Vaccinia Virus LC16m8∆ as a Vaccine Vector for Clinical Applications

Abstract
:1. Introduction
1.1. First-Generation Smallpox Vaccines

{kind=link}
{kind=link}
{kind=link}
| Generation | Product | Platform | Parental Strain |
|---|---|---|---|
| First-generation | Lister/Elstree | Lymph-derived | Lister/Elstree |
| Dryvax | Lymph-derived | NYCBH a | |
| Ikeda | Lymph-derived | Ikeda | |
| Dairen I | Lymph-derived | Dairen I | |
| Second-generation | ACAM1000 | Clonal virus grown in MRC-5 cells | Dryvax |
| ACAM2000 | Clonal virus grown in Vero cells | ACAM1000 | |
| Elstree-BN | Lister/Elstree lymph-derived virus passaged in CEF b | Lister/Elstree | |
| CCSV | NYCBH lymph-derived virus passaged in MRC-5 cells | NYCBH | |
| Third-generation | LC16m8 c | Minute-pock-forming, temperature-sensitive variant virus | Lister/Elstree |
| IMVAMUNE (MVA d) | MVA571 additionally passaged in CEF | MVA571 | |
| DIs e | Minute-pock-forming variant virus passaged in eggs | Dairen I | |
| Fourth-generation | LC16m8∆ | Derived from LC16m8 by deleting the B5R gene | LC16m8 |
| NYVAC | Attenuated clonal Copenhagen strain generated by deleting 18 non-essential genes | Copenhagen |
1.2. Second-Generation Vaccines
1.3. Third-Generation Vaccines
1.4. Fourth-Generation Vaccines
2. LC16m8 and B5R
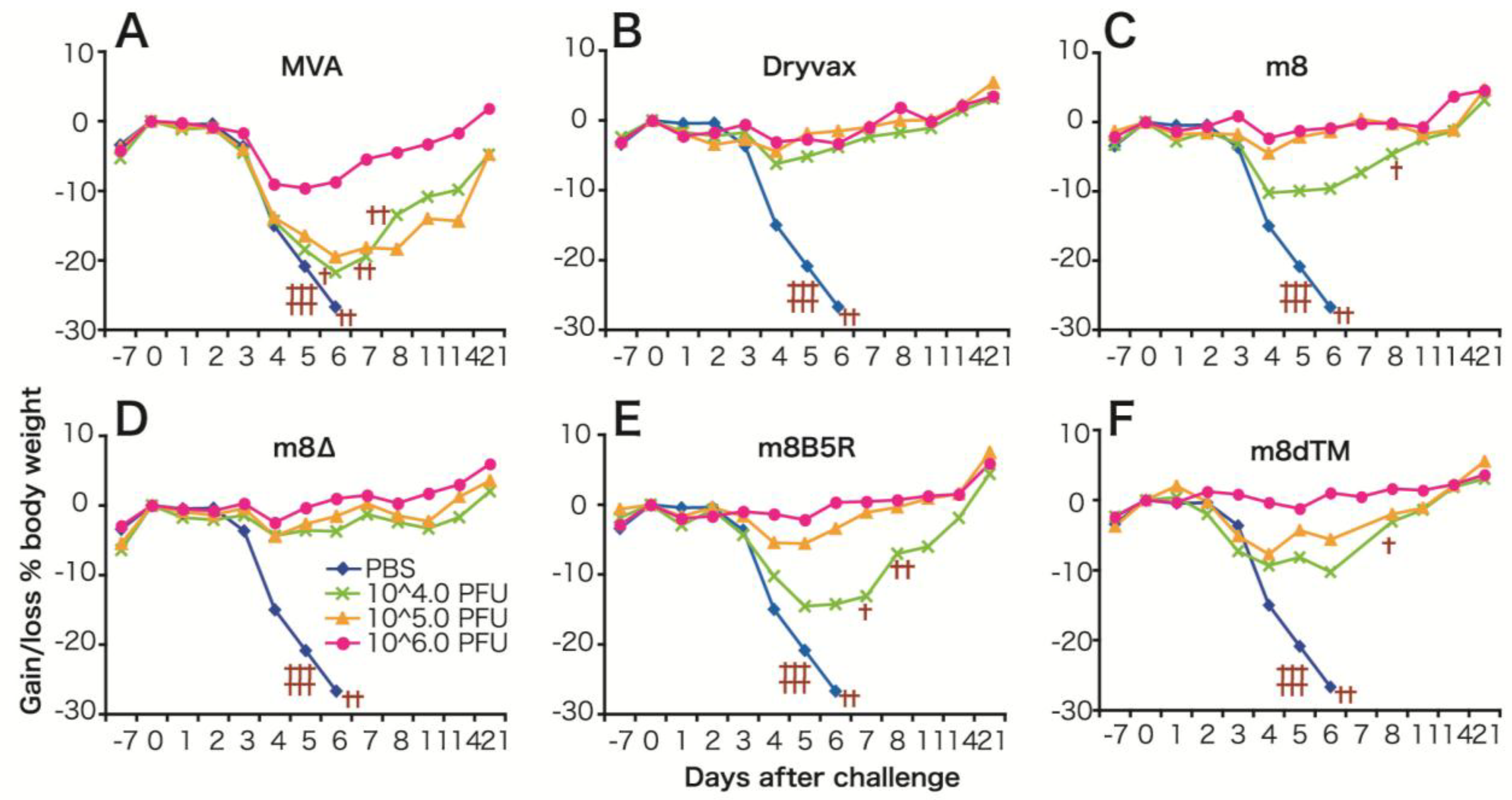
3. LC16m8Δ
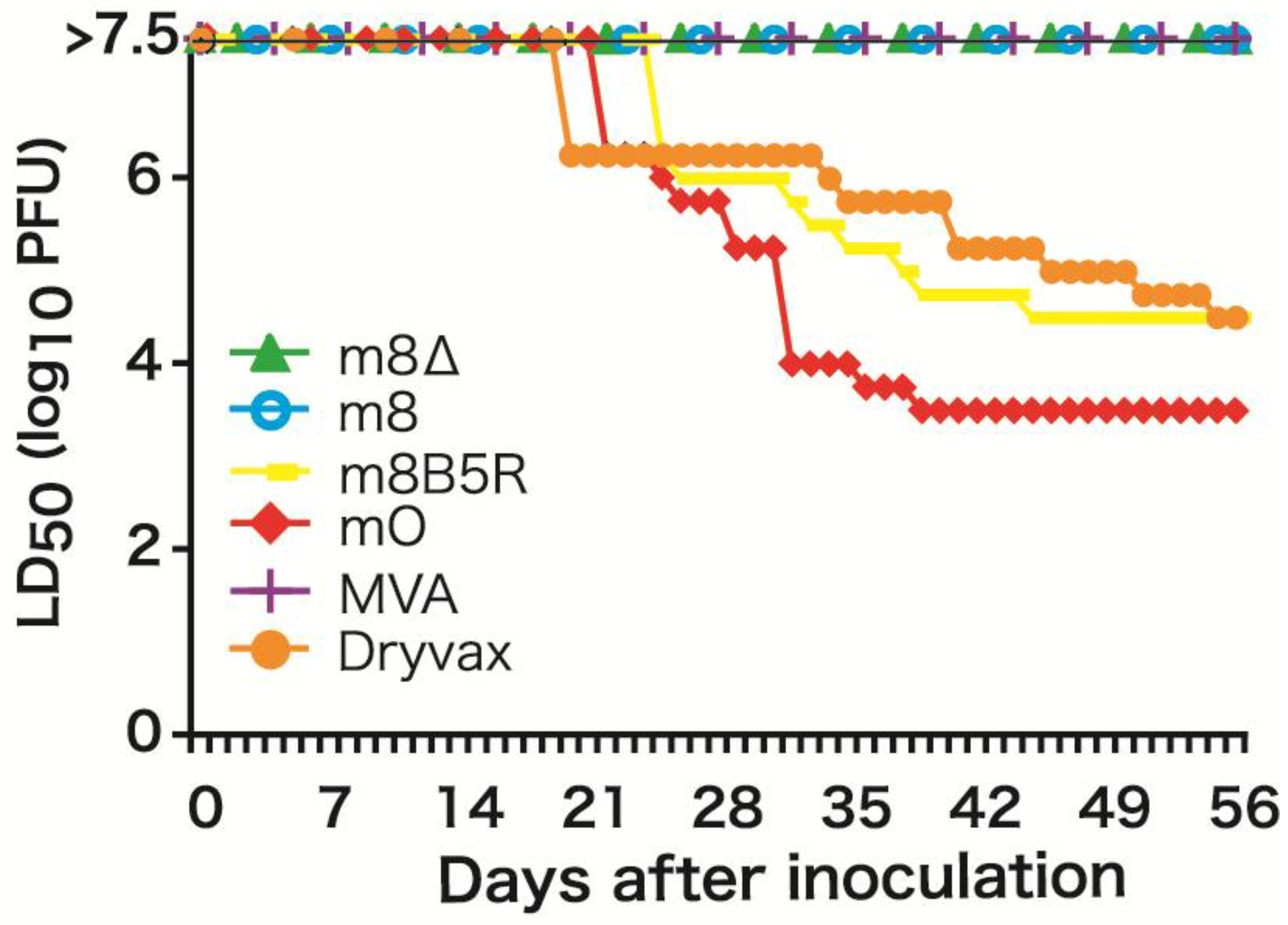
3.1. Safety Profile


3.2. Immunogenicity

4. LC16m8∆ as a Vehicle for Expressing Foreign Genes
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D.; Organization, W.H. Smallpox and Its Eradication; World Health Organization: Geneva, Switzerland, 1988. [Google Scholar]
- Wehrle, P.F. A reality in our time—Certification of the global eradication of smallpox. J. Infect. Dis. 1980, 142, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Henderson, D.A.; Inglesby, T.V.; Bartlett, J.G.; Ascher, M.S.; Eitzen, E.; Jahrling, P.B.; Hauer, J.; Layton, M.; McDade, J.; Osterholm, M.T.; et al. Smallpox as a biological weapon: Medical and public health management. Working group on civilian biodefense. JAMA 1999, 281, 2127–2137. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the western hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [PubMed]
- The Centers for Disease Control and Prevention. Multistate outbreak of monkeypox—Illinois, indiana, and wisconsin, 2003. JAMA 2003, 290, 30–31. [Google Scholar]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.R.; Dolin, R. Vaccinia viruses: Vaccines against smallpox and vectors against infectious diseases and tumors. Expert Rev. Vaccines 2011, 10, 1221–1240. [Google Scholar] [CrossRef] [PubMed]
- Verardi, P.H.; Titong, A.; Hagen, C.J. A vaccinia virus renaissance: New vaccine and immunotherapeutic uses after smallpox eradication. Human Vaccines Immunother. 2012, 8, 961–970. [Google Scholar] [CrossRef]
- Casey, C.G.; Iskander, J.K.; Roper, M.H.; Mast, E.E.; Wen, X.J.; Torok, T.J.; Chapman, L.E.; Swerdlow, D.L.; Morgan, J.; Heffelfinger, J.D.; et al. Adverse events associated with smallpox vaccination in the United States, January–October 2003. JAMA 2005, 294, 2734–2743. [Google Scholar] [CrossRef] [PubMed]
- Sejvar, J.J.; Labutta, R.J.; Chapman, L.E.; Grabenstein, J.D.; Iskander, J.; Lane, J.M. Neurologic adverse events associated with smallpox vaccination in the united states, 2002–2004. JAMA 2005, 294, 2744–2750. [Google Scholar] [CrossRef] [PubMed]
