Witteveldt, J.; Evans, D.J.; Simmonds, P. The influence of CpG and UpA dinucleotide frequencies on RNA virus replication and characterization of the innate cellular pathways underlying virus attenuation and enhanced replication. Nucleic Acids Res. 2014, 42, 4527–4545. [Google Scholar] [CrossRef]
- Munjal, A.; Khandia, R.; Shende, K.K.; Das, J. Mycobacterium lepromatosis genome exhibits unusually high CpG dinucleotide content and selection is key force in shaping codon usage. Infect. Genet. Evol. 2020, 84, 104399. [Google Scholar] [CrossRef]
- Buckingham, R.H. Codon context. Experientia 1990, 46, 1126–1133. [Google Scholar] [CrossRef]
- Chevance, F.F.V.; Le Guyon, S.; Hughes, K.T. The effects of codon context on in vivo translation speed. PLoS Genet. 2014, 10, e1004392. [Google Scholar] [CrossRef] [Green Version]
- Moura, G.; Pinheiro, M.; Silva, R.; Miranda, I.; Afreixo, V.; Dias, G.; Freitas, A.; Oliveira, J.L.; Santos, M.A. Comparative context analysis of codon pairs on an ORFeome scale. Genome Biol. 2005, 6, R28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimpert, J.; Adler, J.M.; Eschke, K.; Abdelgawad, A.; Firsching, T.C.; Ebert, N.; Thao, T.T.N.; Gruber, A.D.; Thiel, V.; Osterrieder, N.; et al. Live attenuated virus vaccine protects against SARS-CoV-2 variants of concern B.1.1.7 (Alpha) and B.1.351 (Beta). Sci. Adv. 2021, 7, eabk0172. [Google Scholar] [CrossRef]
- Jack, B.R.; Boutz, D.R.; Paff, M.L.; Smith, B.L.; Bull, J.J.; Wilke, C.O. Reduced Protein Expression in a Virus Attenuated by Codon Deoptimization. G3 GenesGenomesGenetics 2017, 7, 2957–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosano, G.L.; Ceccarelli, E.A. Rare codon content affects the solubility of recombinant proteins in a codon bias-adjusted Escherichia coli strain. Microb. Cell Factories 2009, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- McNulty, D.E.; Claffee, B.A.; Huddleston, M.J.; Porter, M.L.; Cavnar, K.M.; Kane, J.F. Mistranslational errors associated with the rare arginine codon CGG in Escherichia coli. Protein Expr. Purif. 2003, 27, 365–374. [Google Scholar] [CrossRef]
- Allen, S.R.; Stewart, R.K.; Rogers, M.; Ruiz, I.J.; Cohen, E.; Laederach, A.; Counter, C.M.; Sawyer, J.K.; Fox, D.T. Distinct responses to rare codons in select Drosophila tissues. Elife 2022, 11, e76893. [Google Scholar] [CrossRef]
- Guimaraes, J.C.; Mittal, N.; Gnann, A.; Jedlinski, D.; Riba, A.; Buczak, K.; Schmidt, A.; Zavolan, M. A rare codon-based translational program of cell proliferation. Genome Biol. 2020, 21, 44. [Google Scholar] [CrossRef] [Green Version]
- Le Nouën, C.; Luongo, C.L.; Yang, L.; Mueller, S.; Wimmer, E.; DiNapoli, J.M.; Collins, P.L.; Buchholz, U.J. Optimization of the Codon Pair Usage of Human Respiratory Syncytial Virus Paradoxically Resulted in Reduced Viral Replication in Vivo and Reduced Immunogenicity. J. Virol. 2020, 94, e01296-19. [Google Scholar] [CrossRef] [Green Version]
- Hollams, E.M.; Giles, K.M.; Thomson, A.M.; Leedman, P.J. MRNA stability and the control of gene expression: Implications for human disease. Neurochem. Res. 2002, 27, 957–980. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.-H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki-Katagiri, N.; Lin, B.C.; Simon, J.; Hunt, R.C.; Schiller, T.; Russek-Cohen, E.; Komar, A.A.; Bar, H.; Kimchi-Sarfaty, C. The importance of mRNA structure in determining the pathogenicity of synonymous and non-synonymous mutations in haemophilia. Haemophilia 2017, 23, e8–e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuker, M. Prediction of RNA secondary structure by energy minimization. Comput. Anal. Seq. Data: Part II 1994, 25, 267–294. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Liang, Y.; Zhong, X.; Pan, Z.; Huang, L.; Zhang, H.; Xu, Y.; Zhou, W.; Liu, Z. Codon optimization with deep learning to enhance protein expression. Sci. Rep. 2020, 10, 17617. [Google Scholar] [CrossRef]
- Wagner, A. Robustness, evolvability, and neutrality. FEBS Lett. 2005, 579, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Kulkarni, D.D.; Lee, B.; Kaushik, R.; Bhatia, S.; Sood, R.; Pateriya, A.K.; Bhat, S.; Singh, V.P. Evolution of Codon Usage Bias in Henipaviruses Is Governed by Natural Selection and Is Host-Specific. Viruses 2018, 10, 604. [Google Scholar] [CrossRef] [Green Version]
