
STAT1-Deficient HPV E6/E7-Associated Cancers Maintain Host Immunocompetency against Therapeutic Intervention

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Animal Care
2.2. Plasmid DNA Construction and Preparation
2.3. Generation of Spontaneous Oral Tumor Model
2.4. Bioluminescent Imaging
2.5. DNA Vaccination
2.6. Flow Cytometry Analysis
2.7. Cell Culture
2.8. Tumor Challenge Experiment
2.9. Statistical Analysis
3. Results
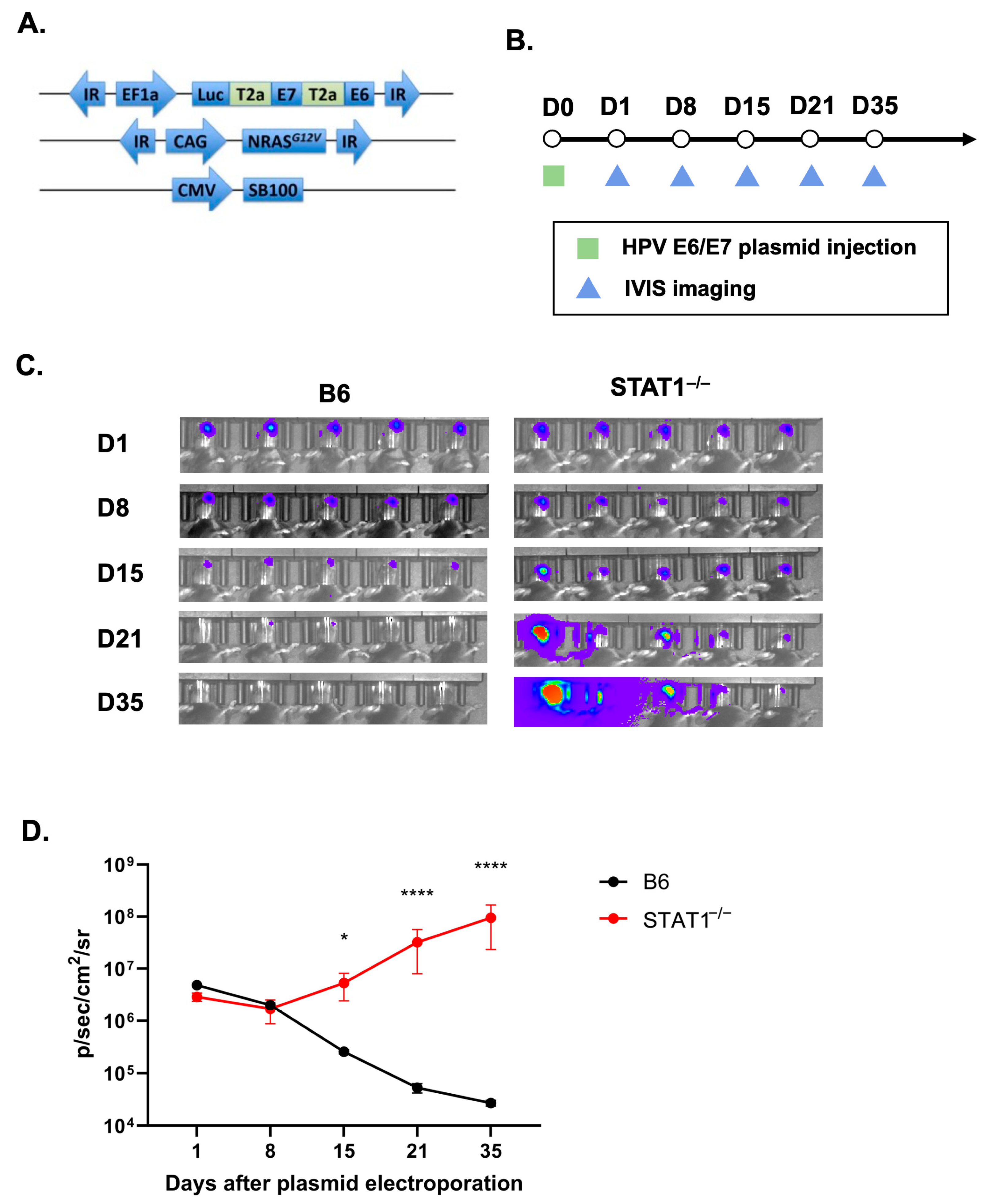
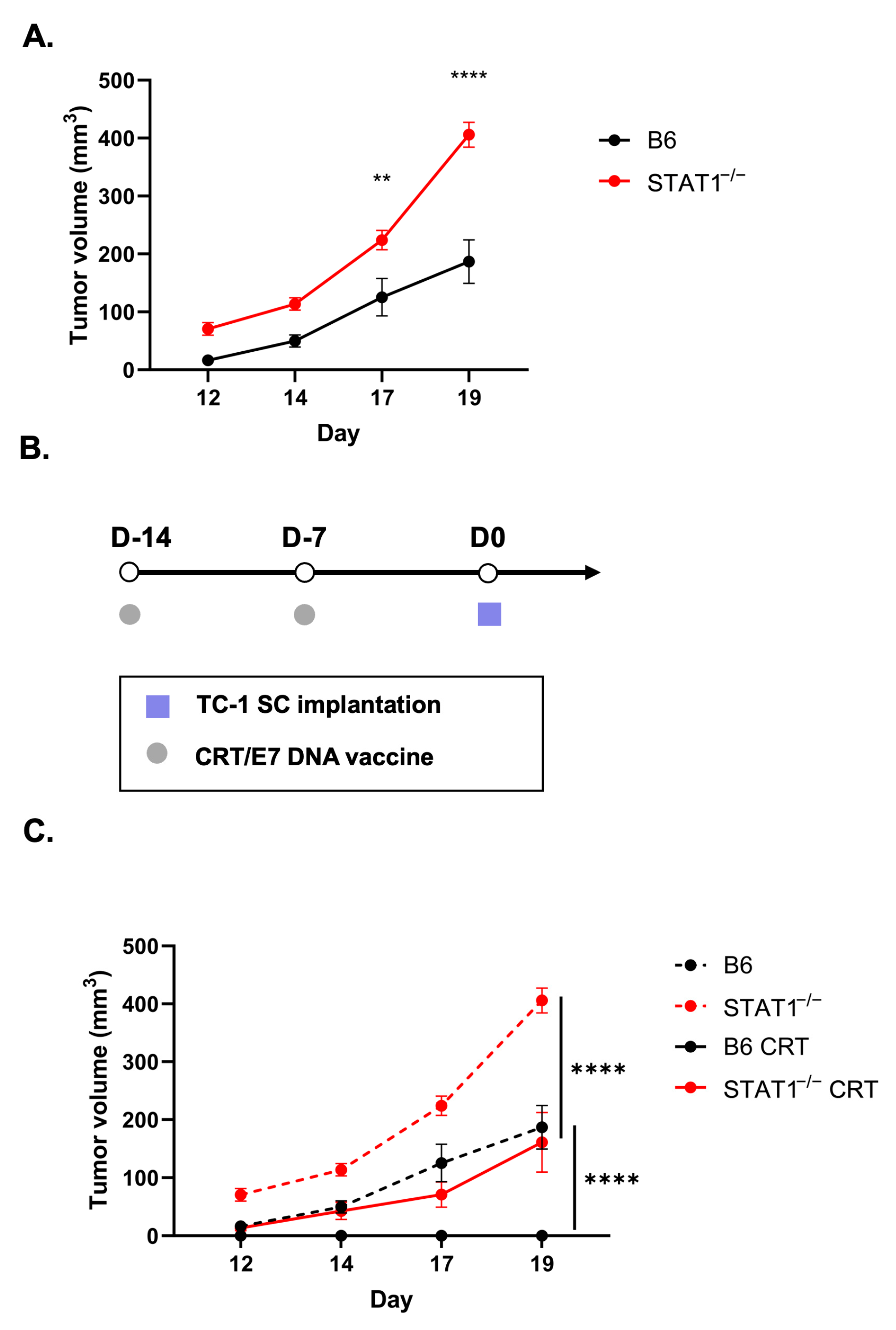
3.1. Formation of Spontaneous HPV+ Oral Tumors in STAT1−/− Mice
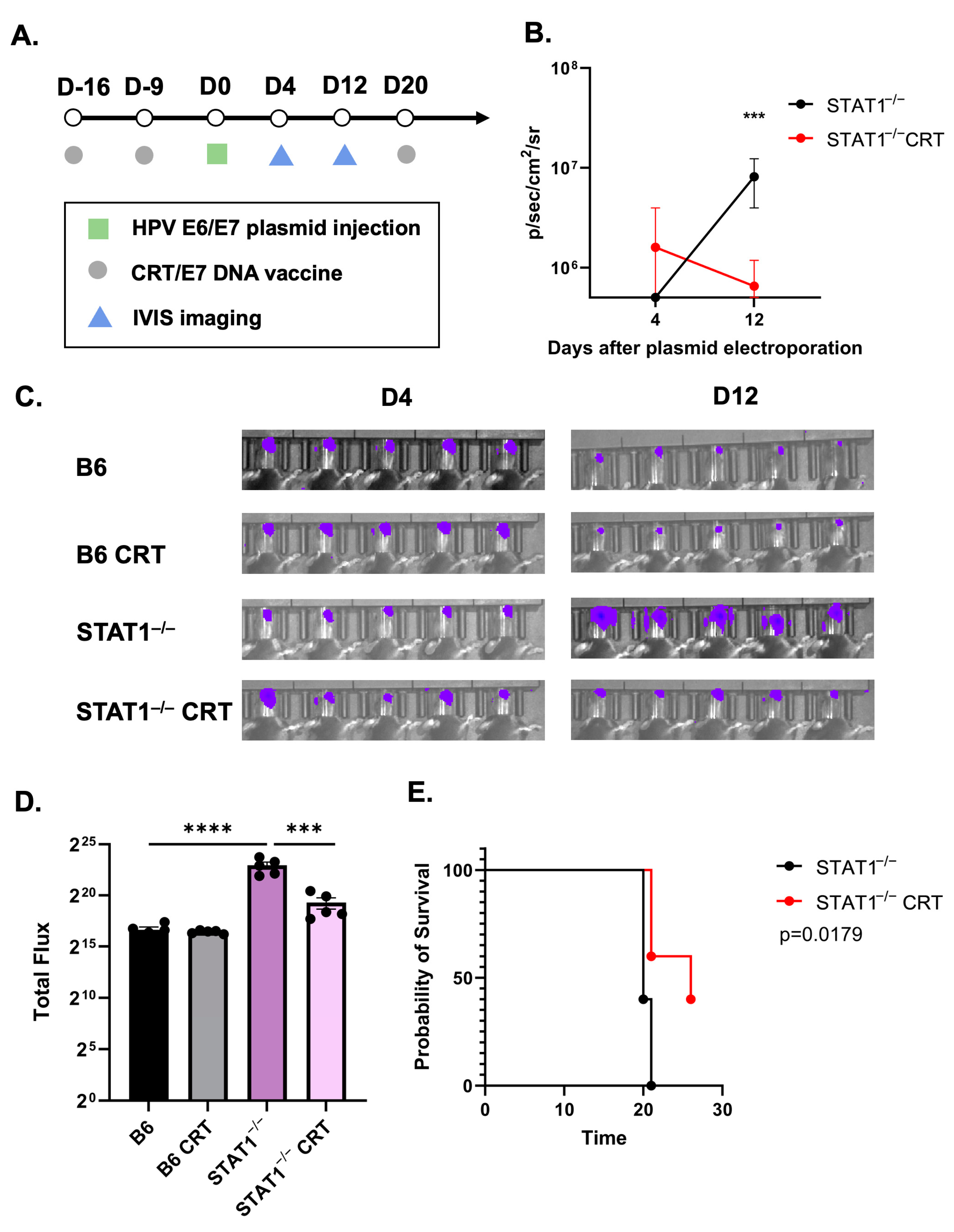
3.2. Vaccination with CRT/E7 DNA Induces Tumor Control in STAT1−/− Mice
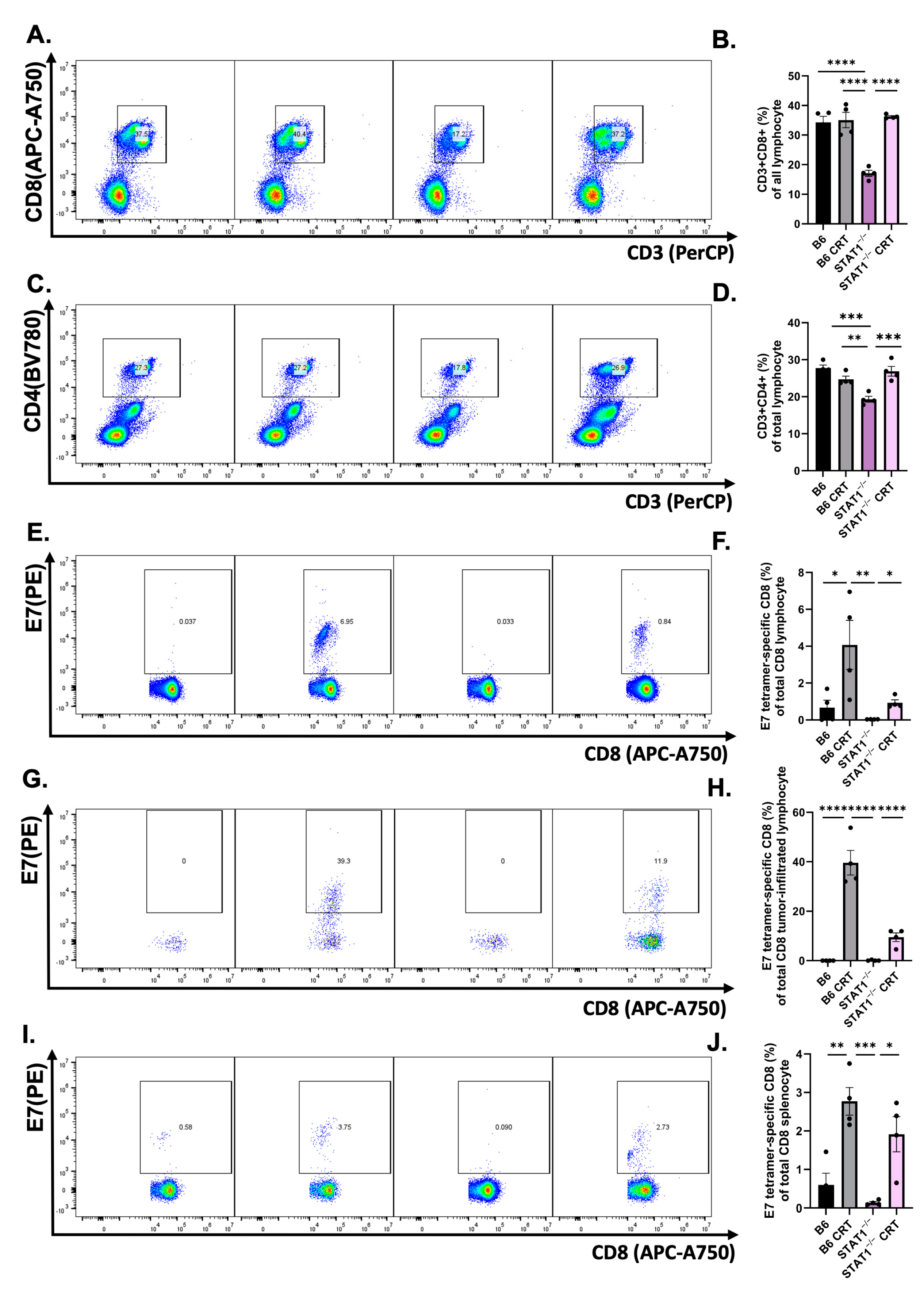
3.3. CRT/E7 Vaccination Contributes to the Enhancement of Effector T-Cell Function in STAT1−/− Mice
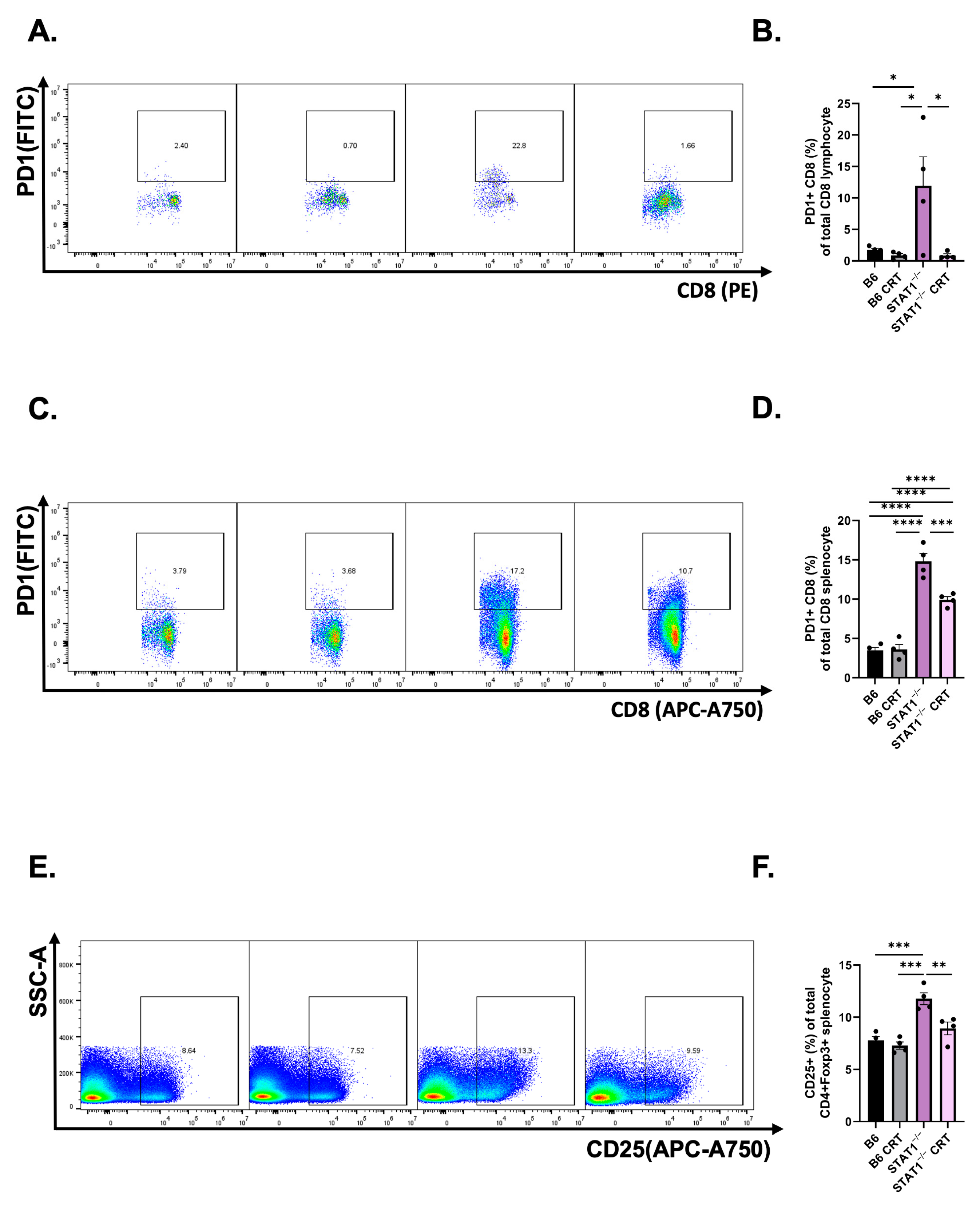
3.4. CRT/E7 Vaccination Contributes to a Reduction in T-Cell Exhaustion in STAT1−/− Mice
3.5. The Host Immune Response Remains Responsive against Tumor Challenge in STAT1−/− Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| CRT/E7 | Calreticulin-E7 |
| EGCG | Epigallocatechin 3-gallate |
| ICB | Immune checkpoint blockade |
| IFN | Interferon |
| IP | Intraperitoneal injection |
| JAK | Janus protein tyrosine kinase |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| HR-HPV | High-risk human papillomavirus |
| MHC | Major histocompatibility complex |
| NK cells | Natural killer cells |
| NSCLC | Non-small-cell lung cancer |
| PBMC | Peripheral blood mononuclear cell |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| RBC | Red blood cell |
| SH2 domain | Src homology 2 domain |
| STAT | Signal transducer and activator of transcription |
| TIL | Tumor-infiltrating lymphocyte |
| Tumor necrosis factor alpha |
References
- Bruni, L.; Albero, G.; Rowley, J.; Alemany, L.; Arbyn, M.; Giuliano, A.R.; Markowitz, L.E.; Broutet, N.; Taylor, M. Global and Regional Estimates of Genital Human Papillomavirus Prevalence among Men: A Systematic Review and Meta-Analysis. Lancet Glob. Health 2023, 11, e1345–e1362. [Google Scholar] [CrossRef] [PubMed]