- Murphy, F.A.; Osburn, B.I. Adventitious agents and smallpox vaccine in strategic national stockpile. Emerg. Infect. Dis. 2005, 11, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Weltzin, R.; Liu, J.; Pugachev, K.V.; Myers, G.A.; Coughlin, B.; Blum, P.S.; Nichols, R.; Johnson, C.; Cruz, J.; Kennedy, J.S.; et al. Clonal vaccinia virus grown in cell culture as a new smallpox vaccine. Nat. Med. 2003, 9, 1125–1130. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.E.; Newman, F.K.; Kennedy, J.S.; Ennis, F.; Abate, G.; Hoft, D.F.; Monath, T.P. Comparison of the safety and immunogenicity of ACAM1000, ACAM2000 and Dryvax® in healthy vaccinia-naive adults. Vaccine 2009, 27, 1637–1644. [Google Scholar] [CrossRef] [PubMed]
- Monath, T.P.; Caldwell, J.R.; Mundt, W.; Fusco, J.; Johnson, C.S.; Buller, M.; Liu, J.; Gardner, B.; Downing, G.; Blum, P.S.; et al. Acam2000 clonal vero cell culture vaccinia virus (New York city board of health strain)—A second-generation smallpox vaccine for biological defense. Int. J. Infect. Dis. 2004, 8, 31–44. [Google Scholar] [CrossRef]
- Greenberg, R.N.; Kennedy, J.S.; Clanton, D.J.; Plummer, E.A.; Hague, L.; Cruz, J.; Ennis, F.A.; Blackwelder, W.C.; Hopkins, R.J. Safety and immunogenicity of new cell-cultured smallpox vaccine compared with calf-lymph derived vaccine: A blind, single-centre, randomised controlled trial. Lancet 2005, 365, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Stittelaar, K.J.; van Amerongen, G.; Kondova, I.; Kuiken, T.; van Lavieren, R.F.; Pistoor, F.H.; Niesters, H.G.; van Doornum, G.; van der Zeijst, B.A.; Mateo, L.; et al. Modified vaccinia virus ankara protects macaques against respiratory challenge with monkeypox virus. J. Virol. 2005, 79, 7845–7851. [Google Scholar] [CrossRef] [PubMed]
- Stickl, H.; Hochstein-Mintzel, V.; Mayr, A.; Huber, H.C.; Schafer, H.; Holzner, A. MVA vaccination against smallpox: Clinical tests with an attenuated live vaccinia virus strain (MVA). Dtsch. Med. Wochenschr. 1974, 99, 2386–2392. (in German). [Google Scholar] [CrossRef] [PubMed]
- Mayr, A.; Stickl, H.; Muller, H.K.; Danner, K.; Singer, H. The smallpox vaccination strain MVA: Marker, genetic structure, experience gained with the parenteral vaccination and behavior in Organisms with a debilitated defence mechanism. Zentralbl. Bakteriol. B 1978, 167, 375–390. (in German). [Google Scholar] [PubMed]
- Kitamura, T.; Kitamura, Y.; Tagaya, I. Immunogenicity of an attenuated strain of vaccinia virus on rabbits and monkeys. Nature 1967, 215, 1187–1188. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, S. Special edition future of smallpox vaccination: Everything about attenuated smallpox vaccines. Basics of new attenuated smallpox vaccine strain LC16m8. Rinshotouirusu 1975, 3, 229–235. [Google Scholar]
- Morita, M.; Aoyama, Y.; Arita, M.; Amona, H.; Yoshizawa, H.; Hashizume, S.; Komatsu, T.; Tagaya, I. Comparative studies of several vaccinia virus strains by intrathalamic inoculation into cynomolgus monkeys. Arch. Virol. 1977, 53, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Arita, M.; Komatsu, T.; Amano, H.; Hashizume, S. A comparison of neurovirulence of vaccinia virus by intrathalamic and/or intracisternal inoculations into cynomolgus monkeys. Microbiol. Immunol. 1977, 21, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, S.; Yoshizawa, H.; Morita, M.; Suzuki, K. Properties of Attenuated Mutant of Vaccinia Virus, LC16m8, Derived from Lister Strain; Elsevier Science Publishing Co. Inc.: New York, NY, USA, 1985. [Google Scholar]