- Seronello, S.; Montanez, J.; Presleigh, K.; Barlow, M.; Park, S.B.; Choi, J. Ethanol and reactive species increase basal sequence heterogeneity of hepatitis C virus and produce variants with reduced susceptibility to antivirals. PLoS ONE 2011, 6, e27436. [Google Scholar] [CrossRef]
- Beutler, E.; Gelbart, T.; Han, J.H.; Koziol, J.A.; Beutler, B. Evolution of the genome and the genetic code: Selection at the dinucleotide level by methylation and polyribonucleotide cleavage. Proc. Natl. Acad. Sci. USA 1989, 86, 192–196. [Google Scholar] [CrossRef] [Green Version]
- Franzo, G.; Segales, J.; Tucciarone, C.M.; Cecchinato, M.; Drigo, M. The analysis of genome composition and codon bias reveals distinctive patterns between avian and mammalian circoviruses which suggest a potential recombinant origin for Porcine circovirus 3. PLoS ONE 2018, 13, e0199950. [Google Scholar] [CrossRef]
- Khandia, R.; Alqahtani, T.; Alqahtani, A.M. Genes Common in Primary Immunodeficiencies and Cancer Display Overrepresentation of Codon CTG and Dominant Role of Selection Pressure in Shaping Codon Usage. Biomedicines 2021, 9, 1001. [Google Scholar] [CrossRef]
- Megremis, S.; Demetriou, P.; Makrinioti, H.; Manoussaki, A.E.; Papadopoulos, N.G. The genomic signature of human rhinoviruses A, B and C. PLoS ONE 2012, 7, e44557. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Fan, R.L.Y.; Wang, D.; Poon, L.L.M. Dinucleotide evolutionary dynamics in influenza A virus. Virus Evol. 2019, 5, vez038. [Google Scholar] [CrossRef] [PubMed]
- Goñi, N.; Iriarte, A.; Comas, V.; Soñora, M.; Moreno, P.; Moratorio, G.; Musto, H.; Cristina, J. Pandemic influenza A virus codon usage revisited: Biases, adaptation and implications for vaccine strain development. Virol. J. 2012, 9, 263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef]
- Jordan-Paiz, A.; Franco, S.; Martínez, M.A. Impact of Synonymous Genome Recoding on the HIV Life Cycle. Front. Microbiol. 2021, 12, 606087. [Google Scholar] [CrossRef]
- Simmonds, P.; Xia, W.; Baillie, J.K.; McKinnon, K. Modelling mutational and selection pressures on dinucleotides in eukaryotic phyla –selection against CpG and UpA in cytoplasmically expressed RNA and in RNA viruses. BMC Genom. 2013, 14, 610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandia, R.; Sharma, A.; Alqahtani, T.; Alqahtani, A.M.; Asiri, Y.I.; Alqahtani, S.; Alharbi, A.M.; Kamal, M.A. Strong Selectional Forces Fine-Tune CpG Content in Genes Involved in Neurological Disorders as Revealed by Codon Usage Patterns. Front. Neurosci. 2022, 16, 596. [Google Scholar] [CrossRef] [PubMed]
- Gun, L.; Yumiao, R.; Haixian, P.; Liang, Z. Comprehensive Analysis and Comparison on the Codon Usage Pattern of Whole Mycobacterium tuberculosis Coding Genome from Different Area. BioMed Res. Int. 2018, 2018, 3574976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, B.D.; Levine, A.J.; Bhanot, G.; Rabadan, R. Patterns of Evolution and Host Gene Mimicry in Influenza and Other RNA Viruses. PLOS Pathog. 2008, 4, e1000079. [Google Scholar] [CrossRef]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.; Tiwari, R.; Dhama, K.; Das, J.; Munjal, A.; Singh, R.K. Analysis of Nipah Virus Codon Usage and Adaptation to Hosts. Front. Microbiol. 2019, 10, 886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Lyu, X.; Cheng, J.; Fu, Y.; Lin, Y.; Abdoulaye, A.H.; Jiang, D.; Xie, J. Codon Usage Provides Insights into the Adaptive Evolution of Mycoviruses in Their Associated Fungi Host. Int. J. Mol. Sci. 2022, 23, 7441. [Google Scholar] [CrossRef]
- Zhou, J.; Su, J.; Chen, H.; Zhang, J.; Ma, L.; Ding, Y.; Stipkovits, L.; Szathmary, S.; Pejsak, Z.; Liu, Y. Clustering of low usage codons in the translation initiation region of hepatitis C virus. Infect. Genet. Evol. 2013, 18, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, M.; Liu, W.; Zhou, J.; Chen, H.; Ma, L.; Ding, Y.; Gu, Y.; Liu, Y. Analysis of codon usage and nucleotide composition bias in polioviruses. Virol. J. 2011, 8, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, R.; Buragohain, L.; Borah, P. Analysis of codon usage of severe acute respiratory syndrome corona virus 2 (SARS-CoV-2) and its adaptability in dog. Virus Res. 2020, 288, 198113. [Google Scholar] [CrossRef]
