
Whole Tumor Antigen Vaccines: Where Are We?

Abstract
:1. Introduction
2. Types of Whole Tumor Antigens
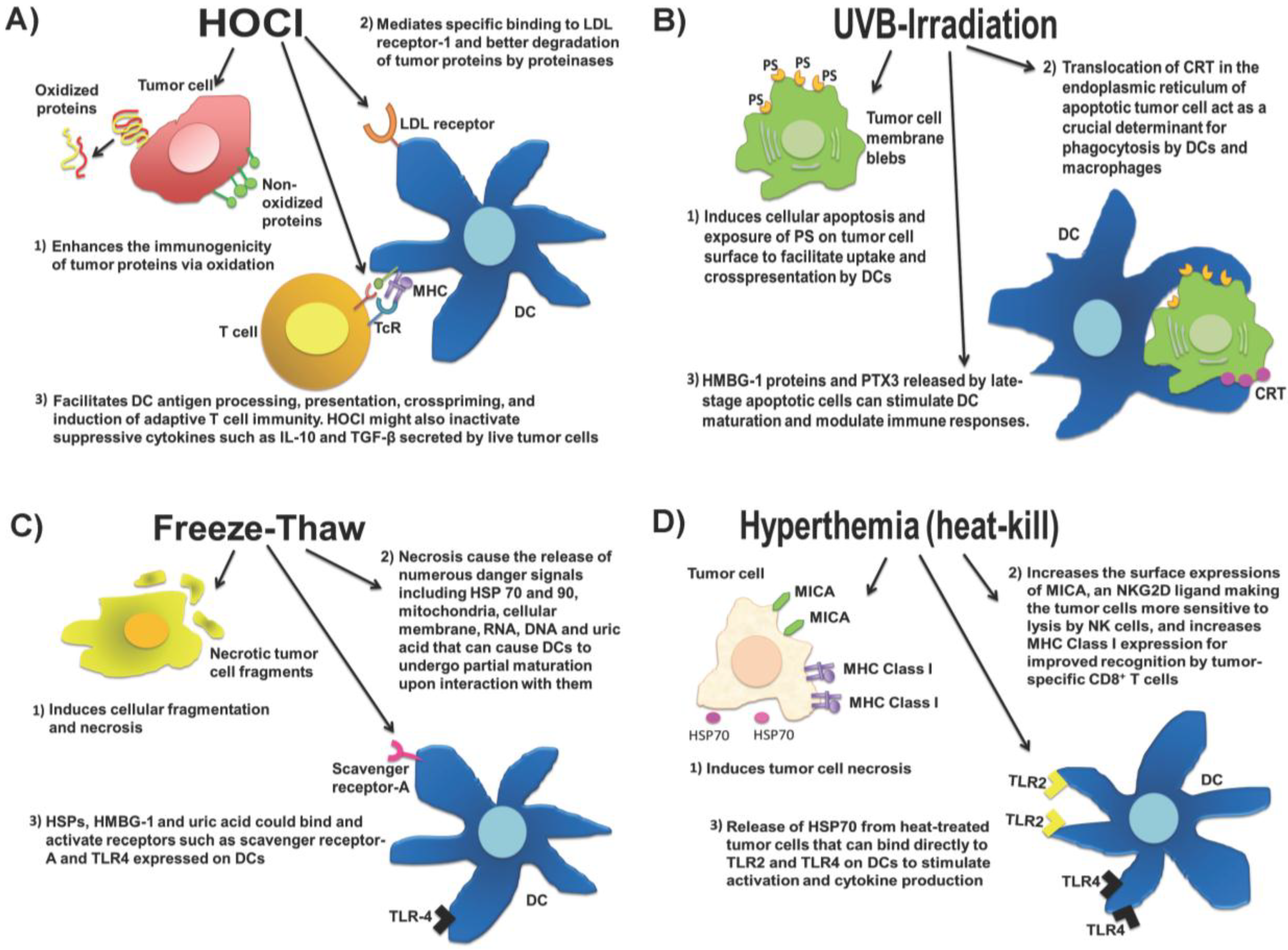
2.1. Whole Tumor Cell Lysates
2.2. Approaches to Preparing Whole Tumor Cell Lysate Vaccines

2.3. Exosomes Derived from Tumor Cells
2.4. Tumor Cell-Derived Messenger Ribonucleic Acid
2.5. Personalized Mutanome-Based Vaccines
2.6. Tumor Stroma-Associated Antigens

{kind=link}
| Source of Whole Tumor Antigen | Method of Whole Tumor Antigen Preparation | Treatment Regimen | Outcome of Study | Reference |
|---|---|---|---|---|
| Whole mouse glioblastoma cell lysate | Freeze-thawed whole tumor lysate administered alone or in combination with type B CpG (ODN 1826) | Glioblastoma-bearing mice were immunized subcutaneously with the tumor lysate-CpG vaccine |
| [88] |
| Whole fibrosarcomas or mammary carcinoma cell lysate | Freeze-thawed whole tumor cell lysate was pulsed onto mouse bone marrow-derived DCs | Fibrosarcomas or mammary carcinoma-bearing mice were vaccinated subcutaneously with the whole tumor lysate-DC vaccine |
| [89] |
| Whole ovarian carcinoma cell lysate | Whole tumor cells were oxidized with HOCl and then frozen and thawed, and pulsed onto mouse bone marrow-derived DCs (i.e., mouse OCDC vaccine) | Ovarian carcinoma-bearing mice were treated intradermally with OCDC vaccine |
| [41] |
| Whole ovarian tumor cells expressing HPV E6 and E7 | UVB-irradiated whole cells pulsed onto mouse bone marrow-derived DCs | Ovarian carcinoma-bearing mice were treated intraperitoneally and subcutaneously with the UVB-irradiated tumor cell-DC vaccine |
| [90] |
| Total RNA of whole ovarian tumor cells expressing HPV E6 and E7 | Total tumor RNA was electroporated into mouse bone marrow-derived DCs | Ovarian cancer-bearing mice were vaccinated with DCs electroporated with total tumor mRNA via the intraperitoneal and subcutaneous routes |
| [90] |
| Mutanome peptides of mouse melanoma cells | Mutated epitopes of tyrosinase-related protein 2 (TRP-2) were identified via sequencing the protein-coding genome of the B16.F10 mouse melanoma cells | Melanome-bearing mice were immunized subcutaneously with the long mutanome peptides and Poly(I:C) as adjuvant |
| [73] |
| Source of Whole Tumor Antigen | Method of Whole Tumor Antigen Preparation | Treatment Regimen | Outcome of Study | Reference |
|---|---|---|---|---|
| Autologous whole ovarian tumor cells | Whole tumor cells were modified with dinitrophenyl (DNP) and UVB-irradiated | Phase I trial in stage III ovarian cancer where patients were immunized intradermally with the vaccine |
| [91] |
| Autologous whole melanoma cells | Whole tumor cells were UVB-irradiated | Phase III/IV metastatic melanoma trial where patients were vaccinated intradermally with UVB-irradiated autologous whole cells and Bacillus Calmette-Guérin (BCG) as adjuvant |
| [92] |
| Allogeneic melanoma cell lysate derived from 3 different melanoma lines (TRIMEL) | TRIMEL was subjected to freeze-thawed cycles and pulsed on autologous monocyte-derived DCs (i.e., TRMEL-DC vaccine) | Phase I trial where stage IV and III melanoma patients were vaccinated intradermally with TRIMEL-DC vaccine and aluminum hydroxide/keyhole limpet haemocyanin (KLH) as an adjuvant |
| [93] |
| Allogeneic whole prostate tumor cells | Prostate tumor cell lines LNCaP and PC-3 were genetically modified to secrete GM-CSF (i.e., GVAX vaccine) and UVB-irradiated | Phase I/II studies in metastatic hormone-refractory prostate cancer (HRPC) whereby patients were immunized intradermally with GVAX vaccine |
| [5,6,7] |
| Autologous whole mesothelioma tumor cells | Autologous tumor cell lysate was administered with recombinant GM-CSF | Phase I trial of mesothelioma patients who were vaccinated subcutaneously with whole tumor lysate vaccine and recombinant GM-CSF |
| [16] |
| Autologous whole ovarian tumor cell lysate | Whole tumor cells were oxidized with HOCl and then frozen and thawed, and pulsed on autologous monocyte-derived DCs (i.e., OCDC vaccine) | Phase I trial of recurrent ovarian cancer whereby patients were vaccinated intranodally with OCDC vaccine |
| [41] |