- Meyer, H.; Sutter, G.; Mayr, A. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 1991, 72, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Carroll, M.W.; Czerny, C.P.; Merchlinsky, M.; Sisler, J.R.; Moss, B. Marker rescue of the host range restriction defects of modified vaccinia virus Ankara. Virology 1998, 251, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Perkus, M.E.; Goebel, S.J.; Davis, S.W.; Johnson, G.P.; Limbach, K.; Norton, E.K.; Paoletti, E. Vaccinia virus host range genes. Virology 1990, 179, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Hochstein-Mintzel, V.; Hanichen, T.; Huber, H.C.; Stickl, H. An attenuated strain of vaccinia virus (MVA). Successful intramuscular immunization against vaccinia and variola. Zentralbl. Bakteriol. Orig. A 1975, 230, 283–297. [Google Scholar] [PubMed]
- Mayr, A. Smallpox vaccination and bioterrorism with pox viruses. Comp. Immunol. Microbiol. Infect. Dis. 2003, 26, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, J.; Arndtz, N.; Eckl, K.M.; Thomsen, T.; Petzold, B.; Mateo, L.; Schlereth, B.; Handley, A.; King, L.; Hulsemann, V.; et al. Safety and immunogenicity of imvamune, a promising candidate as a third generation smallpox vaccine. Vaccine 2006, 24, 2065–2070. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.E.; Newman, F.K.; Kennedy, J.S.; Sobek, V.; Ennis, F.A.; Hill, H.; Yan, L.K.; Chaplin, P.; Vollmar, J.; Chaitman, B.R.; et al. Clinical and immunologic responses to multiple doses of imvamune (modified vaccinia Ankara) followed by dryvax challenge. Vaccine 2007, 25, 8562–8573. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.S.; Greenberg, R.N. Imvamune: Modified vaccinia Ankara strain as an attenuated smallpox vaccine. Expert Rev. Vaccines 2009, 8, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.S.; Wilck, M.B.; Baden, L.R.; Walsh, S.R.; Grandpre, L.E.; Devoy, C.; Giri, A.; Noble, L.C.; Kleinjan, J.A.; Stevenson, K.E.; et al. Effect of vaccination with modified vaccinia Ankara (ACAM3000) on subsequent challenge with Dryvax. J. Infect. Dis. 2010, 201, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Wilck, M.B.; Seaman, M.S.; Baden, L.R.; Walsh, S.R.; Grandpre, L.E.; Devoy, C.; Giri, A.; Kleinjan, J.A.; Noble, L.C.; Stevenson, K.E.; et al. Safety and immunogenicity of modified vaccinia Ankara (ACAM3000): Effect of dose and route of administration. J. Infect. Dis. 2010, 201, 1361–1370. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, M.; Tashiro, M.; Shida, H. Genetically stable and fully effective smallpox vaccine strain constructed from highly attenuated vaccinia LC16m8. Proc. Natl. Acad. Sci. USA 2005, 102, 4152–4157. [Google Scholar] [CrossRef] [PubMed]