- Franzo, G.; Tucciarone, C.M.; Legnardi, M.; Cecchinato, M. Effect of genome composition and codon bias on infectious bronchitis virus evolution and adaptation to target tissues. BMC Genom. 2021, 22, 244. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Shinu, P.; Islam, M.M.; Chohan, M.S.; Rasool, S.T. Analysis of Codon Usage and Nucleotide Bias in Middle East Respiratory Syndrome Coronavirus Genes. Evol. Bioinform. 2020, 16, 1176934320918861. [Google Scholar] [CrossRef]
- Stenico, M.; Lloyd, A.T.; Sharp, P.M. Codon usage in Caenorhabditis elegans: Delineation of translational selection and mutational biases. Nucleic Acids Res. 1994, 22, 2437–2446. [Google Scholar] [CrossRef] [Green Version]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [Green Version]
- Le Nouën, C.; Brock, L.G.; Luongo, C.; McCarty, T.; Yang, L.; Mehedi, M.; Wimmer, E.; Mueller, S.; Collins, P.L.; Buchholz, U.J.; et al. Attenuation of human respiratory syncytial virus by genome-scale codon-pair deoptimization. Proc. Natl. Acad. Sci. USA 2014, 111, 13169–13174. [Google Scholar] [CrossRef] [Green Version]
- Park, C.; Baek, J.H.; Cho, S.H.; Jeong, J.; Chae, C.; You, S.-H.; Cha, S.-H. Field porcine reproductive and respiratory syndrome viruses (PRRSV) attenuated by codon pair deoptimization (CPD) in NSP1 protected pigs from heterologous challenge. Virology 2020, 540, 172–183. [Google Scholar] [CrossRef]
- Lee, M.H.P.; Tan, C.W.; Tee, H.K.; Ong, K.C.; Sam, I.-C.; Chan, Y.F. Vaccine candidates generated by codon and codon pair deoptimization of enterovirus A71 protect against lethal challenge in mice. Vaccine 2021, 39, 1708–1720. [Google Scholar] [CrossRef]
- Stauft, C.B.; Song, Y.; Gorbatsevych, O.; Pantoja, P.; Rodriguez, I.V.; Futcher, B.; Sariol, C.A.; Wimmer, E. Extensive genomic recoding by codon-pair deoptimization selective for mammals is a flexible tool to generate attenuated vaccine candidates for dengue virus 2. Virology 2019, 537, 237–245. [Google Scholar] [CrossRef]
- Mazumdar, P.; Binti Othman, R.; Mebus, K.; Ramakrishnan, N.; Ann Harikrishna, J. Codon usage and codon pair patterns in non-grass monocot genomes. Ann. Bot. 2017, 120, 893–909. [Google Scholar] [CrossRef] [Green Version]
- Yadav, R.; Chaudhary, J.K.; Jain, N.; Chaudhary, P.K.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.-F.; Xu, W.; Liu, S.-W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Tats, A.; Tenson, T.; Remm, M. Preferred and avoided codon pairs in three domains of life. BMC Genom. 2008, 9, 463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komar, A.A. The Yin and Yang of codon usage. Hum. Mol. Genet. 2016, 25, R77–R85. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.F.; Clark, P.L. Increased incidence of rare codon clusters at 5’ and 3’ gene termini: Implications for function. BMC Genom. 2010, 11, 118. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, O.A.; Uppal, S.; Huang, W.; Kane, M.A.; Villasmil, R.; Rogozin, I.B.; Poliakov, E.; Redmond, T.M. The Functional Consequences of the Novel Ribosomal Pausing Site in SARS-CoV-2 Spike Glycoprotein RNA. Int. J. Mol. Sci. 2021, 22, 6490. [Google Scholar] [CrossRef]
- Reid, C.R.; Airo, A.M.; Hobman, T.C. The Virus-Host Interplay: Biogenesis of +RNA Replication Complexes. Viruses 2015, 7, 4385–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moratorio, G.; Henningsson, R.; Barbezange, C.; Carrau, L.; Bordería, A.V.; Blanc, H.; Beaucourt, S.; Poirier, E.Z.; Vallet, T.; Boussier, J.; et al. Attenuation of RNA viruses by redirecting their evolution in sequence space. Nat. Microbiol. 2017, 2, 17088. [Google Scholar] [CrossRef]
- Gao, L.; Wang, L.; Huang, C.; Yang, L.; Guo, X.-K.; Yu, Z.; Liu, Y.; Yang, P.; Feng, W.-H. HP-PRRSV is attenuated by de-optimization of codon pair bias in its RNA-dependent RNA polymerase nsp9 gene. Virology 2015, 485, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Al-Saif, M.; Khabar, K.S.A. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol. Ther. 2012, 20, 954–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odon, V.; Fros, J.J.; Goonawardane, N.; Dietrich, I.; Ibrahim, A.; Alshaikhahmed, K.; Nguyen, D.; Simmonds, P. The role of ZAP and OAS3/RNAseL pathways in the attenuation of an RNA virus with elevated frequencies of CpG and UpA dinucleotides. Nucleic Acids Res. 2019, 47, 8061–8083. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Carneiro, D.; Mastrocola, E.; Lei, X.; DaSilva, J.; Chan, Y.F.; Bieniasz, P.D. Rational attenuation of RNA viruses with zinc finger antiviral protein. Nat. Microbiol. 2022, 7, 1558–1567. [Google Scholar] [CrossRef]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG-dinucleotide suppression enables antiviral defense targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Burns, C.C.; Campagnoli, R.; Shaw, J.; Vincent, A.; Jorba, J.; Kew, O. Genetic inactivation of poliovirus infectivity by increasing the frequencies of CpG and UpA dinucleotides within and across synonymous capsid region codons. J. Virol. 2009, 83, 9957–9969. [Google Scholar] [CrossRef] [Green Version]
