β-Defensins Coordinate In Vivo to Inhibit Bacterial Infections of the Trachea



Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth
2.2. Animals
2.3. Infection of Mice
2.4. Histopathology
2.5. In Vitro Infection of Differentiated Tracheal Epithelial Cells
2.6. Quantification of mRNA Levels
2.7. ELISA for Specific Antibody against B. bronchiseptica
2.8. Bronchoalveolar Lavage (BAL) Cell Morphology
2.9. Statistics
3. Results
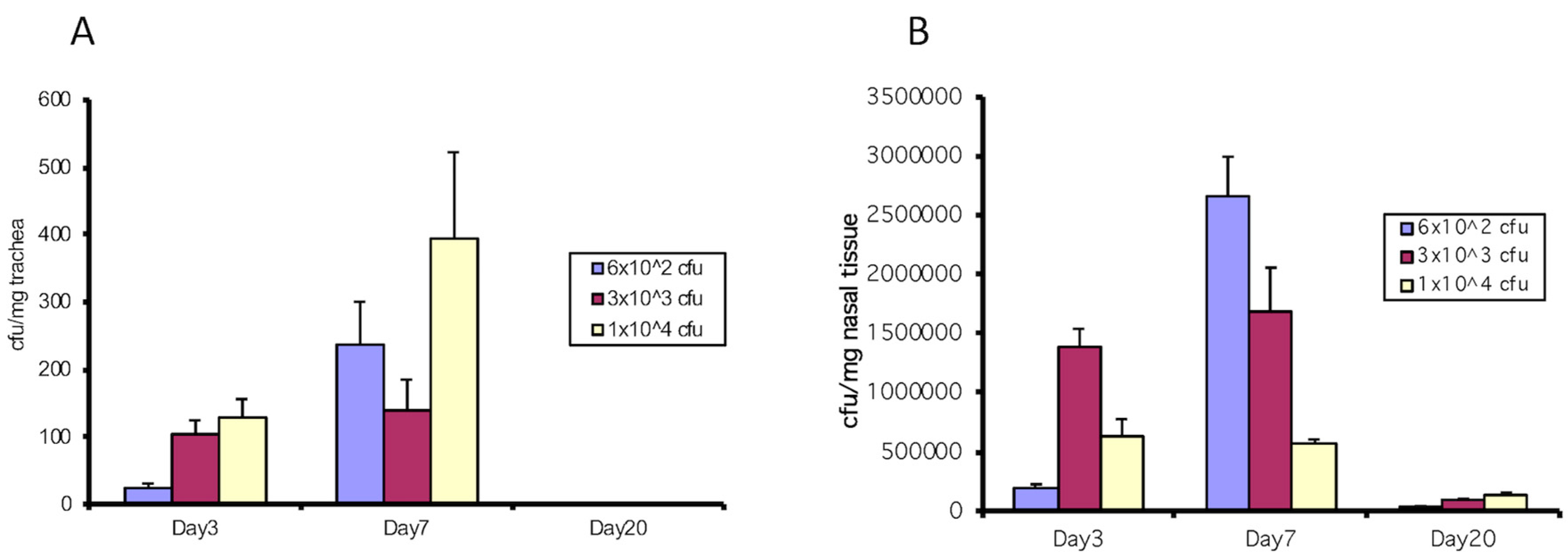
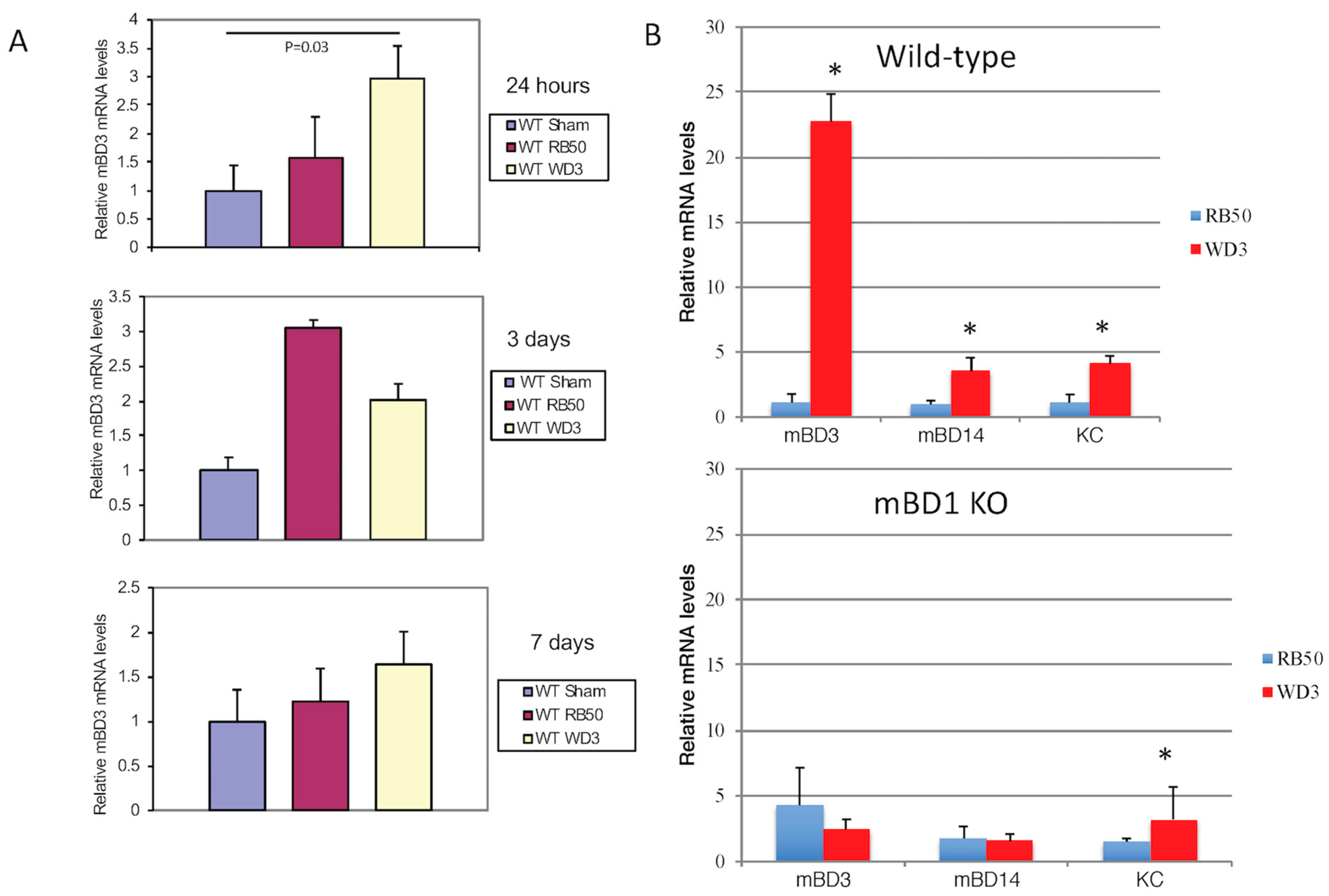
3.1. The Type III Secretion System of B. bronchiseptica Inhibits mBD-3 Expression in Mouse Trachea
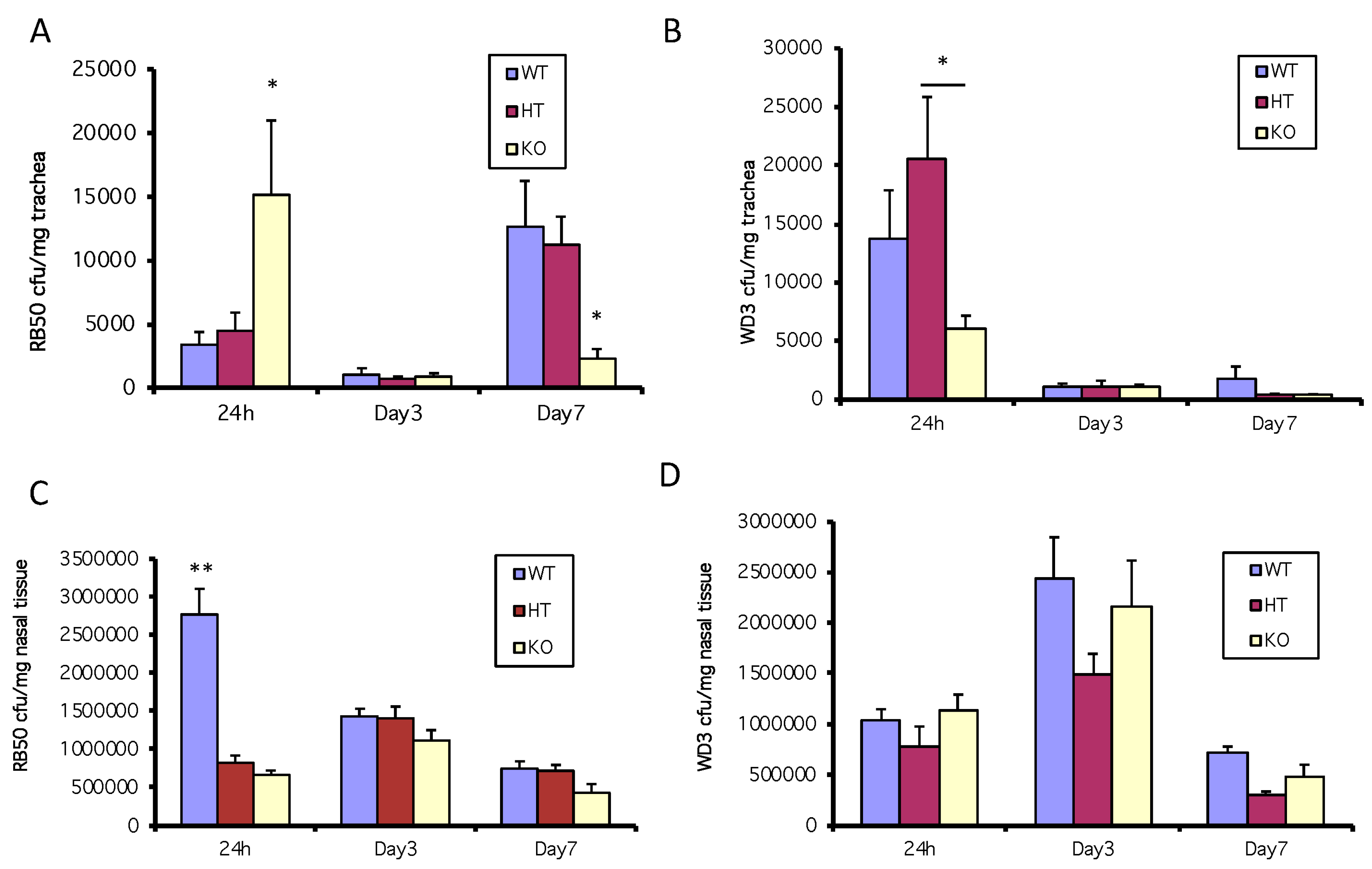
3.2. A Deficiency in mBD-1 Results in an Increased Colonization by Wild-type B. bronchiseptica
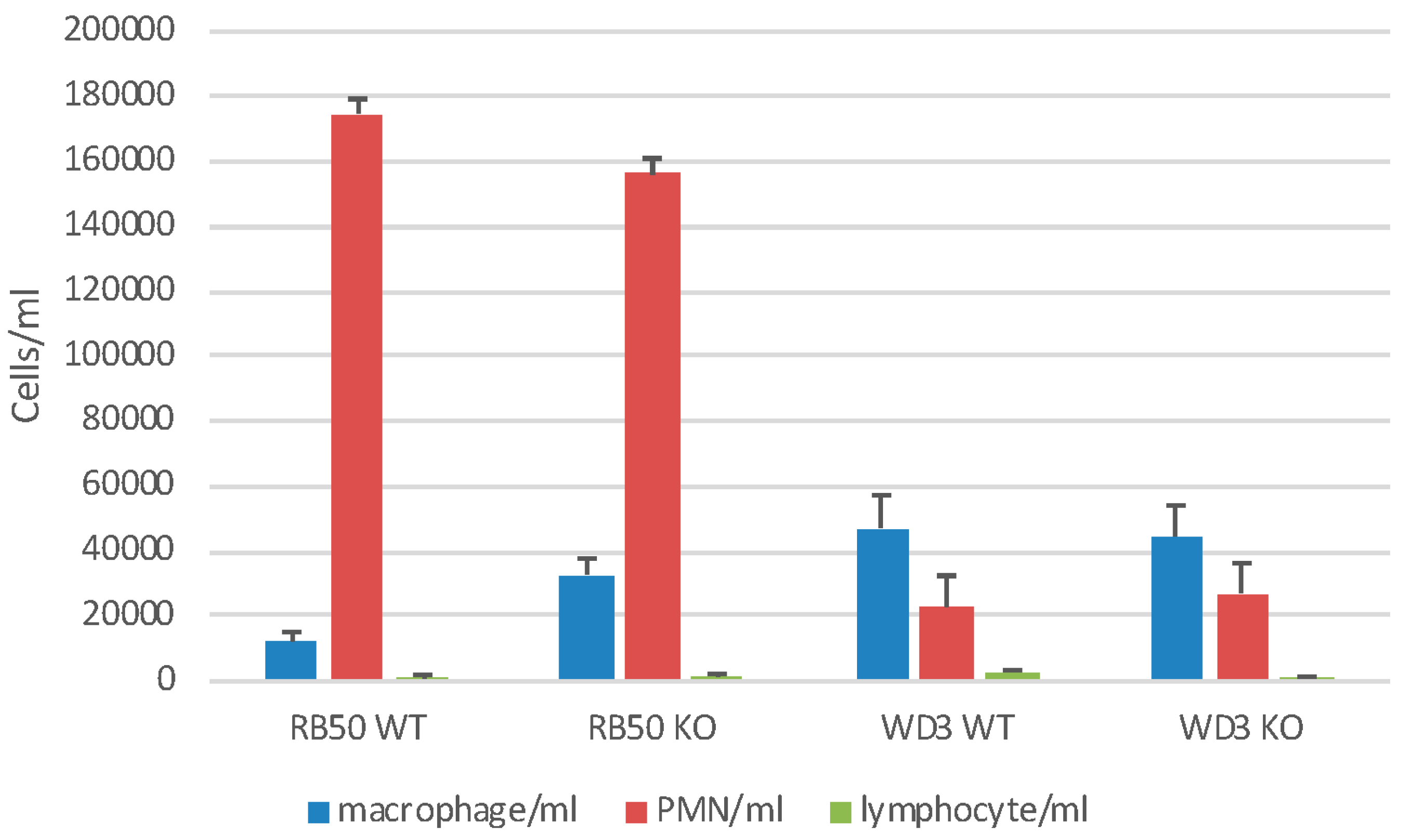
3.3. mBD1-Deficient Mice have a Reduced Neutrophil Accumulation in the Trachea in Response to Bacterial Infection
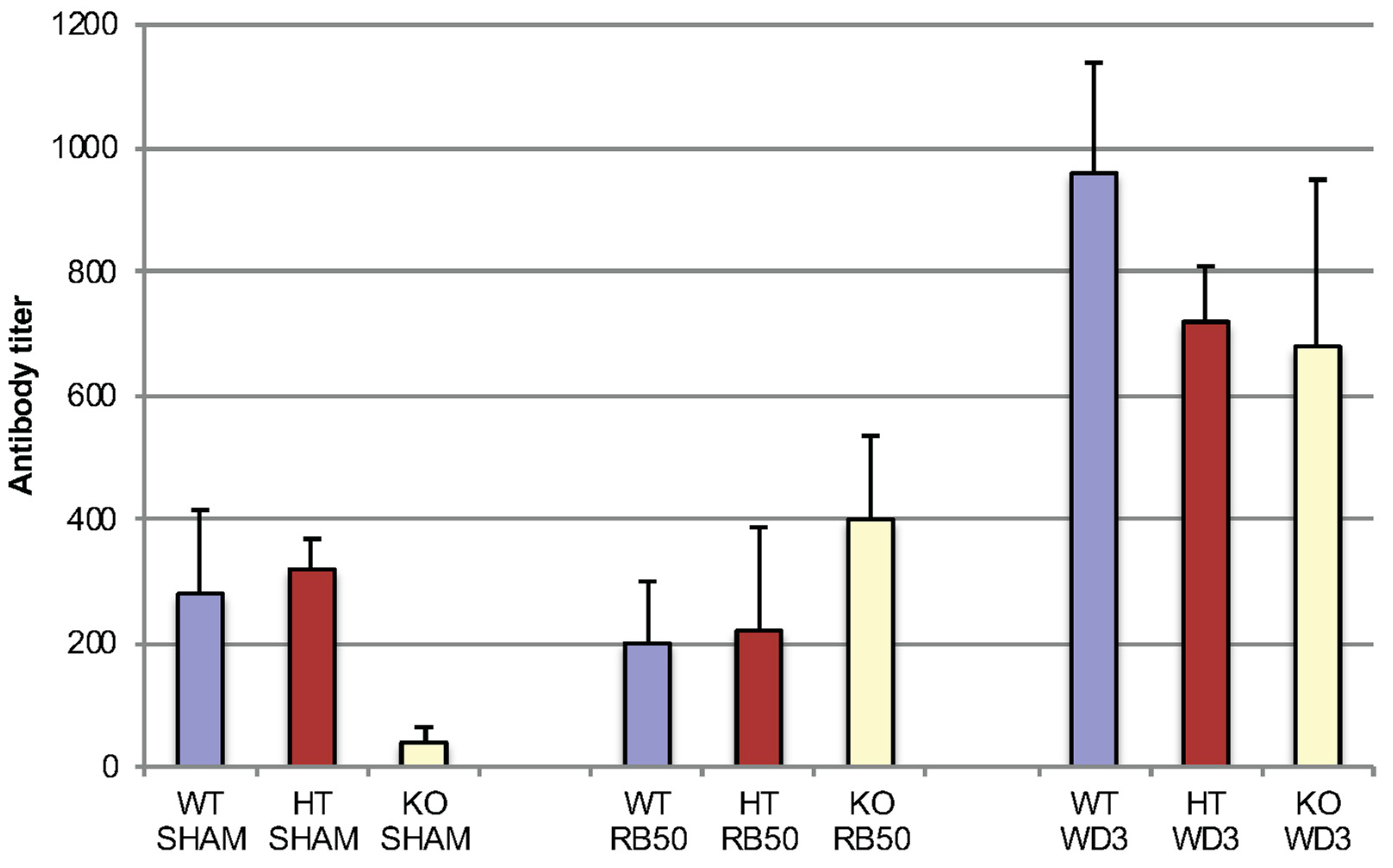
3.4. Early Specific Antibody Formation is not Affected by β-defensin-1
3.5. Variable Induction of β-defensins in Tracheal Epithelial Cells In Vitro
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Diamond, G.; Legarda, D.; Ryan, L.K. The innate immune response of the respiratory epithelium. Immunol. Rev. 2000, 173, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Beckloff, N.; Ryan, L.K. Host defense peptides in the oral cavity and the lung: Similarities and differences. J. Dent. Res. 2008, 87, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, G.; Jones, D.E.; Bevins, C.L. Airway epithelial cells are the site of expression of a mammalian antimicrobial peptide gene. Proc. Nat. Acad. Sci. USA 1993, 90, 4596–4600. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Kaiser, V.; Rhodes, J.; Russell, J.P.; Bevins, C.L. Transcriptional regulation of β-defensin gene expression in tracheal epithelial cells. Infect. Immun. 