
Assaying the Potency of Influenza Vaccines

Abstract
:1. Introduction

2. Measurement of Influenza Antigen Content by Single Radial Immunodiffusion

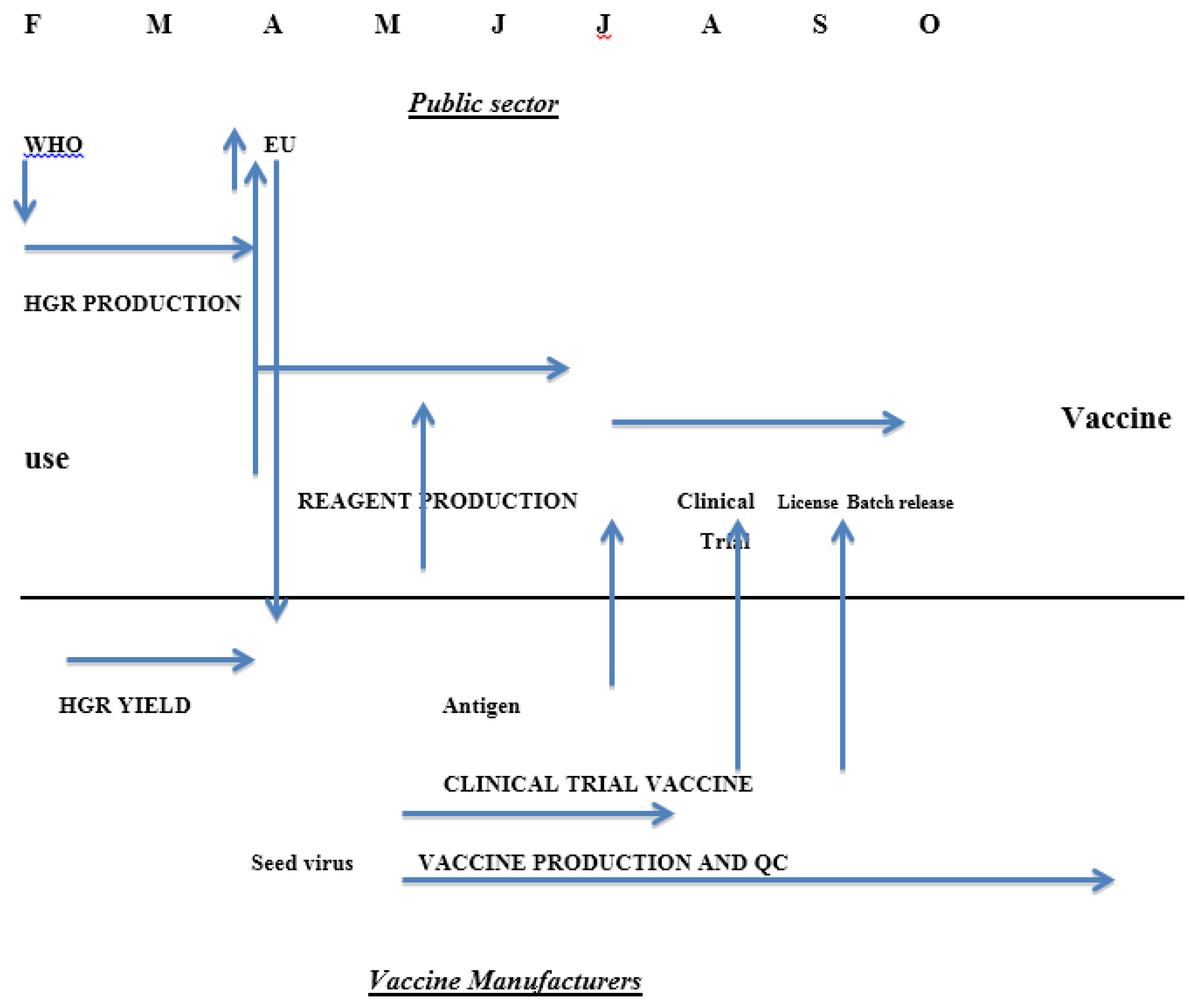
3. Influenza Vaccine Timetable
4. Preparation and Calibration of Reagents for SRD
5. Features of SRD
- It measures the antigen thought to be most relevant to protection, specifically that in the native conformation.