| Autologous Ascites-derived exosomes (Aex) from colorectal tumor cells | Aex administered alone or in combination with recombinant GM-CSF | Phase I colorectal cancer whereby advanced stage patients are vaccinated subcutaneously with Aex ± recombinant GM-CSF. |
| [59] |
| Total mRNA derived from renal tumor cells | Total tumor mRNA was used to transfect autologous monocyte-derived DCs | Phase I trial of metastatic renal cell carcinoma where patients were vaccinated intravenously with the tumor mRNA-expressing DCs |
| [64] |
| Total mRNA derived from melanoma cells | Total tumor mRNA was used to transfect autologous monocyte-derived DCs | Phase I/II trial of advanced melanoma where patients were vaccinated intradermally or intranodally with the tumor mRNA-expressing DCs. No serious adverse effects were observed. |
| [65] |
| Total mRNA derived from glioblastoma cancer stem cells | Total mRNA from cancer stem cells was electroporated into autologous monocyte-derived DCs | Phase I trial where patients were treated intradermally with autologous monocyte-derived DCs that were electroporated with the total mRNA of glioblastoma cancer stem cells |
| [66] |
| Mutanome peptides derived from E6 and E7 of HPV | Synthetic long peptides administered in incomplete Freund’s adjuvant | Phase I study where patients with vulvar intraepithelial neoplasia were treated subcutaneously with the E6 and E7 mutanome peptides |
| [75] |
| Mutanome peptides derived from von Hippel-Lindau (VHL) gene mutations in renal cell carcinoma | Synthetic neo-peptides derived from Hippel-Lindau (VHL) gene mutations in RCC | Pilot clinical trial whereby patients with advanced RCC and mutated VHL genes were vaccinated subcutaneously with the relevant VHL peptide mixed with Montanide |
| [79] |
3. Factors Influencing the Immune Responses to Whole Tumor Antigens
3.1. Immunodominant Antigens versus Mutated Neo-Antigens for Vaccination
3.2. Immune Status of Cancer Patients
4. In Vivo Activation of Dendritic Cells (DCs)
5. In Vivo Tumor Antigen Administration: Intradermal, Intranodal and Intratumoral
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chiang, C.L.-L.; Benencia, F.; Coukos, G. Whole tumor antigen vaccines. Semin. Immunol. 2010, 22, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Neller, M.A.; López, J.A.; Schmidt, C.W. Antigens for cancer immunotherapy. Semin. Immunol. 2008, 20, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, E.C.; Essner, R.; Foshag, L.J.; Ollila, D.W.; Gammon, G.; O’Day, S.J.; Boasberg, P.D.; Stern, S.L.; Ye, X.; Morton, D.L. Prolonged survival after complete resection of disseminated melanoma and active immunotherapy with a therapeutic cancer vaccine. J. Clin. Oncol. 2002, 20, 4549–4554. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Eager, R.; John, N. GM-CSF Gene-transduced tumor vaccines. Mol. Ther. 2005, 12, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Simons, J.; Carducci, M.; Mikhak, B.; Lim, M.; Biedrzycki, B.; Borellini, F.; Clift, S.M.; Hege, K.M.; Ando, D.G.; Piantadosi, S.; et al. Phase I/II trial of an allogeneic cellular immunotherapy in hormone-naïve prostate cancer. Clin. Cancer Res. 2006, 12, 3394–3401. [Google Scholar] [CrossRef] [PubMed]
- Small, E.; Sacks, N.; Nemunaitis, J.; Urba, W.; Dula, E.; Centeno, A.; Nelson, W.G.; Ando, D.; Howard, C.; Borellini, F.; et al. Granulocyte macrophage colony-stimulating factor—Secreting allogeneic cellular immunotherapy for hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 3883–3891. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Macarthur, R.B.; O’Connor, J.; Shelton, G.; Judge, T.; Balog, J.; Pfaff, C.; Bagiella, E.; Heitjan, D.; Fine, R.; et al. Phase I trial of docetaxel with estramustine in androgen-independent prostate cancer. J. Clin. Oncol. 1999, 17, 958–967. [Google Scholar] [PubMed]
- Savarese, D.M.; Halabi, S.; Hars, V.; Akerley, W.L.; Taplin, M.-E.; Godley, P.A.; Hussain, A.; Small, E.J.; Vogelzang, N.J. Phase II study of docetaxel, estramustine, and low-dose hydrocortisone in men with hormone-refractory prostate cancer: A final report of CALGB 9780. J. Clin. Oncol. 2001, 19, 2509–2516. [Google Scholar] [PubMed]
