Protective Cancer Vaccine Using Genetically Modified Hematopoietic Stem Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
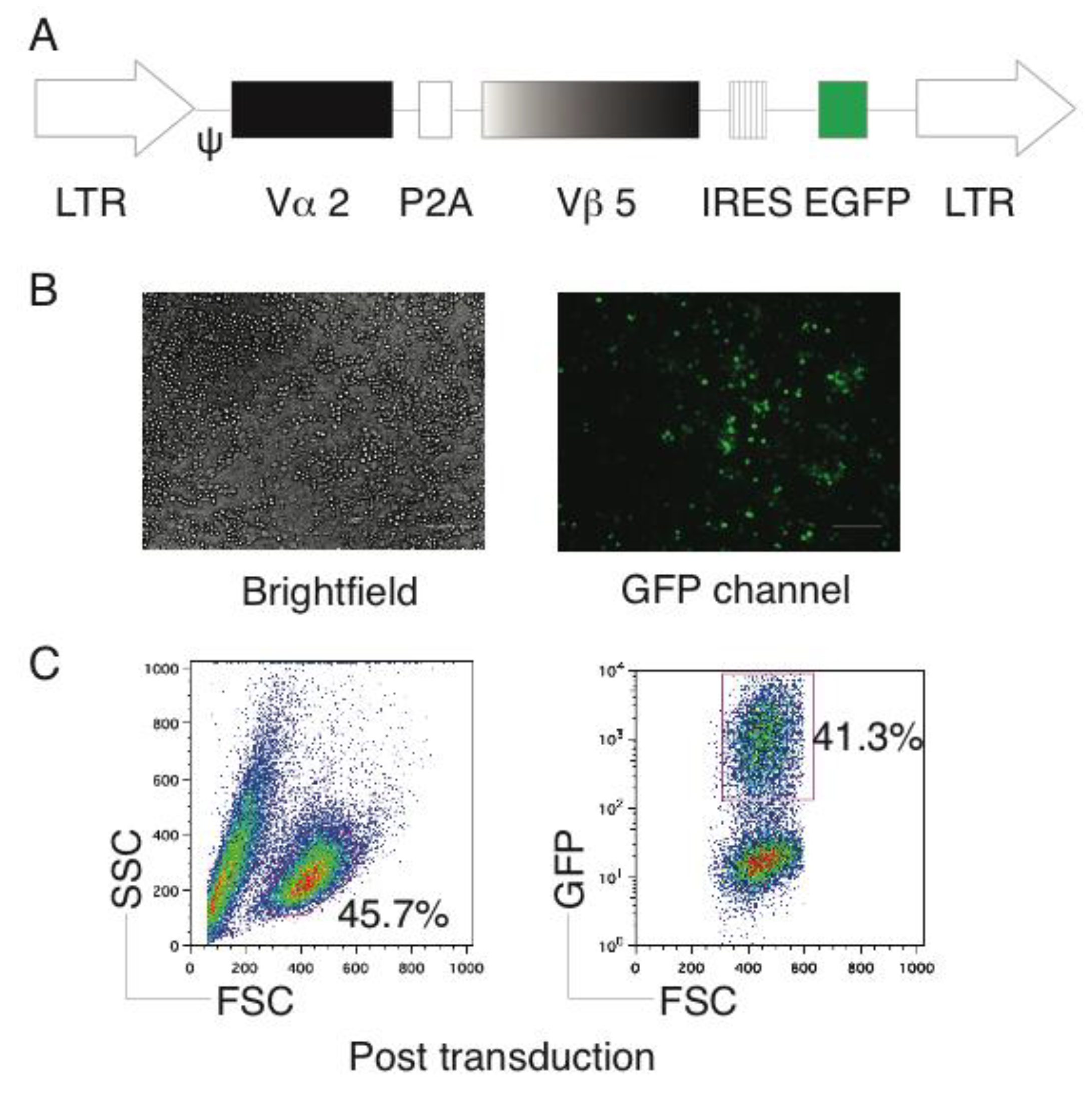
2.1. Generation of OVA-Specific TCR Gene-Transduced HSCs
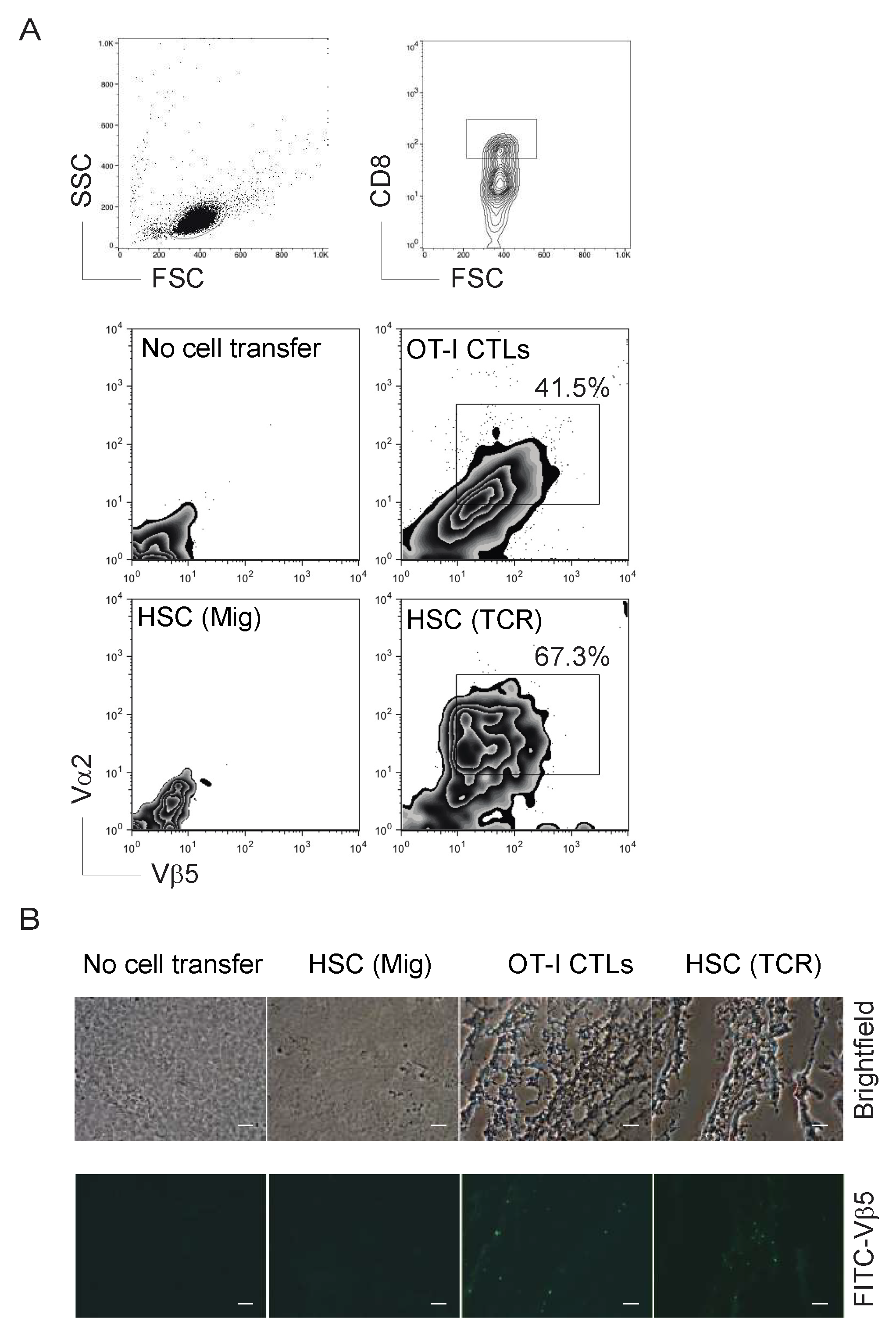
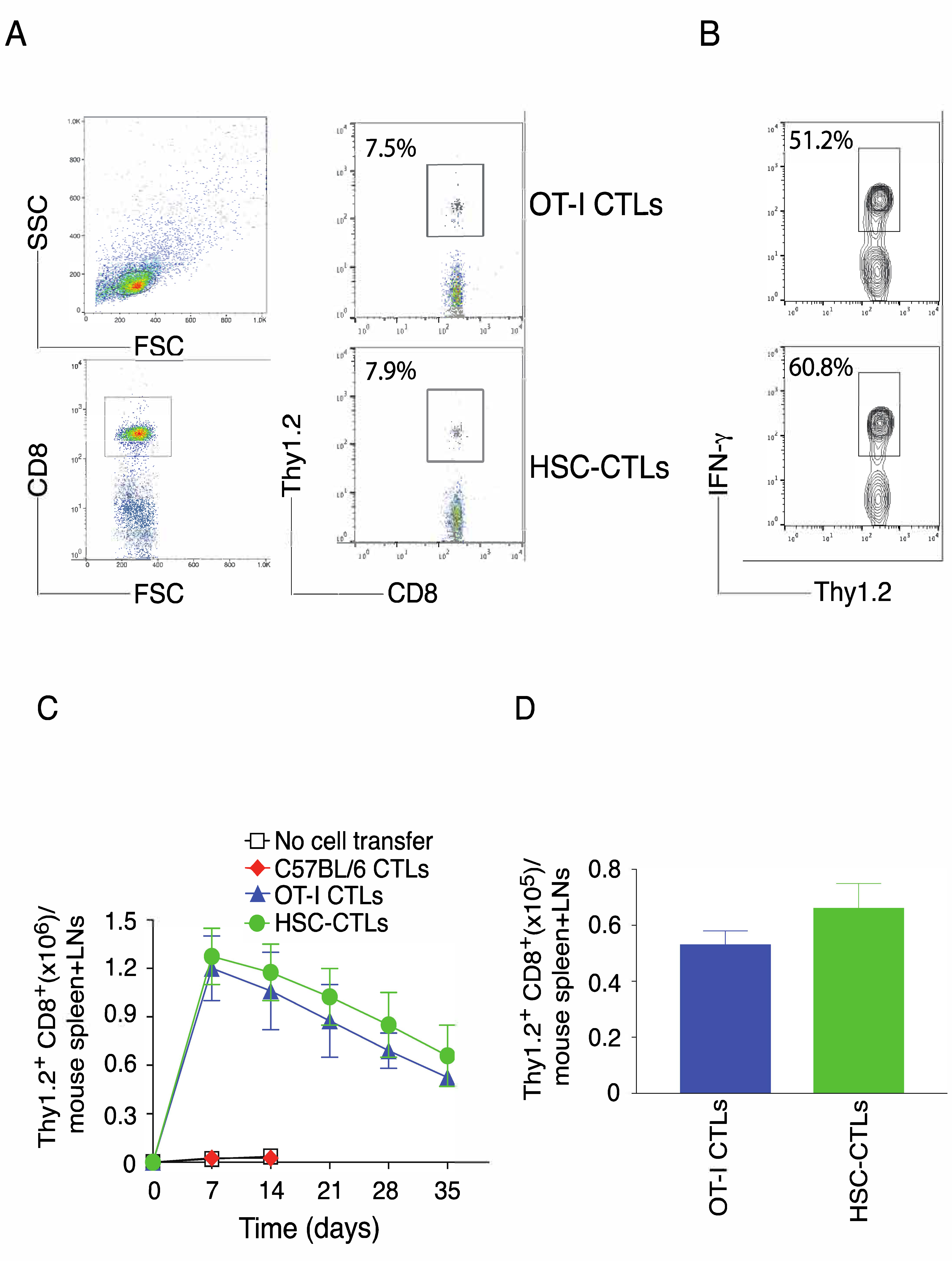
2.2. TCR Gene-Transduced HSCs Differentiated into Ag-Specific CD8+ T Cells In Vivo
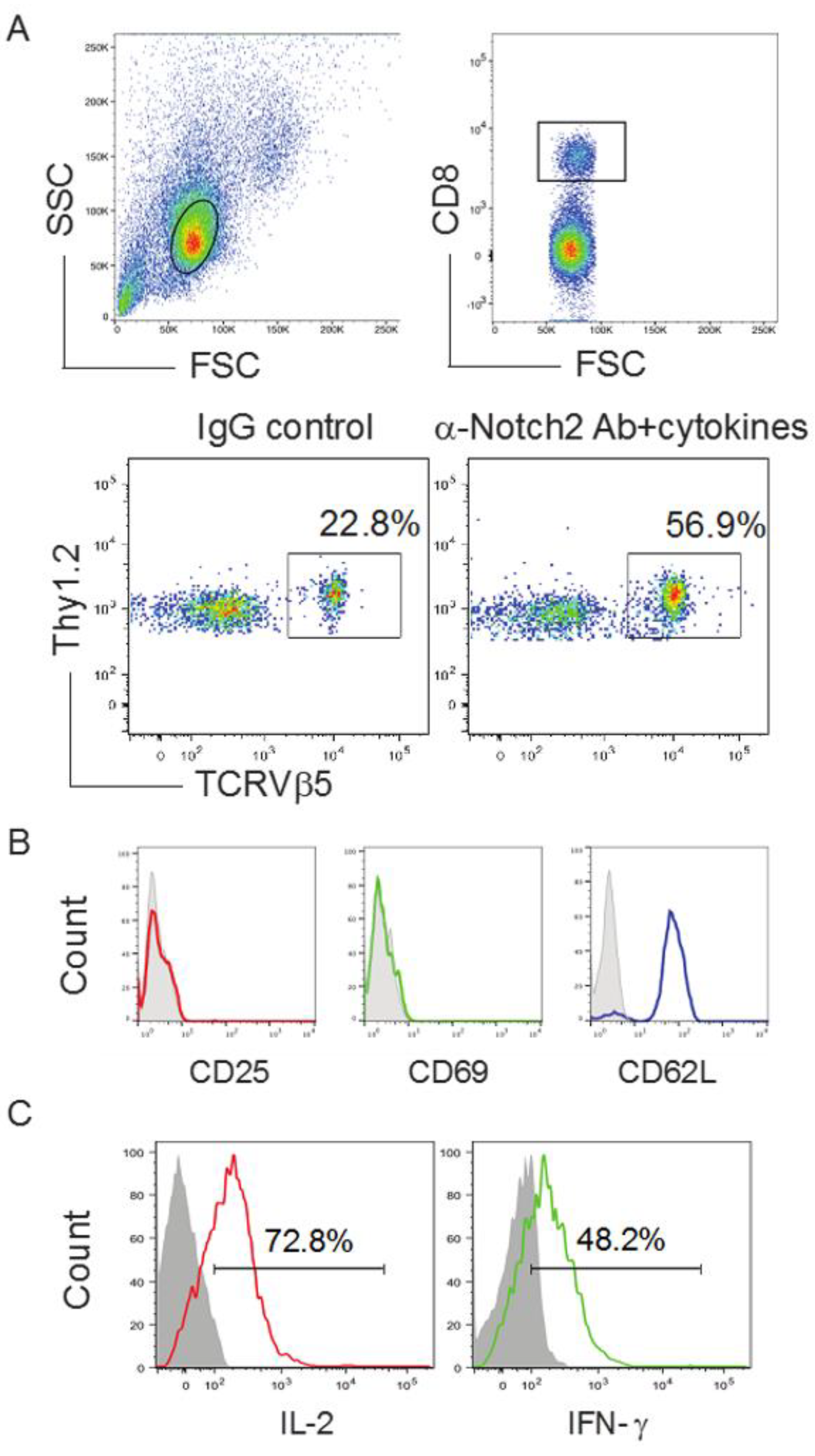
2.3. In Vivo Priming of Ag-Specific HSC-CD8+ T Cells
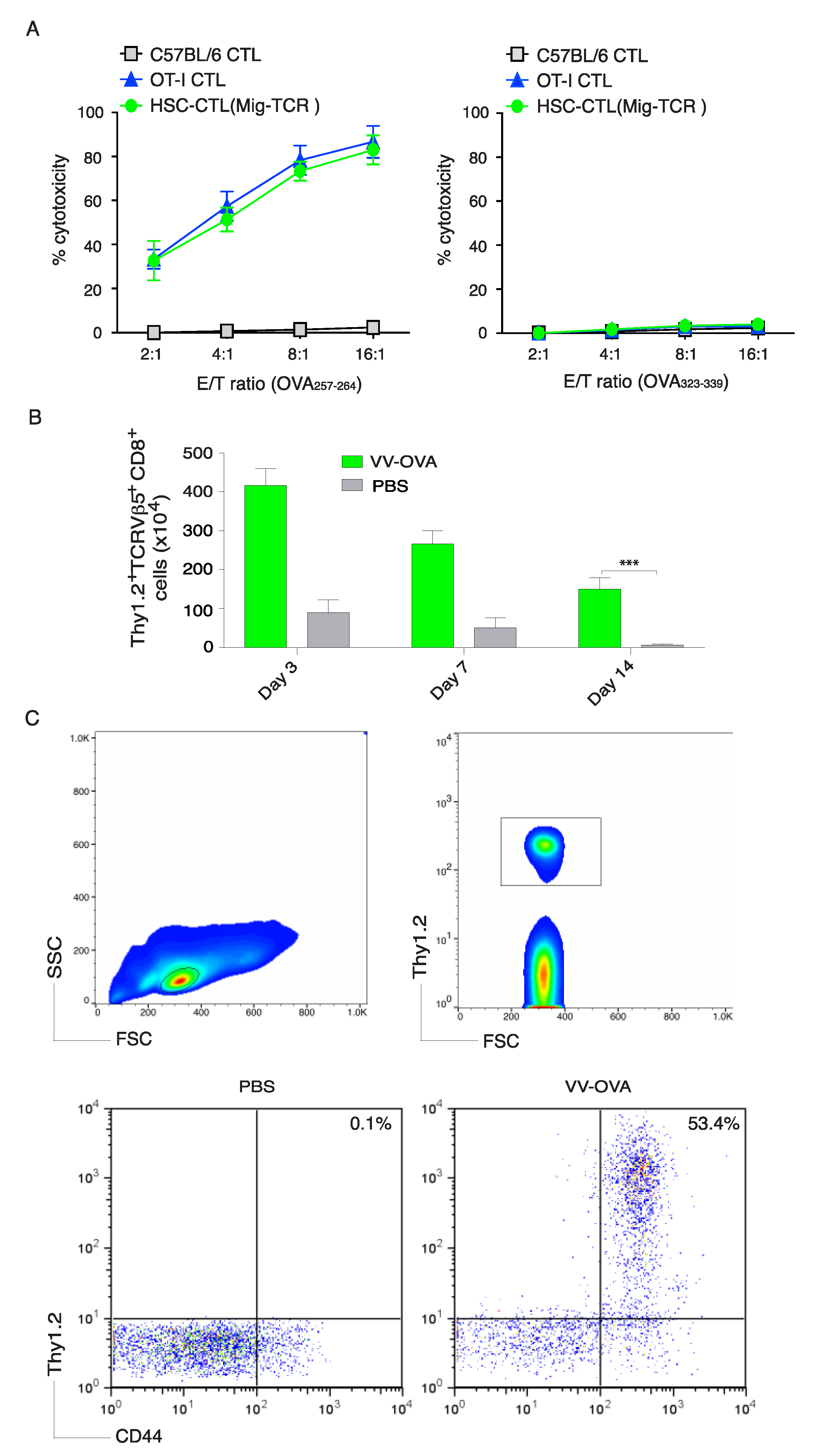
2.4. Genetically Modified HSCs Following In Vivo Priming Induced CTL Infiltration in the Tumor Tissue
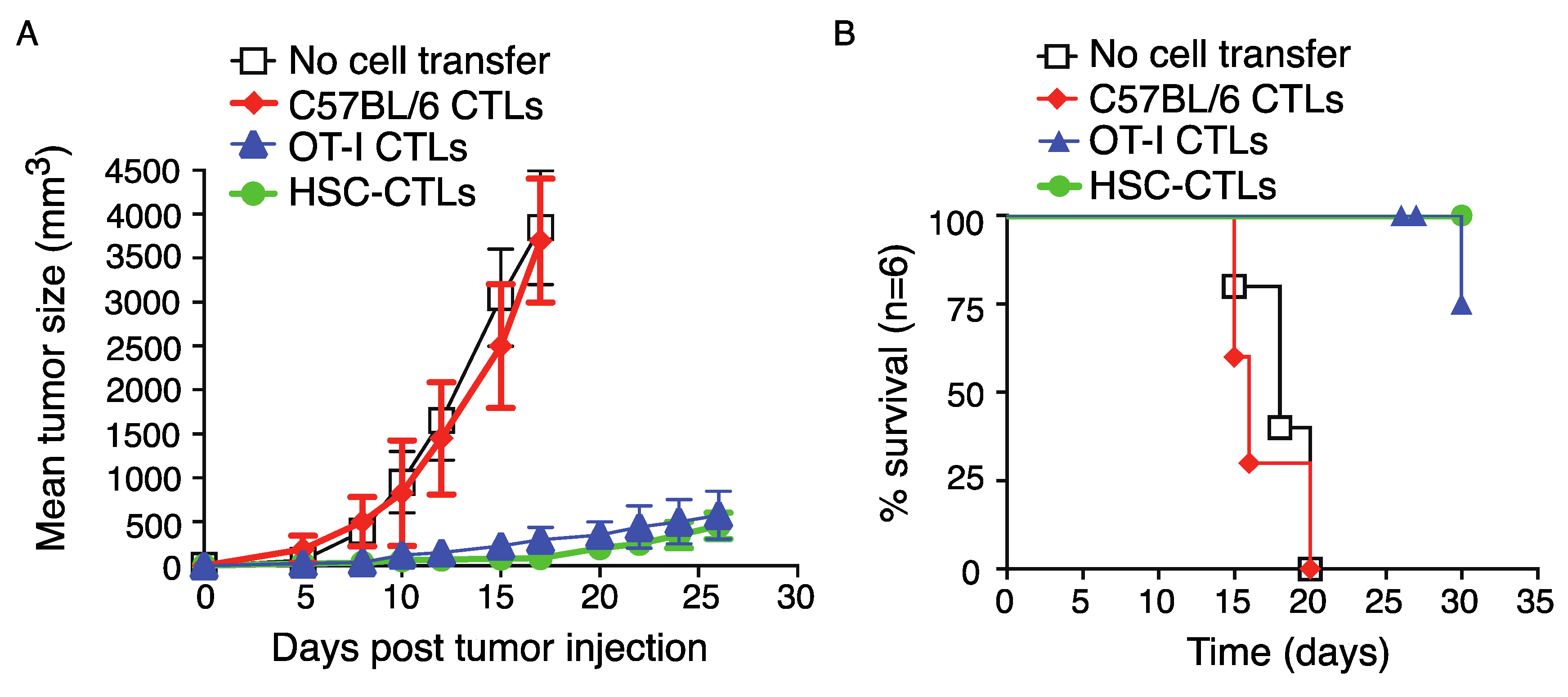
2.5. Immunization Using Genetically Modified HSCs Following In Vivo Priming Protected Mice from Tumor Growth
2.6. Immunization Using Genetically Modified HSCs Following In Vivo Priming Induced the Generation of Tumor Ag-Specific T Cell Memory