- Meseda, C.A.; Garcia, A.D.; Kumar, A.; Mayer, A.E.; Manischewitz, J.; King, L.R.; Golding, H.; Merchlinsky, M.; Weir, J.P. Enhanced immunogenicity and protective effect conferred by vaccination with combinations of modified vaccinia virus Ankara and licensed smallpox vaccine dryvax in a mouse model. Virology 2005, 339, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, L.S.; Earl, P.L.; Eller, L.A.; Moss, B. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc. Natl. Acad. Sci. USA 2004, 101, 4590–4595. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.; Ueda, Y.; Matsuo, K.; Matsuura, Y.; Kitamura, T.; Kato, K.; Izumi, Y.; Someya, K.; Ohsu, T.; Honda, M.; et al. Structural analysis of vaccinia virus Dis strain: Application as a new replication-deficient viral vector. Virology 2002, 302, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Kenner, J.; Cameron, F.; Empig, C.; Jobes, D.V.; Gurwith, M. LC16m8: An attenuated smallpox vaccine. Vaccine 2006, 24, 7009–7022. [Google Scholar] [CrossRef] [PubMed]
- Kempe, C.H.; Fulginiti, V.; Minamitani, M.; Shinefield, H. Smallpox vaccination of eczema patients with a strain of attenuated live vaccinia (CVI-78). Pediatrics 1968, 42, 980–985. [Google Scholar] [PubMed]
- Yamaguchi, M.; Kimura, M.; Hirayama, M. Vaccination research groups research report: Ministry of health and welfare special research: Postvaccination side effects and research regarding treatment of complications. Rinsho Uirusu Clin. Virus 1975, 3, 225–228. (in Japanese). [Google Scholar]
- Tartaglia, J.; Perkus, M.E.; Taylor, J.; Norton, E.K.; Audonnet, J.C.; Cox, W.I.; Davis, S.W.; van der Hoeven, J.; Meignier, B.; Riviere, M.; et al. NYVAC: A highly attenuated strain of vaccinia virus. Virology 1992, 188, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, E.; Tartaglia, J.; Taylor, J. Safe and effective poxvirus vectors—NYVAC and ALVAC. Dev. Biol. Stand. 1994, 82, 65–69. [Google Scholar] [PubMed]
- Holzer, G.W.; Falkner, F.G. Construction of a vaccinia virus deficient in the essential DNA repair enzyme uracil DNA glycosylase by a complementing cell line. J. Virol. 1997, 71, 4997–5002. [Google Scholar] [PubMed]
- Holzer, G.W.; Remp, G.; Antoine, G.; Pfleiderer, M.; Enzersberger, O.M.; Emsenhuber, W.; Hammerle, T.; Gruber, F.; Urban, C.; Falkner, F.G.; et al. Highly efficient induction of protective immunity by a vaccinia virus vector defective in late gene expression. J. Virol. 1999, 73, 4536–4542. [Google Scholar] [PubMed]
- Ober, B.T.; Bruhl, P.; Schmidt, M.; Wieser, V.; Gritschenberger, W.; Coulibaly, S.; Savidis-Dacho, H.; Gerencer, M.; Falkner, F.G. Immunogenicity and safety of defective vaccinia virus lister: Comparison with modified vaccinia virus Ankara. J. Virol. 2002, 76, 7713–7723. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, S.; Bruhl, P.; Mayrhofer, J.; Schmid, K.; Gerencer, M.; Falkner, F.G. The nonreplicating smallpox candidate vaccines defective vaccinia lister (DVV-l) and modified vaccinia Ankara (MVA) elicit robust long-term protection. Virology 2005, 341, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Najera, J.L.; Gomez, C.E.; Domingo-Gil, E.; Gherardi, M.M.; Esteban, M. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J. Virol. 2006, 80, 6033–6047. [Google Scholar] [CrossRef] [PubMed]
- Ferrier-Rembert, A.; Drillien, R.; Tournier, J.N.; Garin, D.; Crance, J.M. Short- and long-term immunogenicity and protection induced by non-replicating smallpox vaccine candidates in mice and comparison with the traditional 1st generation vaccine. Vaccine 2008, 26, 1794–1804. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Nishimaki, F.; Funahashi, S.; Miki, K.; Hashizume, S.; Sugimoto, M. Regulation of plaque size and host range by a vaccinia virus gene related to complement system proteins. Virology 1991, 181, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [PubMed]