- Small, E.; Tchekmedyian, N.; Rini, B.; Fong, L.; Lowy, I.; Allison, J. A pilot trial of CTLA-4 blockade with human anti-CTLA-4 in patients with hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 1810–1815. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J. Granulocyte-macrophage colony-stimulating factor gene-transfected autologous tumor cell vaccine: Focus (correction to focus) on non-small-cell lung cancer. Clin. Lung Cancer 2003, 5, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Jahan, T.; Ross, H.; Sterman, D.; Richards, D.; Fox, B.; Jablons, D.; Aimi, J.; Lin, A.; Hege, K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006, 13, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Tani, K.; Azuma, M.; Nakazaki, Y.; Oyaizu, N.; Hase, H.; Ohata, J.; Takahashi, K.; OiwaMonna, M.; Hanazawa, K.; Wakumoto, Y. Phase I study of autologous tumor vaccines transduced with the GM-CSF gene in four patients with stage IV renal cell cancer in Japan: Clinical and immunological findings. Mol. Ther. 2004, 10, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Borrello, I.M.; Levitsky, H.I.; Stock, W.; Sher, D.; Qin, L.; DeAngelo, D.J.; Alyea, E.P.; Stone, R.M.; Damon, L.E.; Linker, C.A.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting cellular immunotherapy in combination with autologous stem cell transplantation (ASCT) as postremission therapy for acute myeloid leukemia (AML). Blood 2009, 114, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Targets of protective tumor immunity. Cancer Vaccines. Ann. N. Y. Acad. Sci. 2009, 1174, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.; Creaney, J.; Broomfield, S.; van Bruggen, I.; Robinson, B. Recombinant GM-CSF plus autologous tumor cells as a vaccine for patients with mesothelioma. Lung Cancer 2006, 52, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.B.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Larsson, M.; Fonteneau, J.F.; Bhardwaj, N. Dendritic cells resurrect antigens from dead cells. Trends Immunol. 2001, 22, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Jenne, L.; Arrighi, J.-F.; Jonuleit, H.; Saurat, J.-H.; Hauser, C. Dendritic cells containing apoptotic melanoma cells prime human CD8+ T cells for efficient tumor cell lysis. Cancer Res. 2000, 60, 4446–4452. [Google Scholar] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.W.; Jiang, W.; Reich, C.F., III; Pisetsky, D.S. The extracellular release of HMGB1 during apoptotic cell death. Am. J. Physiol. Cell Physiol. 2006, 291, C1318–C1325. [Google Scholar] [CrossRef] [PubMed]
- Rovere, P.; Peri, G.; Fazzini, F.; Bottazzi, B.; Doni, A.; Bondanza, A.; Zimmermann, V.S.; Garlanda, C.; Fascio, U.; Sabbadini, M.G.; et al. The long pentraxin PTX3 binds to apoptotic cells and regulates their clearance by antigen-presenting dendritic cells. Blood 2000, 96, 4300–4306. [Google Scholar] [PubMed]
- Baruah, P.; Propato, A.; Dumitriu, I.E.; Rovere-Querini, P.; Russo, V.; Fontana, R.; Accapezzato, D.; Peri, G.; Mantovani, A.; Barnaba, V.; et al. The pattern recognition receptor PTX3 is recruited at the synapse between dying and dendritic cells, and edits the cross-presentation of self, viral, and tumor antigens. Blood 2006, 107, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: Exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-Y.; Arnouk, H.; Chen, X.; Kazim, L.; Repasky, E.A.; Subjeck, J.R. Extracellular targeting of endoplasmic reticulum chaperone glucose-regulated protein 170 enhances tumor immunity to a poorly immunogenic melanoma. J. Immunol. 2006, 177, 1543–1551. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.-E.; Moore, A.M.; Thomsen, L.L.; Brindle, K.M. Uric Acid Promotes Tumor Immune Rejection. Cancer Res. 2004, 64, 5059–5062. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Evans, J.E.; Rock, K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 2003, 425, 516–521. [Google Scholar] [CrossRef] [PubMed]
- Skitzki, J.J.; Repasky, E.A.; Evans, S.S. Hyperthermia as an immunotherapy strategy for cancer. Curr. Opin. Investig. Drugs 2009, 10, 550–558. [Google Scholar] [PubMed]