2000, 68, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.P.; Diamond, G.; Tarver, A.P.; Scanlin, T.F.; Bevins, C.L. Coordinate induction of two antibiotic genes in tracheal epithelial cells exposed to the inflammatory mediators lipopolysaccharide and tumor necrosis factor alpha. Infect. Immun. 1996, 64, 1565–1568. [Google Scholar] [PubMed]
- McCray, P.B.; Bentley, L. Human airway epithelia express a beta-defensin. Am. J. Respir. Cell Mol. Biol. 1997, 16, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.K.; Jia, H.P.; Wiles, K.; Hesselberth, J.; Liu, L.; Conway, B.A.; Greenberg, E.P.; Valore, E.V.; Welsh, M.J.; Ganz, T.; et al. Production of β-defensins by human airway epithelia. Proc. Natl. Acad. Sci. USA 1998, 95, 14961–14966. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Wang, X.; Meegalla, R.L.; Wattler, S.; Weiner, D.J.; Nehls, M.C.; Wilson, J.M. Mouse β-defensin 3 is an inducible antimicrobial peptide expressed in the epithelia of multiple organs. Infect. Immun. 1999, 67, 3542–3547. [Google Scholar] [PubMed]
- Harder, J.; Meyer-Hoffert, U.; Teran, L.M.; Schwichtenberg, L.; Bartels, J.; Maune, S.; Schroder, J.M. Mucoid Pseudomonas aeruginosa, TNF-α and IL-1β but not IL-6, induce human β-defensin-2 in respiratory epithelia. Am. J. Respir. Cell Mol. Biol. 2000, 22, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Caverly, J.M.; Diamond, G.; Gallup, J.M.; Brogden, K.A.; Dixon, R.A.; Ackermann, M.R. Coordinated expression of tracheal antimicrobial peptide and inflammatory-response elements in the lungs of neonatal calves with acute bacterial pneumonia. Infect. Immun. 2003, 71, 2950–2955. [Google Scholar] [CrossRef] [PubMed]