- Gaunt, E.; Wise, H.M.; Zhang, H.; Lee, L.N.; Atkinson, N.J.; Nicol, M.Q.; Highton, A.J.; Klenerman, P.; Beard, P.M.; Dutia, B.M.; et al. Elevation of CpG frequencies in influenza A genome attenuates pathogenicity but enhances host response to infection. Elife 2016, 5, e12735. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.C.; Young, O.J.; Wintersinger, C.M.; Anastassacos, F.M.; MacDonald, J.I.; Isinelli, G.; Dellacherie, M.O.; Sobral, M.; Bai, H.; Graveline, A.R.; et al. Optimizing CpG spatial distribution with DNA origami for Th1-polarized therapeutic vaccination. BioRxiv 2022. [Google Scholar] [CrossRef]
- Trus, I.; Udenze, D.; Berube, N.; Wheler, C.; Martel, M.-J.; Gerdts, V.; Karniychuk, U. CpG-Recoding in Zika Virus Genome Causes Host-Age-Dependent Attenuation of Infection with Protection Against Lethal Heterologous Challenge in Mice. Front. Immunol. 2019, 10, 3077. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P.; Tulloch, F.; Evans, D.J.; Ryan, M.D. Attenuation of dengue (and other RNA viruses) with codon pair recoding can be explained by increased CpG/UpA dinucleotide frequencies. Proc. Natl. Acad. Sci. USA 2015, 112, E3633–E3634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhenhua, N.; Wanshun, Z.; Zhixun, X. Development of Pasteurella multocida B26-T1200 attenuated vaccine. 1. Isolation of Pasteurella multocida B26-T1200. Zhongguo Shouyi Xuebao China 1998, 18, 248–250. [Google Scholar]
- Xia, X.; Wei, T.; Xie, Z.; Danchin, A. Genomic changes in nucleotide and dinucleotide frequencies in Pasteurella multocida cultured under high temperature. Genetics 2002, 161, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Carneiro, D.; Takata, M.A.; Ong, H.; Shilton, A.; Bieniasz, P.D. Origin and evolution of the zinc finger antiviral protein. PLoS Pathog. 2021, 17, e1009545. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Nishikawa, K.; Ooi, T. Differences in dinucleotide frequencies of human, yeast, and Escherichia coli genes. DNA Res. 1997, 4, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Velazquez-Salinas, L.; Risatti, G.R.; Holinka, L.G.; O’Donnell, V.; Carlson, J.; Alfano, M.; Rodriguez, L.L.; Carrillo, C.; Gladue, D.P.; Borca, M.V. Recoding structural glycoprotein E2 in classical swine fever virus (CSFV) produces complete virus attenuation in swine and protects infected animals against disease. Virology 2016, 494, 178–189. [Google Scholar] [CrossRef]
- Loew, L.; Goonawardane, N.; Ratcliff, J.; Nguyen, D.; Simmonds, P. Use of a small DNA virus model to investigate mechanisms of CpG dinucleotide-induced attenuation of virus replication. J. Gen. Virol. 2020, 101, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Digard, P.; Lee, H.M.; Sharp, C.; Grey, F.; Gaunt, E. Intra-genome variability in the dinucleotide composition of SARS-CoV-2. Virus Evol. 2020, 6, veaa057. [Google Scholar] [CrossRef]
- Wang, W.; Cheng, X.; Buske, P.J.; Suzich, J.A.; Jin, H. Attenuate Newcastle disease virus by codon modification of the glycoproteins and phosphoprotein genes. Virology 2019, 528, 144–151. [Google Scholar] [CrossRef]
- Wang, B.; Yang, C.; Tekes, G.; Mueller, S.; Paul, A.; Whelan, S.P.J.; Wimmer, E. Recoding of the vesicular stomatitis virus L gene by computer-aided design provides a live, attenuated vaccine candidate. Mbio 2015, 6, e00237-15. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Wu, P.; Deng, S.; Zhang, H.; Hou, Y.; Hu, Z.; Zhang, J.; Chen, X.; Yang, J.-R. Dissimilation of synonymous codon usage bias in virus-host coevolution due to translational selection. Nat. Ecol. Evol. 2020, 4, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Pintó, R.M.; Aragonès, L.; Costafreda, M.I.; Ribes, E.; Bosch, A. Codon usage and replicative strategies of hepatitis A virus. Virus Res. 2007, 127, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, L.; Guix, S.; Ribes, E.; Bosch, A.; Pintó, R.M. Fine-tuning translation kinetics selection as the driving force of codon usage bias in the hepatitis A virus capsid. PLoS Pathog. 2010, 6, e1000797. [Google Scholar] [CrossRef] [PubMed] [Green Version]