- Ostberg, J.R.; Dayanc, B.E.; Yuan, M.; Oflazoglu, E.; Repasky, E.A. Enhancement of natural killer (NK) cell cytotoxicity by fever-range thermal stress is dependent on NKG2D function and is associated with plasma membrane NKG2D clustering and increased expression of MICA on target cells. J. Leukoc. Biol. 2007, 82, 1322–1331. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Cao, T.; Connolly, J.E.; Monnet, L.; Bennett, L.; Chapel, S.; Bagnis, C.; Mannoni, P.; Davoust, J.; Palucka, A.K.; et al. Hyperthermia enhances CTL cross-priming. J. Immunol. 2006, 176, 2134–2141. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, J.; Han, C.; Yang, M.; Cao, X. Heat Shock Protein 70, Released from heat-stressed tumor cells, initiates antitumor immunity by inducing tumor cell chemokine production and activating dendritic cells via TLR4 pathway. J. Immunol. 2009, 182, 1449–1459. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.A.; Massagué, J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 2005, 8, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Tsumura, H.; Miwa, M.; Inaba, K. Contrasting effects of TGF-β1 and TNF-α on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997, 15, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Liu, K.; Bonifaz, L.; Bonnyay, D.; Mahnke, K.; Iyoda, T.; Ravetch, J.; Dhodapkar, M.; Inaba, K.; et al. Dendritic cell function in vivo during the steady state: A role in peripheral tolerance. Ann. NY Acad. Sci. 2003, 987, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Rook, A.H.; Kehrl, J.H.; Wakefield, L.M.; Roberts, A.B.; Sporn, M.B.; Burlington, D.B.; Lane, H.C.; Fauci, A.S. Effects of transforming growth factor beta on the functions of natural killer cells: Depressed cytolytic activity and blunting of interferon responsiveness. J. Immunol. 1986, 136, 3916–3920. [Google Scholar] [PubMed]
- Chiang, C.; Ledermann, J.; Rad, A.N.; Katz, D.; Chain, B. Hypochlorous acid enhances immunogenicity and uptake of allogeneic ovarian tumor cells by dendritic cells to cross-prime tumor-specific T cells. Cancer Immunol. Immunother. 2006, 55, 1384–1395. [Google Scholar] [CrossRef]
- Chiang, C.L.; Ledermann, J.A.; Egla, A.; Elizabeth, B.; Katz, R.D.; Chain, B.M. Oxidation of ovarian epithelial cancer cells by hypochlorous acid enhances immunogenicity and stimulates T cells that recognize autologous primary tumor. Clin. Cancer Res. 2008, 14, 4898–4907. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.; Kandalaft, L.; Tanyi, J.L.; Hagemann, A.R.; Motz, G.T.; Svoronos, N.; Montone, K.; Mantia-Smaldone, G.M.; Smith, L.; Nisenbaum, H.L.; et al. A Dendritic cell vaccine pulsed with autologous hypochlorous acid-oxidized ovarian cancer lysate primes effective broad antitumor immunity: From bench to bedside. Clin. Cancer Res. 2013, 19, 4801–4815. [Google Scholar] [CrossRef] [PubMed]
- Prokopowicz, Z.M.; Arce, F.; Biedron, R.; Chiang, C.L.-L.; Ciszek, M.; Katz, D.R.; Nowakowska, M.; Zapotoczny, S.; Marcinkiewicz, J.; Chain, B.M. Hypochlorous acid: A natural adjuvant that facilitates antigen processing, cross-priming, and the induction of adaptive immunity. J. Immunol. 2010, 184, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, A.R.; Cadungog, M.; Hagemann, I.S.; Hammond, R.; Adams, S.F.; Chu, C.S.; Rubin, S.C.; Zhang, L.; Addya, K.; Birrer, M.J.; et al. Tissue-based immune monitoring I: Tumor core needle biopsies allow in-depth interrogation of the tumor microenvironment. Cancer Biol. Ther. 2011, 12, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Merogi, A.J.; Marrogi, A.J.; Ramesh, R.; Robinson, W.R.; Fermin, C.D.; Freeman, S.M. Tumor-host interaction: Analysis of cytokines, growth factors, and tumor-infiltrating lymphocytes in ovarian carcinomas. Hum. Pathol. 1997, 28, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.; Barve, M.; Kuhn, J.; Melnyk, A.; Beitsch, P.; Lazar, M.; Lifshitz, S.; Magee, M.; Oh, J.; Mill, S.W.; et al. Phase I Trial of “bi-shRNAifurin/GMCSF DNA/Autologous Tumor Cell” vaccine (FANG) in advanced cancer. Mol. Ther. 2012, 20, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Fishman, M.; Hunter, T.B.; Soliman, H.; Thompson, P.; Dunn, M.; Smilee, R.; Farmelo, M.J.; Noyes, D.R.; Mahany, J.J.; Lee, J.H.; et al. Phase II trial of B7-1 (CD-86) transduced, cultured autologous tumor cell vaccine plus subcutaneous interleukin-2 for treatment of stage IV renal cell carcinoma. J. Immunother. 