- Legarda, D.; Klein-Patel, M.E.; Yim, S.; Yuk, M.H.; Diamond, G. Suppression of NF-kappaB-mediated β-defensin gene expression in the mammalian airway by the Bordetella type III secretion system. Cell Microbiol. 2005, 7, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Santiago, B.; Schwander, S.K.; Sarabia, C.; Diamond, G.; Klein-Patel, M.E.; Hernandez-Pando, R.; Ellner, J.J.; Sada, E. Human β-defensin 2 is expressed and associated with Mycobacterium tuberculosis during infection of human alveolar epithelial cells. Infect. Immun. 2005, 73, 4505–4511. [Google Scholar] [CrossRef] [PubMed]
- Moranta, D.; Regueiro, V.; March, C.; Llobet, E.; Margareto, J.; Larrarte, E.; Larrate, E.; Garmendia, J.; Bengoechea, J.A. Klebsiella pneumoniae capsule polysaccharide impedes the expression of β-defensins by airway epithelial cells. Infect. Immun. 2010, 78, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Scharf, S.; Zahlten, J.; Szymanski, K.; Hippenstiel, S.; Suttorp, N.; N’Guessan, P.D. Streptococcus pneumoniae induces human β-defensin-2 and -3 in human lung epithelium. Exp. Lung Res. 2012, 38, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Becknell, B.; Spencer, J.D.; Carpenter, A.R.; Chen, X.; Singh, A.; Ploeger, S.; Kline, J.; Ellsworth, P.; Li, B.; Proksch, E.; et al. Expression and antimicrobial function of beta-defensin 1 in the lower urinary tract. PLoS ONE 2013, 8, e77714. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Anderson, G.M.; Stolzenberg, E.D.; Kari, U.P.; Zasloff, M.; Wilson, J.M. Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 1997, 88, 553–560. [Google Scholar] [CrossRef]
- Liu, L.; Wang, L.; Jia, H.P.; Zhao, C.; Heng, H.H.; Schutte, B.C.; McCray, P.B., Jr.; Ganz, T. Structure and mapping of the human β-defensin hBD-2 gene and its expression at sites of inflammation. Gene 1998, 222, 237–244. [Google Scholar] [CrossRef]