| %A | %C | %T | %G | %G+C | %A+T | %A3 | %C3 | %T3 | %G3 | %G3+C3 | %A3+T3 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | Average | 21.56 | 19.43 | 40.48 | 18.52 | 37.96 | 62.04 | 22.18 | 18.49 | 43.06 | 16.27 | 34.76 | 65.24 |
| SD | 0.56 | 0.74 | 0.33 | 0.35 | 0.71 | 0.71 | 2.60 | 0.73 | 4.00 | 1.18 | 1.54 | 1.54 | |
| M | Average | 25.56 | 21.93 | 31.74 | 20.78 | 42.70 | 57.30 | 24.22 | 22.85 | 36.79 | 16.14 | 38.99 | 61.01 |
| SD | 0.03 | 0.08 | 0.08 | 0.02 | 0.09 | 0.09 | 0.06 | 0.10 | 0.12 | 0.05 | 0.12 | 0.12 | |
| NP | Average | 31.75 | 25.01 | 21.25 | 21.98 | 47.00 | 53.00 | 30.58 | 22.58 | 31.72 | 15.12 | 37.70 | 62.30 |
| SD | 0.09 | 0.06 | 0.09 | 0.09 | 0.08 | 0.08 | 0.20 | 0.09 | 0.19 | 0.12 | 0.16 | 0.16 | |
| S | Average | 29.46 | 18.84 | 33.26 | 18.44 | 37.27 | 62.73 | 27.03 | 15.88 | 46.37 | 10.71 | 26.59 | 73.41 |
| 0.04 | 0.05 | 0.03 | 0.04 | 0.05 | 0.05 | 0.05 | 0.09 | 0.11 | 0.06 | 0.13 | 0.13 |
| S.No. | Single Letter Amino Acid | Codon | S | NP | M | E |
|---|---|---|---|---|---|---|
| 1 | F | TTT | 1.532 | 0.462 | 0.909 | 0.8 |
| TTC | 0.468 | 1.538 | 1.091 | 1.2 | ||
| 2 | L | TTA | 1.585 | 0.444 | 0.686 | 0.429 |
| TTG | 1.132 | 2 | 0.686 | 0.857 | ||
| CTT | 2.038 | 1.778 | 2.057 | 3 | ||
| CTC | 0.623 | 0.444 | 1.029 | 0 | ||
| CTA | 0.509 | 0.667 | 0.857 | 0.857 | ||
| CTG | 0.113 | 0.667 | 0.686 | 0.857 | ||
| 3 | I | ATT | 1.737 | 1.929 | 1.737 | 1 |
| ATC | 0.553 | 0.857 | 0.789 | 1 | ||
| ATA | 0.711 | 0.214 | 0.474 | 1 | ||
| 4 | V | GTT | 2.021 | 1 | 1 | 2.154 |
| GTC | 0.825 | 1.5 | 0 | 0.308 | ||
| GTA | 0.619 | 0.5 | 2 | 0.923 | ||
| GTG | 0.536 | 1 | 1 | 0.615 | ||
| 5 | S | TCT | 2.242 | 1.297 | 0.8 | 3 |
| TCC | 0.727 | 0.486 | 1.2 | 0 | ||
| TCA | 1.576 | 1.459 | 1.2 | 0.75 | ||
| TCG | 0.121 | 0.324 | 0.4 | 0.75 | ||
| AGT | 1.03 | 1.459 | 1.6 | 0.75 | ||
| AGC | 0.303 | 0.973 | 0.8 | 0.75 | ||
| 6 | P | CCT | 1.965 | 1.143 | 0.8 | 4 |
| CCC | 0.281 | 1 | 0 | 0 | ||
| CCA | 1.754 | 1.571 | 2.4 | 0 | ||
| CCG | 0 | 0.286 | 0.8 | 0 | ||
| 7 | T | ACT | 1.853 | 2 | 1.429 | 1 |
| ACC | 0.421 | 0.75 | 1.143 | 0 | ||
| ACA | 1.6 | 1 | 0.857 | 2 | ||
| ACG | 0.126 | 0.25 | 0.571 | 1 | ||
| 8 | A | GCT | 2.127 | 2.054 | 2.526 | 1 |
| GCC | 0.405 | 0.757 | 0.421 | 1 | ||
| GCA | 1.367 | 0.865 | 0.842 | 0 | ||
| GCG | 0.101 | 0.324 | 0.211 | 2 | ||
| 9 | Y | TAT | 1.481 | 0.5 | 0.889 | 0 |
| TAC | 0.519 | 1.5 | 1.111 | 2 | ||
| 10 | H | CAT | 1.529 | 1.5 | 1.6 | 0 |
| CAC | 0.471 | 0.5 | 0.4 | 0 | ||
| 11 | Q | CAA | 1.484 | 1.543 | 1 | 0 |
| CAG | 0.516 | 0.457 | 1 | 0 | ||
| 12 | N | AAT | 1.236 | 1.455 | 0.727 | 1.6 |
| AAC | 0.764 | 0.545 | 1.273 | 0.4 | ||
| 13 | K | AAA | 1.258 | 1.375 | 1.143 | 2 |
| AAG | 0.742 | 0.625 | 0.857 | 0 | ||
| 14 | D | GAT | 1.377 | 1.182 | 0.333 | 2 |
| GAC | 0.623 | 0.818 | 1.667 | 0 | ||
| 15 | E | GAA | 1.447 | 1.333 | 1.714 | 1 |
| GAG | 0.553 | 0.667 | 0.286 | 1 | ||
| 16 | C | TGT | 1.4 | 0 | 2 | 0.667 |
| TGC | 0.6 | 0 | 0 | 1.333 | ||
| 17 | R | CGT | 1.364 | 1.333 | 2.143 | 2 |
| CGC | 0.136 | 1.111 | 0.857 | 0 | ||
| CGA | 0 | 1.111 | 0.429 | 2 | ||
| CGG | 0.409 | 0.444 | 0 | 0 | ||
| AGA | 2.727 | 2 | 1.286 | 2 | ||
| AGG | 1.364 | 0 | 1.286 | 0 | ||
| 18 | G | GGT | 2.265 | 0.909 | 1.429 | 4 |
| GGC | 0.723 | 1.545 | 0.857 | 0 | ||
| GGA | 0.867 | 1.182 | 1.714 | 0 | ||
| GGG | 0.145 | 0.364 | 0 | 0 |
| Codons | Amino Acid | Alpha | Beta | Gamma | Omicron | Sarbecoviruses | Delta |
|---|---|---|---|---|---|---|---|
| TTT | F | 1.010 | 0.927 | 0.929 | 0.931 | 0.944 | 0.926 |
| TTC | 0.991 | 1.073 | 1.071 | 1.069 | 1.056 | 1.074 | |
| TTA | L | 0.782 | 0.768 | 0.782 | 0.782 | 0.781 | 0.786 |
| TTG | 1.166 | 1.157 | 1.166 | 1.166 | 1.089 | 1.169 | |
| CTT | 2.214 | 2.245 | 2.200 | 2.214 | 2.082 | 2.218 | |
| CTC | 0.537 | 0.540 | 0.537 | 0.537 | 0.588 | 0.524 | |
| CTA | 0.722 | 0.710 | 0.722 | 0.722 | 0.801 | 0.723 | |
| CTG | 0.581 | 0.581 | 0.595 | 0.581 | 0.660 | 0.581 | |
| ATT | I | 1.616 | 1.597 | 1.583 | 1.609 | 1.634 | 1.601 |
| ATC | 0.799 | 0.813 | 0.826 | 0.796 | 0.745 | 0.800 | |
| ATA | 0.585 | 0.590 | 0.591 | 0.595 | 0.621 | 0.600 | |
| GTT | V | 1.533 | 1.528 | 1.528 | 1.533 | 1.473 | 1.544 |
| GTC | 0.669 | 0.666 | 0.671 | 0.669 | 0.838 | 0.658 | |
| GTA | 1.011 | 1.019 | 1.012 | 1.011 | 0.875 | 1.011 | |