- Kombe Kombe, A.J.; Li, B.; Zahid, A.; Mengist, H.M.; Bounda, G.-A.; Zhou, Y.; Jin, T. Epidemiology and Burden of Human Papillomavirus and Related Diseases, Molecular Pathogenesis, and Vaccine Evaluation. Front. Public Health 2020, 8, 552028. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Park, J.S. Clinical Significance of Human Papillomavirus Genotyping. J. Gynecol. Oncol. 2016, 27, e21. [Google Scholar] [CrossRef] [PubMed]
- Egawa, N.; Egawa, K.; Griffin, H.; Doorbar, J. Human Papillomaviruses; Epithelial Tropisms, and the Development of Neoplasia. Viruses 2015, 7, 3863–3890. [Google Scholar] [CrossRef] [PubMed]
- Gillison, M.L.; Chaturvedi, A.K.; Anderson, W.F.; Fakhry, C. Epidemiology of Human Papillomavirus-Positive Head and Neck Squamous Cell Carcinoma. J. Clin. Oncol. 2015, 33, 3235–3242. [Google Scholar] [CrossRef] [PubMed]
- Forman, D.; de Martel, C.; Lacey, C.J.; Soerjomataram, I.; Lortet-Tieulent, J.; Bruni, L.; Vignat, J.; Ferlay, J.; Bray, F.; Plummer, M.; et al. Global Burden of Human Papillomavirus and Related Diseases. Vaccine 2012, 30 (Suppl. S5), F12–F23. [Google Scholar] [CrossRef] [PubMed]
- Ferrall, L.; Lin, K.Y.; Roden, R.B.S.; Hung, C.-F.; Wu, T.-C. Cervical Cancer Immunotherapy: Facts and Hopes. Clin. Cancer Res. 2021, 27, 4953–4973. [Google Scholar] [CrossRef] [PubMed]
- Oyouni, A.A.A. Human Papillomavirus in Cancer: Infection, Disease Transmission, and Progress in Vaccines. J. Infect. Public. Health 2023, 16, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human Papillomavirus and Rising Oropharyngeal Cancer Incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- Roman, B.R.; Aragones, A. Epidemiology and Incidence of HPV-Related Cancers of the Head and Neck. J. Surg. Oncol. 2021, 124, 920–922. [Google Scholar] [CrossRef]
- Lechner, M.; Liu, J.; Masterson, L.; Fenton, T.R. HPV-Associated Oropharyngeal Cancer: Epidemiology, Molecular Biology and Clinical Management. Nat. Rev. Clin. Oncol. 2022, 19, 306–327. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and Cancer: From Basic Studies to Clinical Application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Mahjoor, M.; Mahmoudvand, G.; Farokhi, S.; Shadab, A.; Kashfi, M.; Afkhami, H. Double-Edged Sword of JAK/STAT Signaling Pathway in Viral Infections: Novel Insights into Virotherapy. Cell Commun. Signal 2023, 21, 272. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT Proteins: A Kaleidoscope of Canonical and Non-Canonical Functions in Immunity and Cancer. J. Hematol. Oncol. 2021, 14, 198. [Google Scholar] [CrossRef] [PubMed]
- Prabhavathy, D.; Vijayalakshmi, R.; Kanchana, M.P.; Karunagaran, D. HPV16 E2 Enhances the Expression of NF-ΚB and STAT3 Target Genes and Potentiates NF-ΚB Activation by Inflammatory Mediators. Cell Immunol. 2014, 292, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 Activation in HPV Positive Cervical Cancer through a Virus-Driven Rac1-NFκB-IL-6 Signalling Axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Shishodia, G.; Mahata, S.; Hedau, S.; Pandey, A.; Bhambhani, S.; Batra, S.; Basir, S.F.; Das, B.C.; Bharti, A.C. Aberrant Expression and Constitutive Activation of STAT3 in Cervical Carcinogenesis: Implications in High-Risk Human Papillomavirus Infection. Mol. Cancer 2010, 9, 282. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Shen, B.; Li, J.; Zhang, H.; Zhang, K.; Yang, Y.; Zu, Z.; Shen, D.; Luo, M. STAT3 Exerts Pro-Tumor and Anti-Autophagy Roles in Cervical Cancer. Diagn. Pathol. 2022, 17, 13. [Google Scholar] [CrossRef] [PubMed]
- Strobel, T.D.; Weber, M.; Heber, N.; Holzer, A.; Hoppe-Seyler, K.; Hoppe-Seyler, F. Revisiting the Role of Endogenous STAT3 in HPV-Positive Cervical Cancer Cells. J. Med. Virol. 2023, 95, e29230. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT Signaling Pathway: Input and Output Integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Meraz, M.A.; White, J.M.; Sheehan, K.C.; Bach, E.A.; Rodig, S.J.; Dighe, A.S.; Kaplan, D.H.; Riley, J.K.; Greenlund, A.C.; Campbell, D.; et al. Targeted Disruption of the Stat1 Gene in Mice Reveals Unexpected Physiologic Specificity in the JAK-STAT Signaling Pathway. Cell 1996, 84, 431–442. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, M.