- It has been successfully used for decades so that there is a great deal of experience in its use and the issues that may arise as it is applied to an ever changing and complex product. Novel methods will almost certainly raise novel issues
- It is of known acceptable accuracy and robustness as demonstrated by a number of unpublished proficiency testing studies by organisations such as EDQM (the European Directive for the Quality of Medicines which incorporates the European Pharmacopoeia) Agreement between competent laboratories assaying the same material is of the order of 5%
- It is by its nature specific for the type and subtype of virus to be assayed, for example H1N1 or H3N2 or B. There may be issues where more than one strain of a particular subtype is included as where B strains of the Yamagata and Victoria lineage are both present and there is extensive cross reaction between them but it can be used to assay the potency of the final finished product which is almost always multivalent.
- It is simple in principle and in application so that sophisticated equipment is not needed and the assay is both flexible and practical. It could be applied to developing country manufacture.
- The production of calibrated reagents takes time; typically the strain selection process in the Northern Hemisphere takes place in February and the final calibrated reagents are released by late June to early July (Table 1) approximately four months later. Process validation by the manufacturer therefore cannot usually be done with fully calibrated reagents. This has been put forward as a major obstacle to the timely production of vaccines in response to the 2009 H1N1 pandemic. Analysis of the events and time course of the global response suggest that while the reagents were not immediately available, other factors were at least equally significant, including the timing of the difficult decision to declare the pandemic so that manufacturers could commit to changing production to the new pandemic strains.
- The reagents and therefore the assays vary from year to year and are not in the full control of either the manufacturers (who depend on the ERLs for calibrated reagents) or the ERLs (who depend on the manufacturers to supply the antigen that will be used to prepare the reagents). This leaves both in vulnerable positions.
- Technically, the dynamic range of the assay is limited, covering 7.5 to 30 micrograms per dose. Where low dose preparations are proposed in pandemics to spare antigen, this has posed a problem. Similarly adjuvants can interfere with the assay although this is not necessarily the case; for example SRD fails to work with alumn-based adjuvants although it will give reliable figures with some squalene-based adjuvants.
- There is a philosophical issue relating to the metrological credibility of the process. All masses expressed in kilograms or its subdivisions are traceable to the standard held in Paris at BIPM, and the process is very sophisticated if done rigorously, including confidence intervals on the accuracy or uncertainty of the measurement. Micrograms of influenza antigen are not traceable in this way and refer only to the mass present in a particular conformation. While this may seem sophistry, it means that the accuracy of the calibration is likely to change from year to year. In particular if a new reagent is required because old stocks are exhausted it will not be identical to the old stock. Usually such differences are expected to fall within the experimental error of the assay or the range of the specifications set; however it is advisable to use one set of reagents throughout one season to avoid discontinuities in the assay results.
- The assay is designed and has been used to assay vaccines derived from virus grown in embryonated hens’ eggs or in cell culture [12]. Novel vaccines have been developed, including antigens expressed in plants or insect cells or which include immunostimulatory molecules where SRD is not satisfactory. SRD has never been appropriate for live attenuated influenza vaccines where assays of infectivity are applied.

{kind=link}
{kind=link}
| Date of Selection | Type | Production Strain | Date of Release of Calibrated Reagent |
|---|---|---|---|
| 24 Feb 2006 | H3N2 | IVR-142 | 23 June 2006 |
| NYMX 161B | 06 July 2006 | ||
| 16 Feb 2007 | H1N1 | IVR-145 | 30 June 2007 |
| 20 Feb 2008 | H1N1 | IVR-148 | 8 June 2008 |
| H3N2 | NYMCK 175 | 13 June 2008 | |
| 16 Feb 2009 | B | B Bris/60 | 11 June 2009 |
| 1 March 2010 | H3N2 | NYMCX 183 | 26 May 2010 |
| NYMCX 187 | 26 May 2010 | ||
| B | NYMC BX35 | 28 May 2010 | |
| 5 April 2011 | No changes | ||
| 8 Feb 2012 | B | NYMC BX39 | 20 June 2012 |
| H3N2 | IVR-165 | 20 June 2012 |
6. Alternative Assays to SRD
- It would measure a biologically relevant parameter to give meaningful measurements of potency, stability and clinical effect.
- It would be accurate and precise although the degree of accuracy and precision required is not specified.
- It would be rapid to allow real time monitoring of processes.