| GTG | 0.788 | 0.787 | 0.789 | 0.788 | 0.813 | 0.788 | |
| TCT | S | 1.841 | 1.835 | 1.879 | 1.835 | 1.770 | 1.835 |
| TCC | 0.605 | 0.603 | 0.600 | 0.603 | 0.518 | 0.603 | |
| TCA | 1.250 | 1.246 | 1.239 | 1.246 | 1.424 | 1.246 | |
| TCG | 0.399 | 0.399 | 0.398 | 0.399 | 0.474 | 0.399 | |
| AGT | 1.182 | 1.210 | 1.179 | 1.210 | 1.071 | 1.210 | |
| AGC | 0.723 | 0.707 | 0.705 | 0.707 | 0.743 | 0.707 | |
| CCT | P | 1.986 | 1.986 | 1.988 | 1.977 | 1.787 | 1.977 |
| CCC | 0.319 | 0.319 | 0.330 | 0.320 | 0.400 | 0.320 | |
| CCA | 1.424 | 1.424 | 1.409 | 1.431 | 1.529 | 1.431 | |
| CCG | 0.272 | 0.272 | 0.274 | 0.272 | 0.284 | 0.272 | |
| ACT | T | 1.562 | 1.572 | 1.583 | 1.559 | 1.523 | 1.571 |
| ACC | 0.538 | 0.527 | 0.512 | 0.581 | 0.496 | 0.579 | |
| ACA | 1.401 | 1.401 | 1.398 | 1.373 | 1.505 | 1.364 | |
| ACG | 0.499 | 0.499 | 0.508 | 0.488 | 0.477 | 0.487 | |
| GCT | A | 1.927 | 1.934 | 1.927 | 1.927 | 1.920 | 1.927 |
| GCC | 0.646 | 0.647 | 0.646 | 0.646 | 0.700 | 0.646 | |
| GCA | 0.769 | 0.760 | 0.769 | 0.769 | 0.718 | 0.769 | |
| GCG | 0.659 | 0.660 | 0.659 | 0.659 | 0.662 | 0.659 | |
| TAT | Y | 0.684 | 0.686 | 0.691 | 0.718 | 0.626 | 0.718 |
| TAC | 1.317 | 1.314 | 1.310 | 1.283 | 1.374 | 1.283 | |
| CAT | H | 1.157 | 1.164 | 1.150 | 1.157 | 0.938 | 1.157 |
| CAC | 0.343 | 0.336 | 0.350 | 0.343 | 0.562 | 0.343 | |
| CAA | Q | 1.007 | 1.013 | 1.007 | 1.007 | 0.977 | 1.007 |
| CAG | 0.493 | 0.487 | 0.493 | 0.493 | 0.523 | 0.493 | |
| AAT | N | 1.252 | 1.252 | 1.247 | 1.255 | 1.198 | 1.255 |
| AAC | 0.748 | 0.748 | 0.753 | 0.746 | 0.802 | 0.746 | |
| AAA | K | 1.436 | 1.444 | 1.449 | 1.439 | 1.462 | 1.444 |
| AAG | 0.564 | 0.556 | 0.551 | 0.561 | 0.538 | 0.556 | |
| GAT | D | 1.219 | 1.214 | 1.217 | 1.223 | 1.158 | 1.223 |
| GAC | 0.781 | 0.786 | 0.783 | 0.777 | 0.842 | 0.777 | |
| GAA | E | 1.366 | 1.363 | 1.363 | 1.381 | 1.236 | 1.374 |
| GAG | 0.634 | 0.637 | 0.637 | 0.619 | 0.764 | 0.627 | |
| TGT | C | 1.021 | 1.017 | 1.017 | 1.517 | 0.885 | 1.017 |
| TGC | 0.480 | 0.483 | 0.483 | 0.483 | 0.615 | 0.483 | |
| CGT | R | 1.660 | 1.675 | 1.665 | 1.698 | 1.606 | 1.710 |
| CGC | 0.508 | 0.509 | 0.501 | 0.516 | 0.722 | 0.526 | |
| CGA | 0.866 | 0.866 | 0.957 | 0.875 | 1.061 | 0.885 | |
| CGG | 0.208 | 0.140 | 0.173 | 0.210 | 0.156 | 0.213 | |
| AGA | 2.037 | 2.071 | 2.053 | 2.039 | 1.652 | 2.003 | |
| AGG | 0.722 | 0.739 | 0.651 | 0.663 | 0.803 | 0.663 | |
| GGT | G | 2.168 | 2.173 | 2.174 | 2.138 | 1.882 | 2.151 |
| GGC | 0.767 | 0.765 | 0.776 | 0.788 | 0.765 | 0.781 | |
| GGA | 0.936 | 0.933 | 0.919 | 0.945 | 1.161 | 0.941 | |
| GGG | 0.129 | 0.129 | 0.132 | 0.129 | 0.191 | 0.127 |
| Gene Name | Envelope | Nucleocapsid | Membrane | Spike | ||||
|---|---|---|---|---|---|---|---|---|
| % frequency of top 20 codon pairs | ||||||||
| 1. | TTA-ATA | 1.46 | CAA-CAA | 0.96 | ATT-GCT | 1.79 | GTT-TAT | 0.54 |
| 2. | TCG-GAA | 1.46 | AAA-GAT | 0.95 | TGT-CTT | 0.89 | GGT-GTT | 0.50 |
| 3. | TAC-TCA | 1.46 | ATT-GGC | 0.72 | GGA-GCT | 0.89 | TTT-GGT | 0.47 |
| 4. | GTT-TCG | 1.46 | AAG-AAG | 0.72 | CTT-GTA | 0.89 | ACT-AAT | 0.45 |
| 5. | GTT-AAT | 1.46 | CAA-GGA | 0.72 | CTT-CTA | 0.89 | GGT-GAT | 0.40 |
| 6. | GTA-CTT | 1.46 | TCA-ACT | 0.71 | CTT-CGT | 0.89 | TTT-AAT | 0.39 |
| 7. | GGT-ACG | 1.46 | CCT-GCT | 0.71 | ATG-TGG | 0.89 | TCT-AAC | 0.39 |
| 8. | GAA-GAG | 1.46 | AGC-AGT | 0.68 | ACT-ATT | 0.89 | AAT-CTT | 0.39 |
| 9. | CTT-TTT | 1.46 | GGA-ACT | 0.61 | GCT-TGT | 0.88 | AAT-GGT | 0.38 |
| 10. | CTT-CTT | 1.46 | CAA-ATT | 0.48 | GAA-GAG | 0.46 | AAT-TTT | 0.32 |
| 11. | ATG-TAC | 1.46 | ACT-CAA | 0.48 | ATA-ATT | 0.46 | AAC-AAA | 0.32 |
| 12. | ATA-GTT | 1.46 | TTG-GAT | 0.48 | TTT-TTG | 0.45 | AAT-GTT | 0.32 |
| 13. | AGC-GTA | 1.46 | TTG-CTG | 0.48 | TTG-CTT | 0.45 | GTT-TTT | 0.32 |
| 14. | ACG-TTA | 1.46 | TAC-TAC | 0.48 | TGG-ATT | 0.45 | TAT-TCT | 0.31 |
| 15. | AAT-AGC | 1.46 | GGC-AGT | 0.48 | CTC-CTT | 0.45 | GTT-GCT | 0.31 |
| 16. | TTT-CTT | 1.44 | GGA-CCC | 0.48 | ATT-ACC | 0.45 | GCA-CAA | 0.31 |
| 17. | TTG-CTA | 1.44 | GCT-GCT | 0.48 | AAT-ATT | 0.45 | ACT-TCT | 0.31 |
| 18. | TTC-TTG | 1.44 | GAC-AAA | 0.48 | TTT-GCT | 0.45 | TAT-AAT | 0.31 |
| 19. | TTC-GTG | 1.44 | CGT-GGT | 0.48 | TTT-GCG | 0.45 | AAT-GAT | 0.31 |
| 20. | GTT-ACA | 1.44 | CGC-ATT | 0.48 | TTT-GCC | 0.45 | GGT-TTT | 0.31 |
| (A) | |||||||||
| Alpha | Delta | Beta | Delta | Gamma | Delta | Omicron | Delta | Sarbecoviruses | Delta |