2008, 31, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Schartz, N.E.C.; Movassagh, M.; Flament, C.; Pautier, P.; Morice, P.; Pomel, C.; Lhomme, C.; Escudier, B.; le Chevalier, T.; et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet 2002, 360, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Mears, R.; Craven, R.A.; Hanrahan, S.; Totty, N.; Upton, C.; Young, S.L.; Patel, P.; Selby, P.J.; Banks, R.E. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 2004, 4, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Flament, C.; Viaud, S.; Taieb, J.; Roux, S.; Spatz, A.; André, F.; LePecq, J.B.; Boussac, M.; Garin, J.; et al. Dendritic cell derived-exosomes: Biology and clinical implementations. J. Leukoc. Biol. 2006, 80, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer Cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodriguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; de Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the Extracellular Environment in a Membrane-Associated Form that Activates Macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wan, T.; Wang, B.; Zhou, X.; Xiu, F.; Chen, T.; Wu, Y.; Cao, X. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef] [PubMed]
- Andreola, G.; Rivoltini, L.; Castelli, C.; Huber, V.; Perego, P.; Deho, P.; Squarcina, P.; Accornero, P.; Lozupone, F.; Lugini, L; et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J. Exp. Med. 2002, 195, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E.; et al. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.-B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, L.E.; Motz, G.T.; Busch, J.; Coukos, G. Angiogenesis and the tumor vasculature as antitumor immune modulators: The role of vascular endothelial growth factor and endothelin. Curr. Top. Microbiol. Immunol. 2011, 344, 129–148. [Google Scholar] [PubMed]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.-P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Jung, G.; Rammensee, H.G.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Carralot, J.-P.; Weide, B.; Schoor, O.; Probst, J.; Scheel, B.; Teufel, R.; Hoerr, I.; Garbe, C.; Rammensee, H.G.; Pascolo, S. Production and characterization of amplified tumor-derived cRNA libraries to be used as vaccines against metastatic melanomas. Genet. Vaccines Ther. 2005. [Google Scholar] [CrossRef]
- Bringmann, A.; Held, S.A.E.; Heine, A.; Brossart, P. RNA Vaccines in cancer treatment. J. Biomed. Biotechnol. 2010. [Google Scholar] [CrossRef]
- Su, Z.; Dannull, J.; Heiser, A.; Yancey, D.; Pruitt, S.; Madden, J.; Coleman, D.; Niedzwiecki, D.; Gilboa, E.; Vieweg, J. Immunological and clinical responses in metastatic renal cancer patients vaccinated with tumor RNA-transfected dendritic cells. Cancer Res. 2003, 63, 2127–2133. [Google Scholar] [PubMed]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I//II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Vik-Mo, E.; Nyakas, M.; Mikkelsen, B.; Moe, M.; Due-Tønnesen, P.; Suso, E.M.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R. Exploring the genomes of cancer cells: Progress and promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Heemskerk, B.; Kvistborg, P.; Schumacher, T.N.M. The cancer antigenome. EMBO J. 2012, 32, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Vallazza, B.; Türeci, Ö.; Sahin, U. Determinants of intracellular RNA pharmacokinetics: Implications for RNA-based immunotherapeutics. RNA Biol. 2011, 8, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Barnea, E.; Beer, I.; Avivi, I.; Katz, T.; Admon, A. Soluble plasma HLA peptidome as a potential source for cancer biomarkers. Proc. Natl. Acad. Sci. USA 2010, 107, 18769–18776. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Konrad, T.; Sester, M.; Huber, C.; Türeci, Ö.; Sahin, U. Simultaneous ex vivo quantification of antigen-specific CD4+ and CD8+ T cell responses using in vitro transcribed RNA. Cancer Immunol. Immunother. 2007, 56, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.-C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Kenter, G.G.; Welters, M.J.P.; Valentijn, A.R.P.M.; Lowik, M.J.G.; Berends-van der Meer, D.M.A.; Vloon, A.P.G.; Essahsah, F.; Fathers, L.M.; Offringa, R.; Drijfhout, J.W.; et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Dittmer, D.; Rea, D.; Tartaglia, J.; Paoletti, E.; Levine, A.J. p53 as a target for cancer vaccines: Recombinant canarypox virus vectors expressing p53 protect mice against lethal tumor cell challenge. Proc. Natl. Acad. Sci. USA 1996, 93, 4781–4786. [Google Scholar] [CrossRef] [PubMed]