- Tecle, T.; Tripathi, S.; Hartshorn, K.L. Review: Defensins and cathelicidins in lung immunity. Innate Immun. 2010, 16, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, P.S.; Amatngalim, G.D.; van der Does, A.M.; Taube, C. Antimicrobial peptides and innate lung defenses: Role in infectious and noninfectious lung diseases and therapeutic applications. Chest 2016, 149, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Weiner, D.J.; Lysenko, E.; Bals, R.; Weiser, J.N.; Wilson, J.M. Beta-defensin 1 contributes to pulmonary innate immunity in mice. Infect. Immun. 2002, 70, 3068–3072. [Google Scholar] [CrossRef] [PubMed]
- Morrison, G.; Kilanowski, F.; Davidson, D.; Dorin, J. Characterization of the mouse beta defensin 1, defb1, mutant mouse model. Infect. Immun. 2002, 70, 3053–3060. [Google Scholar] [CrossRef] [PubMed]
- Semple, C.A.; Gautier, P.; Taylor, K.; Dorin, J.R. The changing of the guard: Molecular diversity and rapid evolution of beta-defensins. Mol. Divers. 2006, 10, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Yuk, M.H.; Harvill, E.T.; Miller, J.F. The BvgAS virulence control system regulates type III secretion in Bordetella bronchiseptica. Mol. Microbiol. 1998, 28, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Yuk, M.H.; Harvill, E.T.; Cotter, P.A.; Miller, J.F. Modulation of host immune responses, induction of apoptosis and inhibition of NF-κB activation by the Bordetella type III secretion system. Mol. Microbiol. 2000, 35, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, S.; Miller, J.F.; Cotter, P.A. Role of Bordetella bronchiseptica fimbriae in tracheal colonization and development of a humoral immune response. Infect. Immun. 