- Schmelz, M.; Sodeik, B.; Ericsson, M.; Wolffe, E.J.; Shida, H.; Hiller, G.; Griffiths, G. Assembly of vaccinia virus: The second wrapping cisterna is derived from the trans golgi network. J. Virol. 1994, 68, 130–147. [Google Scholar] [PubMed]
- Hollinshead, M.; Rodger, G.; van Eijl, H.; Law, M.; Hollinshead, R.; Vaux, D.J.; Smith, G.L. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 2001, 154, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Rietdorf, J.; Ploubidou, A.; Reckmann, I.; Holmstrom, A.; Frischknecht, F.; Zettl, M.; Zimmermann, T.; Way, M. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 2001, 3, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Ward, B.M.; Moss, B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J. Virol. 2001, 75, 4802–4813. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Ward, B.M.; Weisberg, A.S.; Moss, B. Mutations in the vaccinia virus A33R and B5R envelope proteins that enhance release of extracellular virions and eliminate formation of actin-containing microvilli without preventing tyrosine phosphorylation of the A36R protein. J. Virol. 2003, 77, 12266–12275. [Google Scholar] [CrossRef] [PubMed]
- Newsome, T.P.; Scaplehorn, N.; Way, M. Src mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 2004, 306, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.G.; Kristensson, K. Extracellular release of enveloped vaccinia virus from mouse nasal epithelial cells in vivo. J. Gen. Virol. 1985, 66, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Galmiche, M.C.; Goenaga, J.; Wittek, R.; Rindisbacher, L. Neutralizing and protective antibodies directed against vaccinia virus envelope antigens. Virology 1999, 254, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.W.; Custer, D.M.; Thompson, E. Four-gene-combination DNA vaccine protects mice against a lethal vaccinia virus challenge and elicits appropriate antibody responses in nonhuman primates. Virology 2003, 306, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Pulford, D.J.; Gates, A.; Bridge, S.H.; Robinson, J.H.; Ulaeto, D. Differential efficacy of vaccinia virus envelope proteins administered by DNA immunisation in protection of BALB/c mice from a lethal intranasal poxvirus challenge. Vaccine 2004, 22, 3358–3366. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.W.; Thompson, E.; Wilhelmsen, C.; Zimmerman, M.; Ichou, M.A.; Steffen, S.E.; Schmaljohn, C.S.; Schmaljohn, A.L.; Jahrling, P.B. Smallpox DNA vaccine protects nonhuman primates against lethal monkeypox. J. Virol. 2004, 78, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Williamson, J.D.; Reith, R.W.; Jeffrey, L.J.; Arrand, J.R.; Mackett, M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 1990, 71, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, M.S.S.; Ami, Y.; Suzaki, Y.; Nagata, N.; Iwata, N.; Hasegawa, H.; Ogata, M.; Fukushi, H.; Mizutani, T.; Shida, H.; et al. Protective effects of improved smalpox vaccine LC16m8∆ against a lethal monkeypox challenge in cynomolgus monkeys. In Proceedings of the 54th Annual Meeting of the Japanese Society for Virology, Nagoya, Japan, 19–21 November 2006.