- Carbone, D.P.; Ciernik, I.F.; Kelley, M.J.; Smith, M.C.; Nadaf, S.; Kavanaugh, D.; Maher, V.E.; Stipanov, M.; Contois, D.; Johnson, B.E.; et al. Immunization with mutant p53- and K-ras-derived peptides in cancer patients: Immune response and Clinical outcome. J. Clin. Oncol. 2005, 23, 5099–5107. [Google Scholar] [CrossRef] [PubMed]
- Rahma, O.; Ashtar, E.; Ibrahim, R.; Toubaji, A.; Gause, B.; Herrin, V.; Linehan, W.M.; Steinberg, S.M.; Grollman, F.; Grimes, G.; et al. A pilot clinical trial testing mutant von Hippel-Lindau peptide as a novel immune therapy in metastatic Renal Cell Carcinoma. J. Transl. Med. 2010, 8. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.S.; Thrue, C.A.; Junker, N.; Lyngaa, R.; Donia, M.; Ellebæk, E.; Svane, I.M.; Schumacher, T.N.; Thor Straten, P.; Hadrup, S.R. Dissection of T-cell antigen specificity in human melanoma. Cancer Res. 2012, 72, 1642–1650. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, J. Tumor stroma as targets for cancer therapy. Pharmacol. Ther. 2013, 137, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Kono, K.; Matsumoto, Y.; Fujii, H.; Yamane, T.; Mitsumata, M.; Chen, W.T. The expression of a type II transmembrane serine protease (Seprase) in human gastric carcinoma. Oncology 2004, 67, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.A.; Kraut, N.; Park, J.E.; Schubert, R.D.; Rettig, W.J.; Peter, R.U.; Garin-Chesa, P. Fibroblast activation protein: Differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J. Investig. Dermatol. 2003, 120, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Garin-Chesa, P.; Old, L.J.; Rettig, W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA 1990, 87, 7235–7239. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, S.; Kelly, T. Seprase promotes rapid tumor growth and increased microvessel density in a mouse model of human breast cancer. Cancer Res. 2004, 64, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, S.; Jin, X.; Okada, K.; Mitsumata, M.; Ooi, A. Increased expression of seprase, a membrane-type serine protease, is associated with lymph node metastasis in human colorectal cancer. Cancer Lett. 2003, 199, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Oh, S.; Gharagozlou, S.; Chen, W.; Ohlfest, J.R. In vivo vaccination with tumor cell lysate plus CpG oligodeoxynucleotides eradicates murine glioblastoma. J. Immunother. 2007, 30, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.C.; Shimizu, K.; Mulé, J.J. Murine dendritic cells pulsed with whole tumor lysates mediate potent antitumor immune responses in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1998, 95, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Benencia, F.; Courreges, M.C.; Coukos, G. Whole tumor antigen vaccination using dendritic cells: Comparison of RNA electroporation and pulsing with UV-irradiated tumor cells. J. Transl. Med. 2008, 6. [Google Scholar] [CrossRef]
- Berd, D.; Kairys, J.; Dunton, C.; Mastrangelo, M.J.; Sato, T.; Maguire, H.C., Jr. Autologous, hapten-modified vaccine as a treatment for human cancers. Semin. Oncol. 1998, 25, 646–653. [Google Scholar] [PubMed]