| FY | FY | FY | FY | FY | FY | FY | FY | YS | FY |
| FL | LC | FL | LC | FL | LC | FL | LC | VY | LC |
| LC | LL | LC | LL | LC | LL | LC | LL | VK | LL |
| LL | FL | LL | FL | LL | FL | LL | FL | LC | FL |
| FL | FV | FL | FV | FL | FV | FL | FV | LL | FV |
| FV | LI | FV | LI | FV | LI | FV | LI | FL | LI |
| FV | CA | FV | CA | FV | CA | FV | CA | FV | CA |
| LI | CC | LI | CC | LI | CC | LI | CC | LI | CC |
| CA | CN | CA | CN | CA | CN | CA | CN | CA | CN |
| CC | SF | CC | SF | CC | SF | CC | SF | CC | SF |
| CN | SR | CN | SR | CN | SR | CN | SR | CN | SR |
| SF | SE | SF | SE | SF | SE | SF | SE | SS | SE |
| SS | YC | SS | YC | SS | YC | SS | YC | SE | YC |
| SR | YS | SR | YS | SR | YS | SR | YS | SF | YS |
| SR | YV | SR | YV | SR | YV | SR | YV | YC | YV |
| SE | VS | SE | VS | SE | VS | SE | VS | VS | VS |
| SF | VT | SF | VT | SF | VT | SF | VT | VP | VT |
| YC | VN | YC | VN | YC | VN | YC | VN | VN | VN |
| YS | VN | YS | VN | YS | VN | YS | VN | VN | VN |
| YS | VV | YS | VV | YS | VV | YS | VV | VV | VV |
| (B) | |||||||||
| Alpha | Delta | Beta | Delta | Gamma | Delta | Omicron | Delta | Sarbecoviruses | Delta |
| IA | IA | IA | IA | IA | IA | IA | IA | IA | IA |
| CL | CL | CL | CL | CL | CL | CL | CL | CL | CL |
| GA | GA | GA | GA | GA | GA | GA | GA | GA | GA |
| AC | LV | AC | LV | AC | LV | LV | LV | LV | LV |
| LV | LL | LV | LL | LV | LL | LL | LL | LL | LL |
| LL | LR | LL | LR | LL | LR | LR | LR | LR | LR |
| LR | MW | LR | MW | LR | MW | MW | MW | MW | MW |
| MW | TI | MW | TI | MW | TI | TI | TI | TI | TI |
| TI | AC | TI | AC | TI | AC | FL | AC | FL | AC |
| FL | EE | FL | EE | FL | EE | FV | EE | FV | EE |
| FV | II | FV | II | FV | II | FA | II | FA | II |
| FA | FL | FA | FL | FA | FL | FA | FL | FA | FL |
| FA | LL | FA | LL | FA | LL | FA | LL | FA | LL |
| FA | WI | FA | WI | FA | WI | FN | WI | FN | WI |
| FL | LL | LY | LL | LY | LL | LY | LL | LY | LL |
| LY | IT | LG | IT | LG | IT | LG | IT | LG | IT |
| LG | NI | LL | NI | LL | NI | LL | NI | LL | NI |
| LL | FA | LM | FA | LM | FA | LM | FA | LM | FA |
| LM | FA | FL | FA | FL | FA | FL | FA | FL | FA |
| FL | FA | FL | FA | FL | FA | FL | FA | FL | FA |
| (C) | |||||||||
| Alpha | Delta | Beta | Delta | Gamma | Delta | Omicron | Delta | Sarbecoviruses | Delta |
| KD | KD | KD | KD | KD | KD | KD | KD | KK | KD |
| ST | IG | ST | IG | ST | IG | ST | IG | QG | IG |
| PA | KK | PA | KK | PA | KK | PA | KK | IG | KK |
| QG | QG | QG | QG | QG | QG | QG | QG | KD | QG |
| IG | ST | IG | ST | IG | ST | IG | ST | GT | ST |
| SS | PA | SS | PA | KK | PA | KK | PA | KK | PA |
| KK | SS | KK | SS | LD | SS | LD | SS | ST | SS |
| LD | GT | LD | GT | LL | GT | LL | GT | DD | GT |
| LL | QI | LL | QI | FY | QI | FY | QI | PA | QI |
| FY | TQ | FY | TQ | YY | TQ | YY | TQ | GP | TQ |
| YY | LD | YY | LD | YK | LD | YK | LD | DK | LD |
| YK | LL | YK | LL | GK | LL | GK | LL | RI | LL |
| GK | YY | GK | YY | GQ | YY | GQ | YY | PK | YY |
| GQ | GS | GQ | GS | GS | GS | GS | GS | QI | GS |
| GS | GP | GS | GP | GP | GP | GP | GP | YK | GP |
| GP | AA | GP | AA | GT | AA | GT | AA | LP | AA |
| GT | DK | GT | DK | AA | DK | AA | DK | RG | DK |
| AA | RG | AA | RG | AA | RG | AA | RG | KG | RG |
| AA | RI | AA | RI | AN | RI | AN | RI | PQ | RI |
| (D) | |||||||||
| Alpha | Delta | Beta | Delta | Gamma | Delta | Omicron | Delta | Sarbecoviruses | Delta |
| VY | VY | VY | VY | VY | VY | VY | VY | NF | VY |
| YS | YS | YS | YS | AL | YS | YS | YS | YE | YS |
| YN | YN | YN | YN | AQ | YN | YN | YN | VY | YN |
| VF | VF | VF | VF | FG | VF | VA | VF | VF | VF |
| VA | VA | VA | VA | FN | VA | V | VA | TS | VA |
| TS | TS | TS | TS | GA | TS | TS | TS | SN | TS |
| TN | TN | TN | TN | GD | TN | TN | TN | SF | TN |
| SN | SN | SN | SN | GF | SN | SN | SN | PF | SN |
| NL | NV | NV | NV | GV | NV | NL | NV | NV | NV |
| NG | NL | NG | NL | IA | NL | NG | NL | LD | NL |
| IA | NK | NF | NK | IA | NK | IA | NK | IT | NK |
| IA | NG | ND | NG | NF | NG | IA | NG | IA | NG |
| GV | NF | IA | NF | SF | NF | GV | NF | GV | NF |
| GF | ND | IA | ND | SN | ND | GF | ND | GD | ND |
| GD | GV | GV | GV | TN | GV | GD | GV | FN | GV |
| GA | GF | GF | GF | TS | GF | GA | GF | FG | GF |
| FN | GD | GD | GD | VA | GD | FN | GD | DV | GD |
| FG | FN | FN | FN | VF | FN | FG | FN | DI | FN |
| AQ | FG | FG | FG | YN | FG | AQ | FG | AD | FG |
| AL | AQ | AQ | AQ | YS | AQ | AL | AQ | AA | AQ |