-S. STAT1 as a Key Modulator of Cell Death. Cell Signal 2007, 19, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Lesinski, G.B.; Anghelina, M.; Zimmerer, J.; Bakalakos, T.; Badgwell, B.; Parihar, R.; Hu, Y.; Becknell, B.; Abood, G.; Chaudhury, A.R.; et al. The Antitumor Effects of IFN-Alpha Are Abrogated in a STAT1-Deficient Mouse. J. Clin. Investig. 2003, 112, 170–180. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Sexl, V. The Tumor Suppressor Function of STAT1 in Breast Cancer. JAKSTAT 2013, 2, e23353. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Li, C.; Liao, L.; Jiao, J.; Long, L.; Ding, J.; Guo, J.; Li, E.; Xu, L. LncRNA625 Inhibits STAT1-Mediated Transactivation Potential in Esophageal Cancer Cells. Int. J. Biochem. Cell Biol. 2019, 117, 105626. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, J.; Chen, L.; Dong, N.; Ying, Z.; Cai, Z.; Ji, D.; Zhang, Y.; Dong, L.; Li, Y.; et al. STAT1 Modification Improves Therapeutic Effects of Interferons on Lung Cancer Cells. J. Transl. Med. 2015, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Molavi, O.; Su, M.; Lai, R. The Clinical and Biological Significance of STAT1 in Esophageal Squamous Cell Carcinoma. BMC Cancer 2014, 14, 791. [Google Scholar] [CrossRef]
- Anderson, K.; Ryan, N.; Nedungadi, D.; Lamenza, F.; Swingler, M.; Siddiqui, A.; Satoskar, A.; Upadhaya, P.; Pietrzak, M.; Oghumu, S. STAT1 Is Regulated by TRIM24 and Promotes Immunosuppression in Head and Neck Squamous Carcinoma Cells, but Enhances T Cell Antitumour Immunity in the Tumour Microenvironment. Br. J. Cancer 2022, 127, 624–636. [Google Scholar] [CrossRef]
- Hu, X.; Ivashkiv, L.B. Cross-Regulation of Signaling Pathways by Interferon-Gamma: Implications for Immune Responses and Autoimmune Diseases. Immunity 2009, 31, 539–550. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, D.C.; Kim, H.-M.; Kwon, H.-J.; Kwon, S.J.; Kang, S.-J.; Kim, S.C.; Choi, G.-E. STAT1 Deficiency Redirects IFN Signalling toward Suppression of TLR Response through a Feedback Activation of STAT3. Sci. Rep. 2015, 5, 13414. [Google Scholar] [CrossRef]