3. Materials and Methods
3.1. Ethics Statement
3.2. Cells and Mice
3.3. Cell Culture
3.4. Antibodies Used
3.5. Retroviral Transduction
3.6. Adoptive Cell Transfer
3.7. Ex Vivo Peptide Stimulation Assay
3.8. Cytokine Secretion, Cell Recovery and Proliferation
3.9. In Vitro Cytotoxicity Assay
3.10. Xenograft Melanoma Model
3.11. Histology and Immunofluorescence
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Koller, K.M.; Wang, W.; Schell, T.D.; Cozza, E.M.; Kokolus, K.M.; Neves, R.I.; Mackley, H.B.; Pameijer, C.; Leung, A.; Anderson, B.; et al. Malignant melanoma-the cradle of anti-neoplastic immunotherapy. Crit. Rev. Oncol. Hematol. 2016, 106, 25–54. [Google Scholar] [CrossRef] [PubMed]
- Soldner, F.; Hockemeyer, D.; Beard, C.; Gao, Q.; Bell, G.W.; Cook, E.G.; Hargus, G.; Blak, A.; Cooper, O.; Mitalipova, M.; et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 2009, 136, 964–977. [Google Scholar] [CrossRef] [PubMed]
- Raya, A.; Rodriguez-Piza, I.; Guenechea, G.; Vassena, R.; Navarro, S.; Barrero, M.J.; Consiglio, A.; Castella, M.; Rio, P.; Sleep, E.; et al. Disease-corrected haematopoietic progenitors from fanconi anaemia induced pluripotent stem cells. Nature 2009, 460, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Ebert, A.D.; Yu, J.; Rose, F.F., Jr.; Mattis, V.B.; Lorson, C.L.; Thomson, J.A.; Svendsen, C.N. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature 2009, 457, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Zhao, B.; Haque, R.; Xiong, X.; Budgeon, L.; Christensen, N.D.; Wu, Y.; Song, J. In vivo programming of tumor antigen-specific T lymphocytes from pluripotent stem cells to promote cancer immunosurveillance. Cancer Res. 2011, 71, 4742–4747. [Google Scholar] [CrossRef] [PubMed]