- Baars, A.; Claessen, A.M.E.; van den Eertwegh, A.J.M.; Giaccone, G.; Meijer, C.J.; Scheper, R.J.; Wagstaff, J.; et al. Skin tests predict survival after autologous tumor cell vaccination in metastatic melanoma: Experience in 81 patients. Ann. Oncol. 2000, 11, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Lopez, M.N.; Pereda, C.; Segal, G.; Muñoz, L.; Aguilera, R.; González, F.E.; Escobar, A.; Ginesta, A.; Reyes, D.; González, R.; et al. Prolonged survival of dendritic cell-vaccinated melanoma patients correlates with tumor-specific delayed type IV hypersensitivity response and reduction of tumor growth factor beta-expressing T cells. J. Clin. Oncol. 2009, 27, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Croix, B.S.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes Expressed in Human Tumor Endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Carson-Walter, E.B.; Watkins, D.N.; Nanda, A.; Vogelstein, B.; Kinzler, K.W.; St. Croix, B. Cell surface tumor endothelial markers are conserved in mice and humans. Cancer Res. 2001, 61, 6649–6655. [Google Scholar] [PubMed]
- Nanda, A.; Carson-Walter, E.B.; Seaman, S.; Barber, T.D.; Stampfl, J.; Singh, S.; Vogelstein, B.; Kinzler, K.W.; St. Croix, B. TEM8 interacts with the cleaved C5 domain of collagen α3(VI). Cancer Res. 2004, 64, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; O’Keefe, D.S.; Bacich, D.J.; Reuter, V.E.; Heston, W.D.W.; Gaudin, P.B. Prostate-specific membrane antigen is produced in tumor-associated neovasculature. Clin. Cancer Res. 1999, 5, 2674–2681. [Google Scholar] [PubMed]
- Sandberg, J.K.; Grufman, P.; Wolpert, E.Z.; Franksson, L.; Chambers, B.J.; Kärre, K. Superdominance among immunodominant H-2Kb-restricted epitopes and reversal by dendritic cell-mediated antigen delivery. J. Immunol. 1998, 160, 3163–3169. [Google Scholar] [PubMed]
- Derhovanessian, E.; Solana, R.; Larbi, A.; Pawelec, G. Immunity, ageing and cancer. Immun. Ageing 2008. [Google Scholar] [CrossRef]
- Fagnoni, F.F.; Vescovini, R.; Passeri, G.; Bologna, G.; Pedrazzoni, M.; Lavagetto, G.; Casti, A.; Franceschi, C.; Passeri, M.; Sansoni, P. Shortage of circulating naive CD8+ T cells provides new insights on immunodeficiency in aging. Blood 2000, 95, 2860–2868. [Google Scholar] [PubMed]
- Phillips, J.A.; Brondstetter, T.I.; English, C.A.; Lee, H.E.; Virts, E.L.; Thoman, M.L. IL-7 Gene therapy in aging restores early thymopoiesis without reversing involution. J. Immunol. 2004, 173, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Marko, M.G.; Ahmed, T.; Bunnell, S.C.; Wu, D.; Chung, H.; Huber, B.T.; Meydani, S.N. Age-associated decline in effective immune synapse formation of CD4+ T cells is reversed by vitamin E supplementation. J. Immunol. 2007, 178, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Albers, R.; van der Wielen, R.P.J.; Brink, E.J.; Hendriks, H.F.J.; Dorovska-Taran, V.N.; Mohede, I.C.M. Effects of cis-9, trans-11 and trans-10, cis-12 conjugated linoleic acid (CLA) isomers on immune function in healthy men. Eur. J. Clin. Nutr. 2003, 57, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Larbi, A.; Douziech, N.; Dupuis, G.; Khalil, A.; Pelletier, H.; Guerard, K.-P.; Fülöp, T., Jr. Age-associated alterations in the recruitment of signal-transduction proteins to lipid rafts in human T lymphocytes. J. Leukoc. Biol. 2004, 75, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Rivnay, B.; Bergman, S.; Shinitzky, M.; Globerson, A. Correlations between membrane viscosity, serum cholesterol, lymphocyte activation and aging in man. Mech. Ageing Dev. 1980, 12, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Gribben, J.G.; Ryan, D.P.; Boyajian, R.; Urban, R.G.; Hedley, M.L.; Beach, K.; Nealon, P.; Matulonis, U.; Campos, S.; Gilligan, T.D.; et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin. Cancer Res. 2005, 11, 4430–4436. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rubner, Y.; Kulzer, L.; Werthmöller, N.; Weiss, E.-M.; Fietkau, R.; Gaipl, U.S. Antitumor immune responses induced by ionizing irradiation and further immune stimulation. Cancer Immunol. Immunother. 2014, 63, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.L.-L.; Kandalaft, L.E.; Coukos, G. Adjuvants for enhancing the immunogenicity of whole tumor cell vaccines. Int. Rev. Immunol. 2011, 30, 150–182. [Google Scholar] [CrossRef] [PubMed]
- Kushwah, R.; Hu, J. Complexity of dendritic cell subsets and their function in the host immune system. Immunology 2011, 133, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Liu, Y.-J. Development of dendritic-cell lineages. Immunity 2007, 26, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.; Mulligan, M.; Gibbs, R.; Nutman, T.; Francis, D. Intradermal Hepatitis B Virus vaccine: Immunogenicity and side effects in adults. Lancet 1983, 322, 1454–1456. [Google Scholar] [CrossRef]