- Zemek, R.M.; De Jong, E.; Chin, W.L.; Schuster, I.S.; Fear, V.S.; Casey, T.H.; Forbes, C.; Dart, S.J.; Leslie, C.; Zaitouny, A.; et al. Sensitization to Immune Checkpoint Blockade through Activation of a STAT1/NK Axis in the Tumor Microenvironment. Sci. Transl. Med. 2019, 11, eaav7816. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, J.; Heim, L.; Trufa, D.I.; Sirbu, H.; Rieker, R.J.; Chiriac, M.T.; Finotto, S. STAT1 Deficiency Supports PD-1/PD-L1 Signaling Resulting in Dysfunctional TNFα Mediated Immune Responses in a Model of NSCLC. Oncotarget 2018, 9, 37157–37172. [Google Scholar] [CrossRef]
- Liang, Y.-H.; Chen, K.-H.; Tsai, J.-H.; Cheng, Y.-M.; Lee, C.-C.; Kao, C.-H.; Chan, K.-Y.; Chen, Y.-T.; Hsu, W.-L.; Yeh, K.-H. Proteasome Inhibitors Restore the STAT1 Pathway and Enhance the Expression of MHC Class I on Human Colon Cancer Cells. J. Biomed. Sci. 2021, 28, 75. [Google Scholar] [CrossRef]
- Huang, R.; Faratian, D.; Sims, A.H.; Wilson, D.; Thomas, J.S.; Harrison, D.J.; Langdon, S.P. Increased STAT1 Signaling in Endocrine-Resistant Breast Cancer. PLoS ONE 2014, 9, e94226. [Google Scholar] [CrossRef]
- Khodarev, N.N.; Roizman, B.; Weichselbaum, R.R. Molecular Pathways: Interferon/Stat1 Pathway: Role in the Tumor Resistance to Genotoxic Stress and Aggressive Growth. Clin. Cancer Res. 2012, 18, 3015–3021. [Google Scholar] [CrossRef]
- Chang, W.-M.; Chang, Y.-C.; Yang, Y.-C.; Lin, S.-K.; Chang, P.M.-H.; Hsiao, M. AKR1C1 Controls Cisplatin-Resistance in Head and Neck Squamous Cell Carcinoma through Cross-Talk with the STAT1/3 Signaling Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 245. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wu, Y.; Lu, Y.; Yue, Y.; Cui, C.; Yu, M.; Wang, S.; Liu, M.; Zhao, Y.; Sun, Z. STAT1 Expression and HPV16 Viral Load Predict Cervical Lesion Progression. Oncol. Lett. 2020, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, M.S.; Andrade Filho, P.A.; Ferrone, S.; Ferris, R.L. Deficiency of Activated STAT1 in Head and Neck Cancer Cells Mediates TAP1-Dependent Escape from Cytotoxic T Lymphocytes. Cancer Immunol. Immunother. 2011, 60, 525–535. [Google Scholar] [CrossRef]