- It would be specific at least to the level of virus type and subtype.
- It should have a wide dynamic range so as to measure low dosage forms and it should work on different vaccine types such as might be used in pandemics.
- It would either need
- (a)
- no specific reagents
- (b)
- reagents that do not require changing every time there was a strain change or
- (c)
- reagents that are specific, but easier to make, for instance if they could be made in small quantities not requiring production at manufacturing scale.
- It should be readily applicable in developing countries.
- HPLC [24]. HPLC is a well-established technique for separating and quantitating proteins. In the form applied it measures denatured protein rather than the biologically active form but for a well-established process the total protein and the amount of the active form are likely to be fairly closely related. Thus, in the early stages of process evaluation, where yields are being examined, the total protein is likely to be a very useful parameter. HPLC is unlikely to be fully acceptable at the point of release or in stability studies because it does not measure protein in a biologically relevant conformation. It can be accurate and precise, and rapid enough to allow real time monitoring of processes although the dynamic range is not great. There may be an issue in separating the haemagglutinins from different vaccine strains so that applying it to trivalent or tetravalent bulks will not be straightforward. HPLC is a widely used technique throughout the world. Finally accurate quantitation and identification is likely to require a homologous calibrated reference material. HPLC is therefore a useful adjunct to SRD but not a replacement; it is not capable of demonstrating stability except where the instability is due to protein degradation. It is possible that a preparation could be treated to remove the denatured form of the HA protein and the remainder quantitated by HPLC, which would make it stability indicating
- Mass spectroscopy [21]. By including a formally calibrated isotopically labelled peptide of a sequence derived from the haemagglutinin and enzymatically digesting a mixture of the peptide and the haemagglutinin the relative size of the peptide and HA derived peaks can give a very accurate figure for the amount of HA present. In this form it does not measure conformationally correct protein content but could be used to assay the protein content of the primary liquid standard (PLS) used in calibrating reagents. Currently it is unlikely to be an acceptable test for the final product but it could be a useful addition to the methods used by the ERLs in calibrating the reagents.
- A modification of Mass spectroscopy involves capturing the active form of the HA by specific antibodies linked to magnetic beads and measuring the amount caught by mass spectroscopy. As specific antibodies are used the method can be used on formulated final vaccine and measure a biologically relevant protein. The results would be expressed in absolute terms without the need for a homologous influenza virus reference although it does require specific antibodies. The equipment is also potentially not yet wide spread.
- ELISA. Various ELISA formats have been proposed:
- (a)
- The first [25] involves antibodies recognising non-conformational epitopes that will measure total protein but not the biologically active form.
- (b)
- An approach to measuring the biologically active form is to develop a panel of monoclonal antibodies with neutralising or haemagglutination inhibiting activity for a particular strain. The assay therefore measures a biologically relevant property. As described by Bodle et al. [26] it is also very accurate, precise and rapid with a far wider dynamic range than SRD. It is specific by its very nature, can in principle be applied to a multivalent product and the general technique is very widely used across the globe and therefore readily applicable in many settings. In addition when a new strain emerges by mutation, as is the usual seasonal pattern, it will not have mutated all epitopes. Some members of the antibody panel should still react and can be used to measure the amount of haemagglutinin in the new strain. There is therefore no need to generate a new serum immediately. It also seems plausible that the new virus could be measured by reference to an old antigen reagent if the monoclonal antibody reacts equally well with both, so no new reagents would be needed for that season at all although the panel would have to be kept up to date and monitored closely. However Bodle et al. who described the approach [26] report differences in avidity of monoclonal antibodies for haemagglutinins from different strains with which they nonetheless react. This is very likely to affect the potencies measured, so that it is probable that a homologous reference reagent will still be required. It is also not clear how much would be required.
- (c)
- A version of this approach is being developed by InDvr [27] in which panels of antibodies are included on a chip, which can be used to assay all haemagglutinins present in the vaccine at once. Another version of the same approach has also been described [28]. This requires further evaluation from the point of view of the amount of reference antigen required and its suitability for use over multiple seasons. If only a small amount of antigen is required a batch could be made in house by the ERLs so that development of the process at the manufacturing site will not be required and time will be saved. The reagent would still require calibration by a collaborative study among the ERLs however.