- Hooper, J.W.; Custer, D.M.; Schmaljohn, C.S.; Schmaljohn, A.L. DNA vaccination with vaccinia virus L1R and A33R genes protects mice against a lethal poxvirus challenge. Virology 2000, 266, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.R.; Goudsmit, J.; Holterman, L.; Ewald, B.A.; Denholtz, M.; Devoy, C.; Giri, A.; Grandpre, L.E.; Heraud, J.M.; Franchini, G.; et al. Differential antigen requirements for protection against systemic and intranasal vaccinia virus challenges in mice. J. Virol. 2008, 82, 6829–6837. [Google Scholar] [CrossRef] [PubMed]
- Saijo, M.; Ami, Y.; Suzaki, Y.; Nagata, N.; Iwata, N.; Hasegawa, H.; Ogata, M.; Fukushi, S.; Mizutani, T.; Sata, T.; et al. LC16m8, a highly attenuated vaccinia virus vaccine lacking expression of the membrane protein B5R, protects monkeys from monkeypox. J. Virol. 2006, 80, 5179–5188. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, S.; Sakiyama, T.; Hasegawa, H.; Saijo, M.; Maeda, A.; Kurane, I.; Maeno, G.; Kimura, J.; Hirama, C.; Yoshida, T.; et al. An attenuated LC16m8 smallpox vaccine: Analysis of full-genome sequence and induction of immune protection. J. Virol. 2005, 79, 11873–11891. [Google Scholar] [CrossRef] [PubMed]
- Benhnia, M.R.; McCausland, M.M.; Su, H.P.; Singh, K.; Hoffmann, J.; Davies, D.H.; Felgner, P.L.; Head, S.; Sette, A.; Garboczi, D.N.; et al. Redundancy and plasticity of neutralizing antibody responses are cornerstone attributes of the human immune response to the smallpox vaccine. J. Virol. 2008, 82, 3751–3768. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.B.; Keckler, M.S.; Patel, N.; Davies, D.H.; Felgner, P.; Damon, I.K.; Karem, K.L. Humoral immunity to smallpox vaccines and monkeypox virus challenge: Proteomic assessment and clinical correlations. J. Virol. 2013, 87, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Duke-Cohan, J.S.; Wollenick, K.; Witten, E.A.; Seaman, M.S.; Baden, L.R.; Dolin, R.; Reinherz, E.L. The heterogeneity of human antibody responses to vaccinia virus revealed through use of focused protein arrays. Vaccine 2009, 27, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Moss, B. Vaccinia virus: A tool for research and vaccine development. Science 1991, 252, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Perkus, M.E.; Taylor, J.; Tartaglia, J.; Pincus, S.; Kauffman, E.B.; Tine, J.A.; Paoletti, E. Live attenuated vaccinia and other poxviruses as delivery systems: Public health issues. Ann. NY Acad. Sci. 1995, 754, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Thongcharoen, P.; Suriyanon, V.; Paris, R.M.; Khamboonruang, C.; de Souza, M.S.; Ratto-Kim, S.; Karnasuta, C.; Polonis, V.R.; Baglyos, L.; Habib, R.E.; et al. A phase 1/2 comparative vaccine trial of the safety and immunogenicity of a CRF01_AE (subtype E) candidate vaccine: ALVAC-HIV (vCP1521) prime with oligomeric gp160 (92TH023/LAI-DID) or bivalent gp120 (CM235/SF2) boost. J. Acquir. Immune Defic. Syndr. 2007, 46, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Casimiro, D.R.; Wang, F.; Schleif, W.A.; Liang, X.; Zhang, Z.Q.; Tobery, T.W.; Davies, M.E.; McDermott, A.B.; O’Connor, D.H.; Fridman, A.; et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with DNA and recombinant adenoviral vaccine vectors expressing Gag. J. Virol. 2005, 79, 15547–15555. [Google Scholar] [CrossRef] [PubMed]