2000, 68, 2024–2033. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.K.; Diamond, G. Modulation of human β-defensin-1 production by viruses. Viruses 2017, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Bals, R.; Goldman, M.J.; Wilson, J.M. Mouse β-defensin 1 is a salt-sensitive antimicrobial peptide present in epithelia of the lung and urogenital tract. Infect. Immun. 1998, 66, 1225–1232. [Google Scholar] [PubMed]
- Semple, F.; Webb, S.; Li, H.N.; Patel, H.B.; Perretti, M.; Jackson, I.J.; Gray, M.; Davidson, D.J.; Dorin, J.R. Human β-defensin 3 has immunosuppressive activity in vitro and in vivo. Eur. J. Immunol. 2010, 40, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Diamond, G.; Zasloff, M.; Eck, H.; Brasseur, M.; Maloy, W.L.; Bevins, C.L. Tracheal antimicrobial peptide, a novel cysteine-rich peptide from mammalian tracheal mucosa: Peptide isolation and cloning of a cDNA. Proc. Natl. Acad. Sci. USA 1991, 88, 3952–3956. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.A.; Miller, J.F. BvgAS-mediated signal transduction: Analysis of phase-locked regulatory mutants of Bordetella bronchiseptica in a rabbit model. Infect. Immun. 1994, 62, 3381–3390. [Google Scholar] [PubMed]
- You, Y.; Richer, E.J.; Huang, T.; Brody, S.L. Growth and differentiation of mouse tracheal epithelial cells: Selection of a proliferative population. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 283, L1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, D.N.; Kirimanjeswara, G.S.; Harvill, E.T. Clearance of Bordetella parapertussis from the lower respiratory tract requires humoral and cellular immunity. Infect. Immun. 2005, 73, 6508–6513. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schroeder, J.M.; Wang, J.M.; Howard, O.M.; et al. β-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Ogawa, H.; Nagaoka, I. Human β-defensin-2 functions as a chemotactic agent for tumour necrosis factor-α-treated human neutrophils. Immunology 2004, 111, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef] [PubMed]
- Braida, L.; Boniotto, M.; Pontillo, A.; Tovo, P.A.; Amoroso, A.; Crovella, S. A single-nucleotide polymorphism in the human β-defensin 1 gene is associated with HIV-1 infection in italian children. AIDS 2004, 18, 1598–1600. [Google Scholar] [CrossRef] [PubMed]
- Jurevic, R.J.; Bai, M.; Chadwick, R.B.; White, T.C.; Dale, B.A. Single-nucleotide polymorphisms (SNPs) in human β-defensin 1: High-throughput SNP assays and association with Candida carriage in type I diabetics and nondiabetic controls. J. Clin. Microbiol. 2003, 41, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, I.; Hasegawa, K.; Nakata, K.; Yasuda, K.; Tokunaga, K.; Keicho, N. Genetic variants of human β-defensin-1 and chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2002, 291, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Milanese, M.; Segat, L.; Pontillo, A.; Arraes, L.C.; de Lima Filho, J.L.; Crovella, S. DEFB1 gene polymorphisms and increased risk of HIV-1 infection in Brazilian children. AIDS 2006, 20, 1673–1675. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, A.; Famili, P.; Vieira, A.R. The antimicrobial peptide DEFB1 is associated with caries. J. Dent. Res. 2010, 89, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Hollox, E.J.; Barber, J.C.; Brookes, A.J.; Armour, J.A. Defensins and the dynamic genome: What we can learn from structural variation at human chromosome band 8p23.1. Genome Res. 2008, 18, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Bakar, S.; Hollox, E.J.; Armour, J.A. Allelic