- (d)
- A related alternative to the use of a panel of antibodies directed at the variable globular head of the HA is the use of conformation dependent cross-reactive antibodies directed at the stalk region [29,30]. These are being investigated in the same way [31]. An ELISA based on attachment of the native conformation to synthetic receptors has been described which would not require specific reagents provided the avidity was constant between strains [32]
- Antibody dependent Surface Plasmon Resonance [33]. When an incident beam of p-polarized light strikes an electrically conducting gold layer at the interface of a glass sensor with high RI (Refractive Index) and a medium with low RI at a given angle, excitation of surface plasmons takes place and the intensity of the reflected light is reduced. A slight change at the interface, for instance binding of an antibody or binding of an antigen to an immobilised antibody leads to a change in SPR signal. Binding can therefore be measured precisely on very small quantities of material in a strain or type specific manner. The method would require both antibodies and reference antigen, but use of reagents may be small so that small batches produced at the ERLs are a possibility. The same considerations apply as to the ELISA approaches outlined above.
- A modification of one of the ELISA methods uses SPR to detect binding rather than ELISA by coating the chip with synthetic glycans linked to sialic acid imitating the influenza receptor. This has the advantage of eliminating the need for an antibody reagent and using the same glycan reagent year on year. It requires a reference antigen but possibly not in large amounts as above. It has been successfully applied to a number of strains [34] It is rapid, accurate, relates to SRD and assays only the active form of the HA. The fact that it uses binding to the receptor that all influenza viruses use means that it cannot be used to assay multivalent products as all will bind. It is also possible that differences in the avidity of binding could affect the use of the method. The equipment is currently sophisticated and expensive.
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Shortridge, K.F. Poultry and the influenza H5N1 outbreak in Hong Kong, 1997: Abridged chronology and virus isolation. Vaccine 1999, 17, S26–S29. [Google Scholar]
- Chan, P.K. Outbreak of avian influenza A(H5N1) virus infection in Hong Kong in 1997. Clin. Infect. Dis. 2002, 34, S58–S64. [Google Scholar] [PubMed]
- Centers for Disease Control. The 2009 H1N1 Pandemic: Summary Highlights, April 2009–April 2010. Available online: http://www.cdc.gov/h1n1flu/cdcresponse.htm (accessed on 9 September 2014).
- Plennevaux, E.; Sheldon, E.; Blatter, M.; Reeves-Hoché, M.-K.; Denis, M. Immune response after a single vaccination against 2009 influenza A H1N1 in USA: A preliminary report of two randomised controlled phase 2. Lancet 2010, 375, 41–48. [Google Scholar] [PubMed]
- Nicholson, K.G.; Colegate, A.E.; Podda, A.; Stephenson, I.; Wood, J.; Ypma, E.; Zambon, M.C. Safety and antigenicity of non-adjuvanted and MF59-adjuvanted influenza A/Duck/Singapore/97 (H5N3) vaccine: A randomised trial of two potential vaccines against H5N1 influenza. Lancet 2001, 357, 1937–1943. [Google Scholar] [PubMed]
- Nolan, T.M.; Richmond, P.C.; Skeljo, M.V.; Pearce, G.; Hartel, G.; Formica, N.T.; Höschler, K.; Bennet, J.; Ryan, D.; Papanaoum, K.; et al. Phase I and II randomised trials of the safety and immunogenicity of a prototype adjuvanted inactivated split-virus influenza A (H5N1) vaccine in healthy adults. Vaccine 2008, 26, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Richmond, P.C.; Formica, N.T.; Höschler, K.; Skeljo, M.V.; Stoney, T.; McVernon, J.; Hartel, G.; Sawlwin, D.C.; Bennet, J.; et al. Safety and immunogenicity of a prototype adjuvanted inactivated split-virus influenza A (H5N1) vaccine in infants and children. Vaccine 2008, 26, 6383–6391. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Regnerb, S.; Vaman, T.; Devaster, J.-M.; Rombo, L. Safety and immunogenicity of an AS03-adjuvanted A (H1N1) pmd09 vaccine administered simultaneously or sequentially with a seasonal trivalent vaccine in adults 61 years or older: Data from two multicentre randomised trials. Vaccine 2012, 30, 6483–6491. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Dunleavy, U.; Newman, R.W.; Riley, A.M.; Robertson, J.S.; Minor, P.D. The influence of the host cell on standardisation of influenza vaccine potency. Dev. Biol. Stand. 1999, 98, 183–188. [Google Scholar] [PubMed]