- Ryan, N.; Anderson, K.; Volpedo, G.; Hamza, O.; Varikuti, S.; Satoskar, A.R.; Oghumu, S. STAT1 Inhibits T-Cell Exhaustion and Myeloid Derived Suppressor Cell Accumulation to Promote Antitumor Immune Responses in Head and Neck Squamous Cell Carcinoma. Int. J. Cancer 2020, 146, 1717–1729. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Yang, M.-C.; Tseng, S.-H.; Jiang, R.; Yang, A.; Farmer, E.; Peng, S.; Henkle, T.; Chang, Y.-N.; Hung, C.-F.; et al. Integration of Oncogenes via Sleeping Beauty as a Mouse Model of HPV16+ Oral Tumors and Immunologic Control. Cancer Immunol. Res. 2018, 6, 305–319. [Google Scholar] [CrossRef]
- Wiesner, S.M.; Decker, S.A.; Larson, J.D.; Ericson, K.; Forster, C.; Gallardo, J.L.; Long, C.; Demorest, Z.L.; Zamora, E.A.; Low, W.C.; et al. De Novo Induction of Genetically Engineered Brain Tumors in Mice Using Plasmid DNA. Cancer Res. 2009, 69, 431–439. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular Evolution of a Novel Hyperactive Sleeping Beauty Transposase Enables Robust Stable Gene Transfer in Vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.F.; Hung, C.F.; Chai, C.Y.; Hsu, K.F.; He, L.; Ling, M.; Wu, T.C. Tumor-Specific Immunity and Antiangiogenesis Generated by a DNA Vaccine Encoding Calreticulin Linked to a Tumor Antigen. J. Clin. Investig. 2001, 108, 669–678. [Google Scholar] [CrossRef]
- Cheng, W.-F.; Lee, C.-N.; Chang, M.-C.; Su, Y.-N.; Chen, C.-A.; Hsieh, C.-Y. Antigen-Specific CD8+ T Lymphocytes Generated from a DNA Vaccine Control Tumors through the Fas-FasL Pathway. Mol. Ther. 2005, 12, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.-F.; Hung, C.-F.; Chen, C.-A.; Lee, C.-N.; Su, Y.-N.; Chai, C.-Y.; Boyd, D.A.K.; Hsieh, C.-Y.; Wu, T.-C. Characterization of DNA Vaccines Encoding the Domains of Calreticulin for Their Ability to Elicit Tumor-Specific Immunity and Antiangiogenesis. Vaccine 2005, 23, 3864–3874. [Google Scholar] [CrossRef]
- Jenkins, E.; Whitehead, T.; Fellermeyer, M.; Davis, S.J.; Sharma, S. The Current State and Future of T-Cell Exhaustion Research. Oxf. Open Immunol. 2023, 4, iqad006. [Google Scholar] [CrossRef]