- WHO Publication. Hepatitis B vaccines: WHO position paper—Recommendations. Vaccine 2010, 28, 589–590. [Google Scholar]
- Wu, X.; Franka, R.; Svoboda, P.; Pohl, J.; Rupprecht, C.E. Development of combined vaccines for rabies and immunocontraception. Vaccine 2009, 27, 7202–7209. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.L.; Bertholet, S.; Kahn, M.; Zharkikh, I.; Ireton, G.C.; Vedvick, T.S.; Reed, S.G.; Coler, R.N. Intradermal immunization improves protective efficacy of a novel TB vaccine candidate. Vaccine 2009, 27, 3063–3071. [Google Scholar] [CrossRef] [PubMed]
- Cutts, F.T.; Clements, C.J.; Bennett, J.V. Altenative Routes of measles immunization: A Review. Biologicals 1997, 25, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, S.; Cherian, T.; Samuel, B.; Rajasingh, J.; Raghupathy, P.; John, T. Immune response of infants to fractional doses of intradermally administered inactivated poliovirus vaccine. Vaccine 1998, 23, 3063–3071. [Google Scholar]
- Chowell, G.; Viboud, C.; Wang, X.; Bertozzi, S.M.; Miller, M.A. Adaptive vaccination strategies to mitigate pandemic influenza: Mexico as a case study. PLOS ONE 2009, 4, e8164. [Google Scholar] [CrossRef] [PubMed]
- King, J.W.; Taylor, E.M.; Crow, S.D.; White, M.C.; Todd, J.R.; Poe, M.B.; Conrad, S.A.; Gelder, F.B. Comparison of the Immunogenicity of Hepatitis B Vaccine Administered Intradermally and Intramuscularly. Rev. Infect. Dis. 1990, 12, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Vien, N.C.; Feroldi, E.; Lang, J. Long-term anti-rabies antibody persistence following intramuscular or low-dose intradermal vaccination of young Vietnamese children. Trans. R. Soc. Trop. Med. Hyg. 2008, 102, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Karthaus, N.; Torensma, R.; Tel, J. Deciphering the Message Broadcast by Tumor-Infiltrating Dendritic Cells. Am. J. Pathol. 2012, 181, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Herr, H.W.; Schwalb, D.M.; Zhang, Z.F.; Sogani, P.C.; Fair, W.R.; Whitmore, W.F., Jr.; Oettgen, H.F. Intravesical bacillus Calmette-Guérin therapy prevents tumor progression and death from superficial bladder cancer: Ten-Year follow-up of a prospective randomized trial. J. Clin. Oncol. 1995, 13, 1404–1408. [Google Scholar] [PubMed]
- Carpentier, A.; Laigle-Donadey, F.; Zohar, S.; Capelle, L.; Behin, A.; Tibi, A.; Martin-Duverneuil, N.; Sanson, M.; Lacomblez, L.; Taillibert, T.; et al. Phase 1 trial of a CpG oligodeoxynucleotide for patients with recurrent glioblastoma. Neuro Oncol. 2006, 8, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Gross, B.; Wongrakpanich, A.; Francis, M.; Salem, A.; Norian, L. A Therapeutic microparticle-based tumor lysate vaccine reduces spontaneous metastases in murine breast cancer. AAPS J. 2014, 16, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.; Reichardt, W.; Koerner, J.; Groettrup, M. Coencapsulation of tumor lysate and CpG-ODN in PLGA-microspheres enables successful immunotherapy of prostate carcinoma in TRAMP mice. J. Control. Release 2012, 162, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Li, W.A.; Choi, Y.; Lewin, S.A.; Verbeke, C.S.; Dranoff, G.; Mooney, D.J. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 2015, 33, 64–72. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, C.L.-L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344-372. https://doi.org/10.3390/vaccines3020344
Chiang CL-L, Coukos G, Kandalaft LE. Whole Tumor Antigen Vaccines: Where Are We? Vaccines. 2015; 3(2):344-372. https://doi.org/10.3390/vaccines3020344
Chicago/Turabian StyleChiang, Cheryl Lai-Lai, George Coukos, and Lana E. Kandalaft. 2015. "Whole Tumor Antigen Vaccines: Where Are We?" Vaccines 3, no. 2: 344-372. https://doi.org/10.3390/vaccines3020344
APA StyleChiang, C. L. -L., Coukos, G., & Kandalaft, L. E. (2015). Whole Tumor Antigen Vaccines: Where Are We? Vaccines, 3(2), 344-372. https://doi.org/10.3390/vaccines3020344