| tRNA Isotype in Human | Total Count | Most Preferred Codon | ||||
|---|---|---|---|---|---|---|
| S | NP | M | E | |||
| Phe (F) | AAA(0), GAA(10) | 10 | TTT | TTC | TTC | TTC |
| Leu (L) | AAG(9), GAG(0), CAG(9),TAG(3), CAA(6), TAA(4) | 31 | CTT | TTG | CTT | CTT |
| Ile (I) | AAT(14), GAT(3), CAT(0), TAT(5) | 22 | ATT | ATT | ATT | ATT ATC ATA |
| Val (V) | AAC(9), GAC(0), CAC(11), TAC(5) | 25 | GTT | GTC | GTA | GTT |
| Ser (S) | AGA(9), GGA(0), CGA(4), TGA(4), ACT (8),GCT(8) | 25 | TCT | TCA AGT | AGT | TCT |
| Pro (P) | AGG(9), GGG(0), CGG(4), TGG(7) | 20 | CCT | CCA | CCA | CCT |
| Thr (T) | AGT(9),GGT(0), CGT(5), TGT(6) | 20 | ACA | ACT | ACT | ACA |
| Ala (A) | AGC(22), GGC(0), CGC(4), TGC(8) | 34 | GCT | GCT | GCT | GCG |
| Tyr (Y) | ATA(0), GTA(13), | 13 | TAT | TAC | TAC | TAC |
| His (H) | ATG(0), GTG(10) | 10 | CAT | CAT | CAT | * |
| Gln (Q) | CTG(13), TTG(6) | 19 | CAA | CAA | CAA CAG | * |
| Asp (N) | ATT(0), GTT(20) | 20 | AAT | AAT | AAC | AAT |
| Lys (K) | CTT(15), TTT(12) | 27 | AAA | AAA | AAA | AAA |
| Asp (D) | ATC(0), GTC(13) | 13 | GAT | GAT | GAC | GAT |
| Glu (E) | CTC(8), TTC(7) | 15 | GAA | GAA | GAA | GAA GAG |
| Cys (C) | ACA(0), GCA(29), | 29 | TGT | * | TGT | TGC |
| Arg (R) | ACG(7), GCG(0), CCG(4), TCG(6), CCT(5), TCT(6) | 28 | AGA | CGT | CGT | AGA CGA AGA |
| Gly (G) | ACC(0), GCC(14), CCC(5), TCC(9) | 28 | GGT | GGC | GGA | GGT |
| From Codon | Frequency | To Codon | Frequency | CAI | Nc | CPS | MFE (kcal/mol) | %G+C | Intracodon CpG | CpG at p3-1 Unction | Total CpG | ∆CpG | Intracodon TpA | TpA at p3-1 Unction | Total TpA | ∆TpA | CpG-O/E | TpA-O/E | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Wild-type SARS-CoV-2 Delta strain | - | - | - | - | 0.699 | 48.6 | 0.158 | −1776.90 | 40.1 | 66 | 34 | 100 | -- | 181 | 192 | 373 | -- | 0.268 | 1.005 |
| 2. | Overrepresented codons to TA ending codons leading to TpT dimer to TpA (Construct 1) | CTT | 31.6 | CTA | 10 | 0.666/659 | 45 | 0.158 | −1684.40 | 39.98 | 66 | 34 | 100 | 0 | 421 | 144 | 565 | 192 | 0.268 | 1.386 |
| ATT | 32.6 | ATA | 11.5 | |||||||||||||||||
| GTT | 30.1 | GTA | 12.5 | |||||||||||||||||
| 3. | Introduction of rare codons (Construct 2) | CCT | 19.5 | CCG | 1.5 | 0.558 | 43.4 | 0.143 | −1801.30 | 43.89 | 194 | 43 | 237 | 137 | 181 | 149 | 330 | −43 | 0.635 | 0.874 |
| CAT | 10 | CAC | 3.5 | |||||||||||||||||
| CGT | 10.5 | CGG | 2 | |||||||||||||||||
| GGT | 32.1 | GGG | 3.5 | |||||||||||||||||
| CCA | 19.5 | CCC | 1.5 | |||||||||||||||||
| TCT | 25.1 | TCG | 3 | |||||||||||||||||
| 4. | Disruption of favored codon pairs at the 5′ end (Construct 3) | ACT | 33.1 | ACG | 4 | 0.577 | 41 | 0.152 | −1747.80 | 45.41 | 137 | 90 | 227 | 127 | 241 | 105 | 346 | −27 | 0.63 | 0.938 |
| AAT | 36.6 | AAC | 24.1 | |||||||||||||||||
| TTT | 32.6 | TTC | 18.5 | |||||||||||||||||
| TTG | 17.5 | CTG | 6 | |||||||||||||||||
| GTT | 30.1 | GTA | 12.5 | |||||||||||||||||
| AGG | 6.5 | CGG | 2 | |||||||||||||||||
| CTT | 31.6 | CTG | 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurjar, P.; Karuvantevida, N.; Rzhepakovsky, I.V.; Khan, A.A.; Khandia, R. A Synthetic Biology Approach for Vaccine Candidate Design against Delta Strain of SARS-CoV-2 Revealed Disruption of Favored Codon Pair as a Better Strategy over Using Rare Codons. Vaccines 2023, 11, 487. https://doi.org/10.3390/vaccines11020487
Gurjar P, Karuvantevida N, Rzhepakovsky IV, Khan AA, Khandia R. A Synthetic Biology Approach for Vaccine Candidate Design against Delta Strain of SARS-CoV-2 Revealed Disruption of Favored Codon Pair as a Better Strategy over Using Rare Codons. Vaccines. 2023; 11(2):487. https://doi.org/10.3390/vaccines11020487
Chicago/Turabian StyleGurjar, Pankaj, Noushad Karuvantevida, Igor Vladimirovich Rzhepakovsky, Azmat Ali Khan, and Rekha Khandia. 2023. "A Synthetic Biology Approach for Vaccine Candidate Design against Delta Strain of SARS-CoV-2 Revealed Disruption of Favored Codon Pair as a Better Strategy over Using Rare Codons" Vaccines 11, no. 2: 487. https://doi.org/10.3390/vaccines11020487
APA StyleGurjar, P., Karuvantevida, N., Rzhepakovsky, I. V., Khan, A. A., & Khandia, R. (2023). A Synthetic Biology Approach for Vaccine Candidate Design against Delta Strain of SARS-CoV-2 Revealed Disruption of Favored Codon Pair as a Better Strategy over Using Rare Codons. Vaccines, 11(2), 487. https://doi.org/10.3390/vaccines11020487