- Tietscher, S.; Wagner, J.; Anzeneder, T.; Langwieder, C.; Rees, M.; Sobottka, B.; de Souza, N.; Bodenmiller, B. A Comprehensive Single-Cell Map of T Cell Exhaustion-Associated Immune Environments in Human Breast Cancer. Nat. Commun. 2023, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Huff, A.L.; Longway, G.; Mitchell, J.T.; Andaloori, L.; Davis-Marcisak, E.; Chen, F.; Lyman, M.R.; Wang, R.; Mathew, J.; Barrett, B.; et al. CD4 T Cell-Activating Neoantigens Enhance Personalized Cancer Vaccine Efficacy. JCI Insight 2023, 8. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-M.; Compans, R.W. Host Responses from Innate to Adaptive Immunity after Vaccination: Molecular and Cellular Events. Mol. Cells 2009, 27, 5–14. [Google Scholar] [CrossRef]
- Leopold Wager, C.M.; Hole, C.R.; Wozniak, K.L.; Olszewski, M.A.; Wormley, F.L. STAT1 Signaling Is Essential for Protection against Cryptococcus neoformans Infection in Mice. J. Immunol. 2014, 193, 4060–4071. [Google Scholar] [CrossRef]
- Hofer, M.J.; Li, W.; Manders, P.; Terry, R.; Lim, S.L.; King, N.J.C.; Campbell, I.L. Mice Deficient in STAT1 but Not STAT2 or IRF9 Develop a Lethal CD4+ T-Cell-Mediated Disease Following Infection with Lymphocytic Choriomeningitis Virus. J. Virol. 2012, 86, 6932–6946. [Google Scholar] [CrossRef] [PubMed]
- Bente, D.A.; Alimonti, J.B.; Shieh, W.-J.; Camus, G.; Ströher, U.; Zaki, S.; Jones, S.M. Pathogenesis and Immune Response of Crimean-Congo Hemorrhagic Fever Virus in a STAT-1 Knockout Mouse Model. J. Virol. 2010, 84, 11089–11100. [Google Scholar] [CrossRef] [PubMed]
- Bradfute, S.B.; Stuthman, K.S.; Shurtleff, A.C.; Bavari, S. A STAT-1 Knockout Mouse Model for Machupo Virus Pathogenesis. Virol. J. 2011, 8, 300. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cavalli, A.; Cascio, A. STAT1 and Its Crucial Role in the Control of Viral Infections. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, B.; Stoiber, D.; Moriggl, R.; Weisz, E.; Ott, R.G.; Kreibich, R.; Levy, D.E.; Beug, H.; Freissmuth, M.; Sexl, V. STAT1 Acts as a Tumor Promoter for Leukemia Development. Cancer Cell 2006, 10, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.R.; Vermi, W.; Luo, J.; Lucini, L.; Rickert, C.; Fowler, A.M.; Lonardi, S.; Arthur, C.; Young, L.J.; Levy, D.E.; et al. STAT1-Deficient Mice Spontaneously Develop Estrogen Receptor α-Positive Luminal Mammary Carcinomas. Breast Cancer Res. 2012, 14, R16. [Google Scholar] [CrossRef] [PubMed]