- Keitel, W.; Groth, N.; Lattanzi, M.; Praus, M.; Hilbert, A.K.; Borkowski, A.; Tsai, T.F. Dose ranging of adjuvant and antigen in a cell culture H5N1 influenza vaccine: Safety and immunogenicity of a phase 1/2 trial. Vaccine 2010, 28, 840–848. [Google Scholar] [CrossRef] [PubMed]
- Keitel, W.A.; Dekker, C.L.; Mink, C.A.; Campbell, J.D.; Edwards, K.M.; Patel, S.M.; Ho, D.Y.; Talbot, H.K.; Guo, K.; Noah, D.L.; et al. Safety and immunogenicity of inactivated, Vero cell culture-derived whole virus influenza A/H5N1 vaccine given alone or with aluminum hydroxide adjuvant in healthy adults. Vaccine 2009, 27, 6642–6648. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.L.; Sajkov, D.; Woodman, R.J.; Honda-Okubo, Y.; Cox, M.M.J.; Heinzel, S.; Petrovsky, N. Randomized clinical trial of immunogenicity and safety of a recombinant H1N1/2009 pandemic influenza vaccine containing AdvaxTM polysaccharide adjuvant. Vaccine 2012, 30, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Kelly, H.; Steffens, I. Complexities in assessing the effectiveness of inactivated influenza vaccines. Available online: http://www.eurosurveillance.org/ViewArticle.aspx?ArticleId=20403 (accessed on 30 January 2015).
- Nicoll, A.; Sprenger, M. Low effectiveness undermines promotion of seasonal influenza vaccine. Lancet Inf. Dis. 2013, 13, 7–9. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Explanatory Note on the Withdrawal of the Note for Guidance on Harmonisation of Requirements for Influenza Vaccines and of the Core SmPC/PL for Inactivated Seasonal Influenza Vaccines. Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/08/WC500147010.pdf (accessed on 30 January 2015).
- Stephenson, I.; Das, R.G.; Wood, J.M.; Katz, J.M. Comparison of neutralising antibody assays for detection of antibody to influenza A/H3N2 viruses: an international collaborative study. Vaccine 2007, 25, 4056–4063. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; Schild, G.C.; Seagroatt, V. An improved single radial immunodiffusion technique for the assay of influenza haemagglutinin antigen: application for potency determinations of inactivated whole virus and subunit vaccines. J. Biol. Stand. 1977, 5, 237–247. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Expert committee on Biological Standardization. Thirteenth Report; World Health Organization: Geneva, Switzerland, 1979; Annex 3 (WHO Technical Report Series No 638). [Google Scholar]
- Wood, J.M.; Mumford, J.A.; Dunleavy, U.; Seagroatt, V.; Newman, R.W.; Thornton, D.; Schild, G.C. Single radial immunodiffusion potency tests for inactivated equine influenza vaccines. In Equine Infection Disease V. Proceedings of the Fifth International Conference; Powell, D.G., Ed.; The University Press of Kentucky: KY, USA, 1988; pp. 74–79. [Google Scholar]
- WHO: Generic Protocol for the Calibration of Seasonal and Pandemic Influenza Antigen Working Reagents by WHO Essential Regulatory Laboratories World Health Organization Technical Report Series 979 Annex 5 2012. Available online: http://www.who.int/biologicals/areas/vaccines/TRS_979_Annex_5.pdf?ua=1 (accessed on 8 September 2014).
- Williams, T.L.; Luna, L.; Guo, Z.; Cox, N.J.; Pirkle, N.J.; Donis, R.O.; Barr, J.R. Quantification of influenza virus hemagglutinins in complex mixtures using isotope dilution tandem mass spectrometry. Vaccine 2008, 20, 2510–2520. [Google Scholar] [CrossRef]
- Harvey, R.; Hamill, M.; Robertson, J.R.; Minor, P.D.; Vodeiko, G.M.; Weir, J.P. Application of deglycosylation to SDS PAGE analysis improves calibration of influenza antigen standards. Biologicals 2012, 40, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, O.; Minor, P.; Dunleavy, U.; Division of Virology, National Institute of Biological Standards and Control, South Mimms, Herts , UK. Unpublished data. 2014.