- Vogel, T.U.; Reynolds, M.R.; Fuller, D.H.; Vielhuber, K.; Shipley, T.; Fuller, J.T.; Kunstman, K.J.; Sutter, G.; Marthas, M.L.; Erfle, V.; et al. Multispecific vaccine-induced mucosal cytotoxic T lymphocytes reduce acute-phase viral replication but fail in long-term control of simian immunodeficiency virus SIVmac239. J. Virol. 2003, 77, 13348–13360. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.S.; Clair, J.H.; Prokop, M.T.; Sykes, K.J.; Dubey, S.A.; Shiver, J.W.; Robertson, M.N.; Casimiro, D.R. DNA gag/adenovirus type 5 (Ad5) gag and Ad5 gag/Ad5 gag vaccines induce distinct T-cell response profiles. J. Virol. 2008, 82, 8161–8171. [Google Scholar] [CrossRef] [PubMed]
- Goonetilleke, N.; Moore, S.; Dally, L.; Winstone, N.; Cebere, I.; Mahmoud, A.; Pinheiro, S.; Gillespie, G.; Brown, D.; Loach, V.; et al. Induction of multifunctional human immunodeficiency virus type 1 (HIV-1)-specific T cells capable of proliferation in healthy subjects by using a prime-boost regimen of DNA- and modified vaccinia virus ankara-vectored vaccines expressing HIV-1 gag coupled to CD8+ T-cell epitopes. J. Virol. 2006, 80, 4717–4728. [Google Scholar] [CrossRef]
- Harari, A.; Bart, P.A.; Stohr, W.; Tapia, G.; Garcia, M.; Medjitna-Rais, E.; Burnet, S.; Cellerai, C.; Erlwein, O.; Barber, T.; et al. An HIV-1 clade C DNA prime, NYVAC boost vaccine regimen induces reliable, polyfunctional, and long-lasting T cell responses. J. Exp. Med. 2008, 205, 63–77. [Google Scholar] [PubMed]
- Tameris, M.D.; Hatherill, M.; Landry, B.S.; Scriba, T.J.; Snowden, M.A.; Lockhart, S.; Shea, J.E.; McClain, J.B.; Hussey, G.D.; Hanekom, W.A.; et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: A randomised, placebo-controlled phase 2b trial. Lancet 2013, 381, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.D.; Burton, D.R. Toward an AIDS vaccine. Science 2008, 320, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Kitabatake, M.; Inoue, S.; Yasui, F.; Yokochi, S.; Arai, M.; Morita, K.; Shida, H.; Kidokoro, M.; Murai, F.; Le, M.Q.; et al. SARS-CoV spike protein-expressing recombinant vaccinia virus efficiently induces neutralizing antibodies in rabbits pre-immunized with vaccinia virus. Vaccine 2007, 25, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Viner, K.M.; Girgis, N.; Kwak, H.; Isaacs, S.N. B5-deficient vaccinia virus as a vaccine vector for the expression of a foreign antigen in vaccinia immune animals. Virology 2007, 361, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.Y.; Funahashi, S.; Shida, H. Constructions of vaccinia virus A-type inclusion body protein, tandemly repeated mutant 7.5 kDa protein, and hemagglutinin gene promoters support high levels of expression. Arch. Virol. 1994, 138, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kidokoro, M.; Aoki, A.; Horiuchi, K.; Shida, H. Large-scale preparation of biologically active measles virus haemagglutinin expressed by attenuated vaccinia virus vectors. Microbes Infect. 2002, 4, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Funahashi, S.; Sato, T.; Shida, H. Cloning and characterization of the gene encoding the major protein of the A-type inclusion body of cowpox virus. J. Gen. Virol. 1988, 69, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kidokoro, M.; Fofana, I.B.; Ohashi, T.; Okamura, T.; Matsuo, K.; Yamamoto, N.; Shida, H. Immunogenicity of newly constructed attenuated vaccinia strain LC16m8delta that expresses SIV gag protein. Vaccine 2009, 27, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Kiepiela, P.; Ngumbela, K.; Thobakgale, C.; Ramduth, D.; Honeyborne, I.; Moodley, E.; Reddy, S.; de Pierres, C.; Mncube, Z.; Mkhwanazi, N.; et al. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 2007, 13, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Matano, T.; Kobayashi, M.; Igarashi, H.; Takeda, A.; Nakamura, H.; Kano, M.; Sugimoto, C.; Mori, K.; Iida, A.; Hirata, T.; et al. Cytotoxic T lymphocyte-based control of simian immunodeficiency virus replication in a preclinical AIDS vaccine trial. J. Exp. Med. 2004, 199, 1709–1718. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Someya, K.; Matsuo, K.; Hasegawa, A.; Yamamoto, N.; Honda, M. Recombinant vaccinia Dis expressing simian immunodeficiency virus gag and pol in mammalian cells induces efficient cellular immunity as a safe immunodeficiency virus vaccine candidate. Microbiol. Immunol. 2006, 50, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Mackett, M.; Smith, G.L.; Moss, B. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J. Virol. 1984, 49, 857–864. [Google Scholar] [PubMed]
- Hansen, S.G.; Vieville, C.; Whizin, N.; Coyne-Johnson, L.; Siess, D.C.; Drummond, D.D.; Legasse, A.W.; Axthelm, M.K.; Oswald, K.; Trubey, C.M.; et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 2009, 15, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Ford, J.C.; Lewis, M.S.; Ventura, A.B.; Hughes, C.M.; Coyne-Johnson, L.; Whizin, N.; Oswald, K.; Shoemaker, R.; Swanson, T.; et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 2011, 473, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Jing, C.; Isshiki, M.; Matsuo, K.; Kidokoro, M.; Takamura, S.; Zhang, X.; Ohashi, T.; Shida, H. Immunogenicity and safety of the vaccinia virus LC16m8delta vector expressing SIV Gag under a strong or moderate promoter in a recombinant BCG prime-recombinant vaccinia virus boost protocol. Vaccine 2013, 31, 3549–3557. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Elizaga, M.L.; Seaton, K.; Tomaras, G.D.; Montefiori, D.C.; Sato, A.; Hural, J.; Derosa, S.C.; Kalams, S.A.; McElrath, M.J.; et al. Specificity and 6-month durability of immune responses induced by DNA and recombinant modified vaccinia Ankara vaccines expressing HIV-1 virus-like particles. J. Infect. Dis. 2014, 210, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sobue, T.; Isshiki, M.; Makino, S.; Inoue, M.; Kato, K.; Shioda, T.; Ohashi, T.; Sato, H.; Komano, J.; et al. Elicitation of both anti HIV-1 Env humoral and cellular immunities by replicating vaccinia prime sendai virus boost regimen and boosting by CD40Lm. PLoS One 2012, 7, e51633. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kidokoro, M.; Shida, H. Vaccinia Virus LC16m8∆ as a Vaccine Vector for Clinical Applications. Vaccines 2014, 2, 755-771. https://doi.org/10.3390/vaccines2040755
Kidokoro M, Shida H. Vaccinia Virus LC16m8∆ as a Vaccine Vector for Clinical Applications. Vaccines. 2014; 2(4):755-771. https://doi.org/10.3390/vaccines2040755
Chicago/Turabian StyleKidokoro, Minoru, and Hisatoshi Shida. 2014. "Vaccinia Virus LC16m8∆ as a Vaccine Vector for Clinical Applications" Vaccines 2, no. 4: 755-771. https://doi.org/10.3390/vaccines2040755
APA StyleKidokoro, M., & Shida, H. (2014). Vaccinia Virus LC16m8∆ as a Vaccine Vector for Clinical Applications. Vaccines, 2(4), 755-771. https://doi.org/10.3390/vaccines2040755