- Palakurthi, B.; Fross, S.R.; Guldner, I.H.; Aleksandrovic, E.; Liu, X.; Martino, A.K.; Wang, Q.; Neff, R.A.; Golomb, S.M.; Lewis, C.; et al. Targeting CXCL16 and STAT1 Augments Immune Checkpoint Blockade Therapy in Triple-Negative Breast Cancer. Nat. Commun. 2023, 14, 2109. [Google Scholar] [CrossRef]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 Expression in the Tumor Microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Liu, F.; Liu, J.; Zhang, J.; Shi, J.; Gui, L.; Xu, G. Expression of STAT1 Is Positively Correlated with PD-L1 in Human Ovarian Cancer. Cancer Biol. Ther. 2020, 21, 963–971. [Google Scholar] [CrossRef]
- Seamons, A.; Treuting, P.M.; Meeker, S.; Hsu, C.; Paik, J.; Brabb, T.; Escobar, S.S.; Alexander, J.S.; Ericsson, A.C.; Smith, J.G.; et al. Obstructive Lymphangitis Precedes Colitis in Murine Norovirus-Infected Stat1-Deficient Mice. Am. J. Pathol. 2018, 188, 1536–1554. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, L.; Hu, M.-H.; Fan, D.; Tu, H.-F.; Tsai, Y.-C.; Cheng, M.; Wang, S.; Chang, C.-L.; Wu, T.-C.; Hung, C.-F. STAT1-Deficient HPV E6/E7-Associated Cancers Maintain Host Immunocompetency against Therapeutic Intervention. Vaccines 2024, 12, 430. https://doi.org/10.3390/vaccines12040430
Lim L, Hu M-H, Fan D, Tu H-F, Tsai Y-C, Cheng M, Wang S, Chang C-L, Wu T-C, Hung C-F. STAT1-Deficient HPV E6/E7-Associated Cancers Maintain Host Immunocompetency against Therapeutic Intervention. Vaccines. 2024; 12(4):430. https://doi.org/10.3390/vaccines12040430
Chicago/Turabian StyleLim, Ling, Ming-Hung Hu, Darrell Fan, Hsin-Fang Tu, Ya-Chea Tsai, Michelle Cheng, Suyang Wang, Chih-Long Chang, Tzyy-Choou Wu, and Chien-Fu Hung. 2024. "STAT1-Deficient HPV E6/E7-Associated Cancers Maintain Host Immunocompetency against Therapeutic Intervention" Vaccines 12, no. 4: 430. https://doi.org/10.3390/vaccines12040430
APA StyleLim, L., Hu, M. -H., Fan, D., Tu, H. -F., Tsai, Y. -C., Cheng, M., Wang, S., Chang, C. -L., Wu, T. -C., & Hung, C. -F. (2024). STAT1-Deficient HPV E6/E7-Associated Cancers Maintain Host Immunocompetency against Therapeutic Intervention. Vaccines, 12(4), 430. https://doi.org/10.3390/vaccines12040430