- Lorbetskie, B.; Wang, J.; Gravel, C.; Allen, C.; Walsh, M.; Rinfret, A.; Li, X.; Girard, M. Optimization and qualification of a quantitative reversed phase HPLC method for hemagglutinin in influenza preparations and its comparative evaluation with biochemical assays. Vaccine 2011, 29, 3377–3389. [Google Scholar] [CrossRef] [PubMed]
- Chuna, S.; Li, C.; Domselaar, G.V.; Wang, J.; Farnsworth, A.; Cuib, X.; Rode, H.; Cyr, T.D.; He, R.; Li, X. Universal antibodies and their applications to the quantitative determination of virtually all subtypes of the influenza A viral hemagglutinins. Vaccine 2008, 26, 6068–6076. [Google Scholar] [CrossRef] [PubMed]
- Bodle, J.; Verity, E.E.; Ong, C.; Vandenberg, K.; Shaw, R.; Barr, I.G.; Rockman, S. Development of an enzyme-linked immunoassay for the quantitation of influenza haemagglutinin: An alternative method to single radial immunodiffusion. Influenza Other Respir. Viruses 2013, 7, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Indevr Website. Available online: http://indevr.com/2014/07/press-release-2-9-m-award-advance-titer-chip/ (accessed on 9 September 2014).
- Schmeisser, F.; Vasudevan, A.; Soto, S.; Kumar, A.; Williams, O.; Weir, J.P. A monoclonal antibody-based immunoassay for measuring the potency of 2009 pandemic influenza H1N1 vaccines. Influenza Other Respir. Viruses 2014, 8, 587–595. [Google Scholar] [CrossRef]
- Ekiert, D.C.; Friesen, R.H.E.; Bhabha, G.; Kwaks, T.; Jongeneelen, M.; Yu, W.; Ophorst, C.; Cox, F.; Korse, H.J.; Brandenburg, B.; et al. A Highly Conserved Neutralizing Epitope on Group 2 Influenza A Viruses. Science 2011, 333, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Hufton, S.E.; Risley, P.; Ball, C.R.; Major, D.; Engelhardt, O.G. The breadth of cross sub-type neutralisation activity of a single domain antibody to influenza hemagglutinin can be increased by antibody valency. PLOS ONE 2014, 9, e103294. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.; Hufton, S.; Engelhardt, O.; Division of Virology, National Institute of Biological Standards and Control, South Mimms, Herts, UK. Personal Communication, 2014.
- Hashem, A.; Gravel, C.; Farnsworth, A.; Zou, W.; Lemieux, M.; Xu, K.; Li, C.; Wang, J.; Goneau, M.-F.; Merziotis, M.; et al. A novel synthetic receptor-based immunoassay for influenza vaccine quantification. PLOS ONE 2013, 8, e55428. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, C.E.; Abbas, S.; Bennemo, M.; Larsson, A.; Hämäläinen, M.D.; Frostell-Karlsson, Å. A novel assay for influenza virus quantification using surface plasmon resonance. Vaccine 2010, 28, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; King, L.R.; Manischewitz, J.; Coyle, E.; Golding, H. Novel rapid antibody-independent receptor binding SPR-based high-throughput assay for measurement of influenza vaccine potency. Vaccine 2014, 32, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Larkin, C.; Verma, S.; Joshi, M.B.; Fontana, J.; Steven, A.C.; King, L.R.; Manischewitz, J.; McCormick, W.; Gupta, R.K.; et al. Recombinant HA1 produced in E.coli forms functional oligomers and generates strain-specific SRID potency antibodies for pandemic influenza vaccines. Vaccine 2011, 29, 5657–5665. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minor, P.D. Assaying the Potency of Influenza Vaccines. Vaccines 2015, 3, 90-104. https://doi.org/10.3390/vaccines3010090
Minor PD. Assaying the Potency of Influenza Vaccines. Vaccines. 2015; 3(1):90-104. https://doi.org/10.3390/vaccines3010090
Chicago/Turabian StyleMinor, Philip D. 2015. "Assaying the Potency of Influenza Vaccines" Vaccines 3, no. 1: 90-104. https://doi.org/10.3390/vaccines3010090
APA StyleMinor, P. D. (2015). Assaying the Potency of Influenza Vaccines. Vaccines, 3(1), 90-104. https://doi.org/10.3390/vaccines3010090