recombination between distinct genomic locations generates copy number diversity in human beta-defensins. Proc. Natl. Acad. Sci. USA 2009, 106, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Ouellette, A.J.; Satchell, D.P.; Ayabe, T.; López-Boado, Y.S.; Stratman, J.L.; Hultgren, S.J.; Matrisian, L.M.; Parks, W.C. Regulation of intestinal α-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999, 286, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Beckloff, N.; Diamond, G. Computational analysis suggests β-defensins are processed to mature peptides by signal peptidase. Protein Pept. Lett. 2008, 15, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Amid, C.; Rehaume, L.M.; Brown, K.L.; Gilbert, J.G.; Dougan, G.; Hancock, R.E.; Harrow, J.L. Manual annotation and analysis of the defensin gene cluster in the C57Bl/6J mouse reference genome. BMC Genomics 2009, 10, 606. [Google Scholar] [CrossRef] [PubMed]
- Kolar, S.S.; Baidouri, H.; Hanlon, S.; McDermott, A.M. Protective role of murine β-defensins 3 and 4 and cathelin-related antimicrobial peptide in Fusarium solani keratitis. Infect. Immun. 2013, 81, 2669–2677. [Google Scholar] [CrossRef] [PubMed]
- Ryan, L.K.; Dai, J.; Yin, Z.; Megjugorac, N.; Uhlhorn, V.; Yim, S.; Schwartz, K.D.; Abrahams, J.M.; Diamond, G.; Fitzgerald-Bocarsly, P. Modulation of human β-defensin-1 (hBD-1) in plasmacytoid dendritic cells (PDC), monocytes and epithelial cells by influenza virus, Herpes Simplex virus and Sendai virus and its possible role in innate immunity. J. Leukoc. Biol. 2011, 90, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, M.S.; Lebreton, F.; Van Tyne, D. Dual defensin strategy for targeting Enterococcus faecalis. Proc Natl. Acad. Sci. USA 2013, 110, 19980–19981. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, M.H.; Diamond, G.; Ryan, L.K. Synergy of antimicrobial peptides. In Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies, 2nd ed.; Wang, G., Ed.; CAB International: Boston, MA, USA, 2017; pp. 118–201. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequences | Product Size |
|---|---|---|
| mBD-3 | Forward: GCATTTCTCCTGGTGCTGCTGTCTC | 128 bp |
| Reverse: CTGCCAATCTGACGAGTGTTGCC | ||
| mBD-4 | Forward: CTCACTTGCAGCCTTTACC | 202 bp |
| Reverse: CATGGAGGAGCAAATTCTGG | ||
| mBD-14 | Forward: CCTCATCTTGTTCTTGGTGCCTGCTG | 102 bp |
| Reverse: TTAAGTACAGCACACCGGCCACCTCT | ||
| KC | Forward: ATGGCTGGGATTCACCTC | 166 bp |
| Reverse: CACCTTTTAGCATCTTTTGGA | ||
| β-actin | Forward: ATCCTGAAAGACCTCTATGC | 287 bp |
| Reverse: AACGCAGCTCAGTAACAGTC | ||
| mβ2-µglobulin | Forward: CTCCGTGGCCTTAGCTGTG | 69 bp |
| Reverse: TTTGGAGTACGCTGGATAGCCT |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryan, L.K.; Wu, J.; Schwartz, K.; Yim, S.; Diamond, G. β-Defensins Coordinate In Vivo to Inhibit Bacterial Infections of the Trachea. Vaccines 2018, 6, 57. https://doi.org/10.3390/vaccines6030057
Ryan LK, Wu J, Schwartz K, Yim S, Diamond G. β-Defensins Coordinate In Vivo to Inhibit Bacterial Infections of the Trachea. Vaccines. 2018; 6(3):57. https://doi.org/10.3390/vaccines6030057
Chicago/Turabian StyleRyan, Lisa Kathleen, Jichuan Wu, Kyell Schwartz, Sunghan Yim, and Gill Diamond. 2018. "β-Defensins Coordinate In Vivo to Inhibit Bacterial Infections of the Trachea" Vaccines 6, no. 3: 57. https://doi.org/10.3390/vaccines6030057
APA StyleRyan, L. K., Wu, J., Schwartz, K., Yim, S., & Diamond, G. (2018). β-Defensins Coordinate In Vivo to Inhibit Bacterial Infections of the Trachea. Vaccines, 6(3), 57. https://doi.org/10.3390/vaccines6030057