- Haque, R.; Lei, F.; Xiong, X.; Bian, Y.; Zhao, B.; Wu, Y.; Song, J. Programming of regulatory T cells from pluripotent stem cells and prevention of autoimmunity. J. Immunol. 2012, 189, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Song, J.; Fino, K.; Sandhu, P.; Wang, Y.; Ni, B.; Fang, D.; Song, J. Melanoma immunotherapy in mice using genetically engineered pluripotent stem cells. Cell Transplant. 2016, 25, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Song, J.; Fino, K.; Sandhu, P.; Song, X.; Lei, F.; Zheng, S.; Ni, B.; Fang, D.; Song, J. Stem cell-derived tissue-associated regulatory T cells ameliorate the development of autoimmunity. Sci. Rep. 2016, 6, 20588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Yang, C.S.; Nakashima, K.; Rana, T.M. Small rna-mediated regulation of ips cell generation. EMBO J. 2011, 30, 823–834. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Zhang, Z.N.; Rong, Z.; Xu, Y. Immunogenicity of induced pluripotent stem cells. Nature 2011, 474, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.; Song, J.; Fino, K.; Wang, Y.; Sandhu, P.; Song, X.; Norbury, C.; Ni, B.; Fang, D.; Salek-Ardakani, S.; et al. C-myc regulation by costimulatory signals modulates the generation of cd8+ memory T cells during viral infection. Open Biol. 2016, 6, 150208. [Google Scholar] [CrossRef] [PubMed]
- Sekine, C.; Koyanagi, A.; Koyama, N.; Hozumi, K.; Chiba, S.; Yagita, H. Differential regulation of osteoclastogenesis by notch2/delta-like 1 and notch1/jagged1 axes. Arthritis Res. Ther. 2012, 14, R45. [Google Scholar] [CrossRef] [PubMed]
- Kijima, M.; Yamaguchi, T.; Ishifune, C.; Maekawa, Y.; Koyanagi, A.; Yagita, H.; Chiba, S.; Kishihara, K.; Shimada, M.; Yasutomo, K. Dendritic cell-mediated nk cell activation is controlled by jagged2-notch interaction. Proc. Natl. Acad. Sci. USA 2008, 105, 7010–7015. [Google Scholar] [CrossRef] [PubMed]
- Frankel, T.L.; Burns, W.R.; Peng, P.D.; Yu, Z.; Chinnasamy, D.; Wargo, J.A.; Zheng, Z.; Restifo, N.P.; Rosenberg, S.A.; Morgan, R.A. Both cd4 and cd8 T cells mediate equally effective in vivo tumor treatment when engineered with a highly avid tcr targeting tyrosinase. J. Immunol. 2010, 184, 5988–5998. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Song, A.; Haque, R.; Lei, F.; Weiler, L.; Xiong, X.; Wu, Y.; Croft, M.; Song, J. Cooperation between molecular targets of costimulation in promoting T cell persistence and tumor regression. J. Immunol. 2009, 182, 6744–6752. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, X.; Das, J.K.; Song, J.; Ni, B.; Ren, X.; Yang, J.-M.; Song, J. Protective Cancer Vaccine Using Genetically Modified Hematopoietic Stem Cells. Vaccines 2018, 6, 40. https://doi.org/10.3390/vaccines6030040
Xiong X, Das JK, Song J, Ni B, Ren X, Yang J-M, Song J. Protective Cancer Vaccine Using Genetically Modified Hematopoietic Stem Cells. Vaccines. 2018; 6(3):40. https://doi.org/10.3390/vaccines6030040
Chicago/Turabian StyleXiong, Xiaofang, Jugal Kishor Das, Jianyong Song, Bing Ni, Xingcong Ren, Jin-Ming Yang, and Jianxun Song. 2018. "Protective Cancer Vaccine Using Genetically Modified Hematopoietic Stem Cells" Vaccines 6, no. 3: 40. https://doi.org/10.3390/vaccines6030040
APA StyleXiong, X., Das, J. K., Song, J., Ni, B., Ren, X., Yang, J. -M., & Song, J. (2018). Protective Cancer Vaccine Using Genetically Modified Hematopoietic Stem Cells. Vaccines, 6(3), 40. https://doi.org/10.3390/vaccines6030040