From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies


Abstract
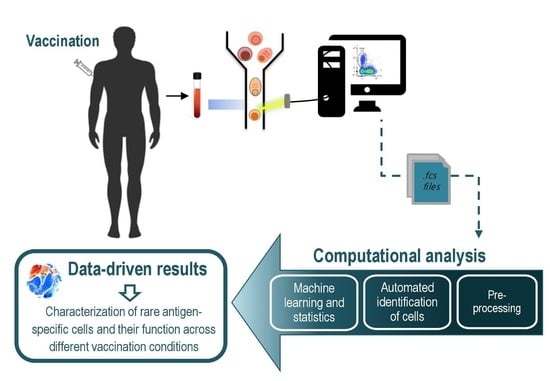
:
1. Introduction
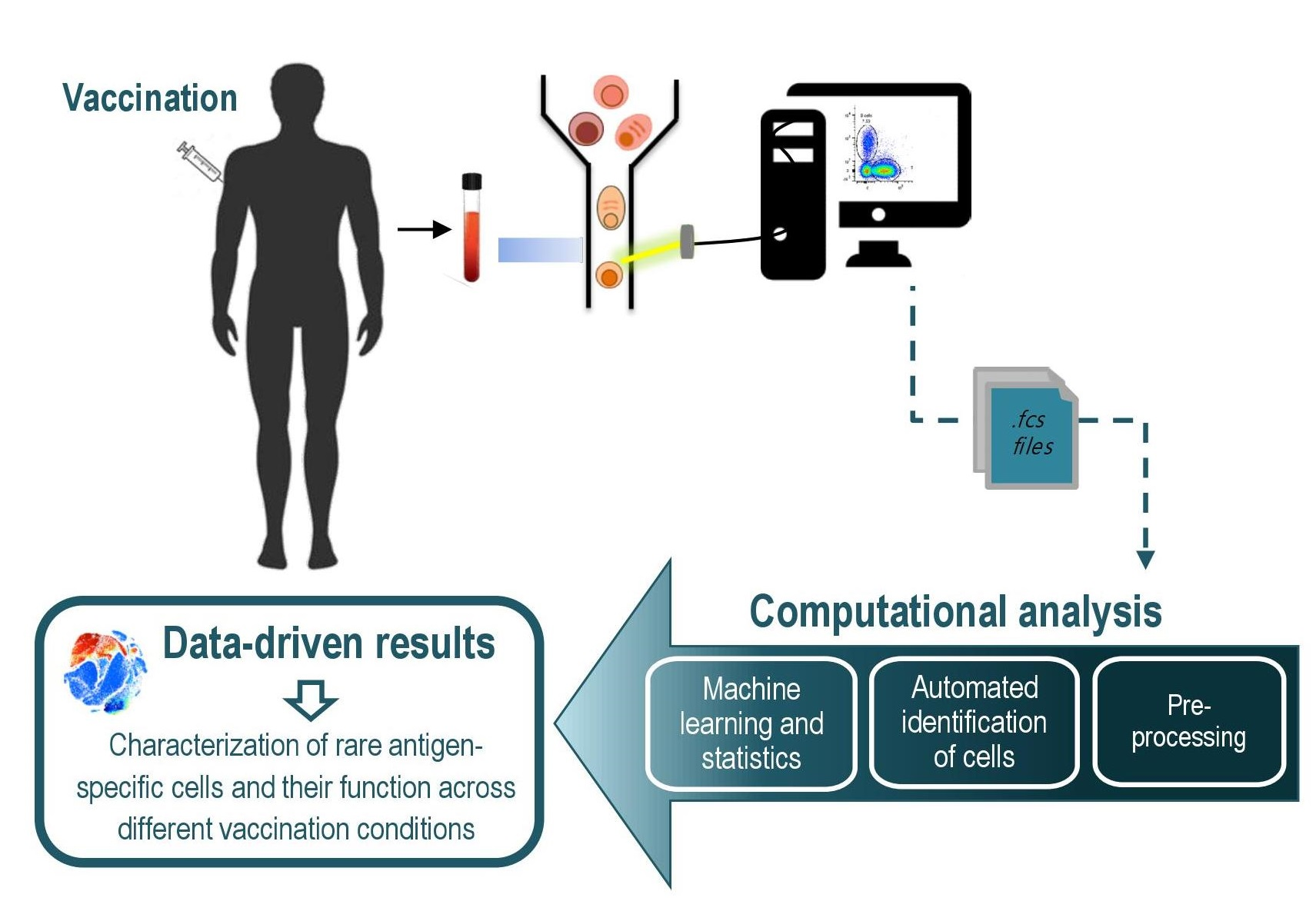
2. Automated Cytometry Data Analysis Workflow
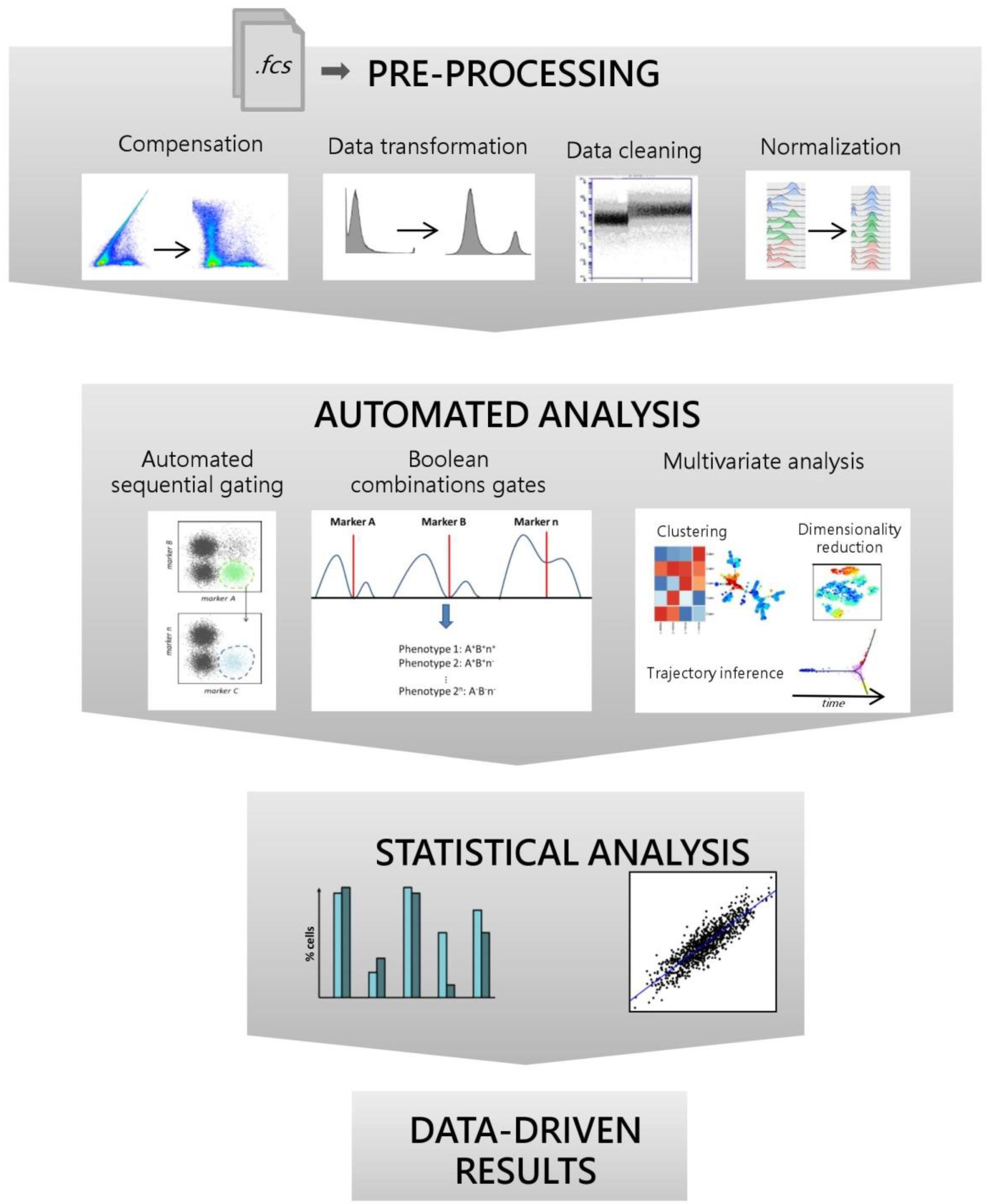
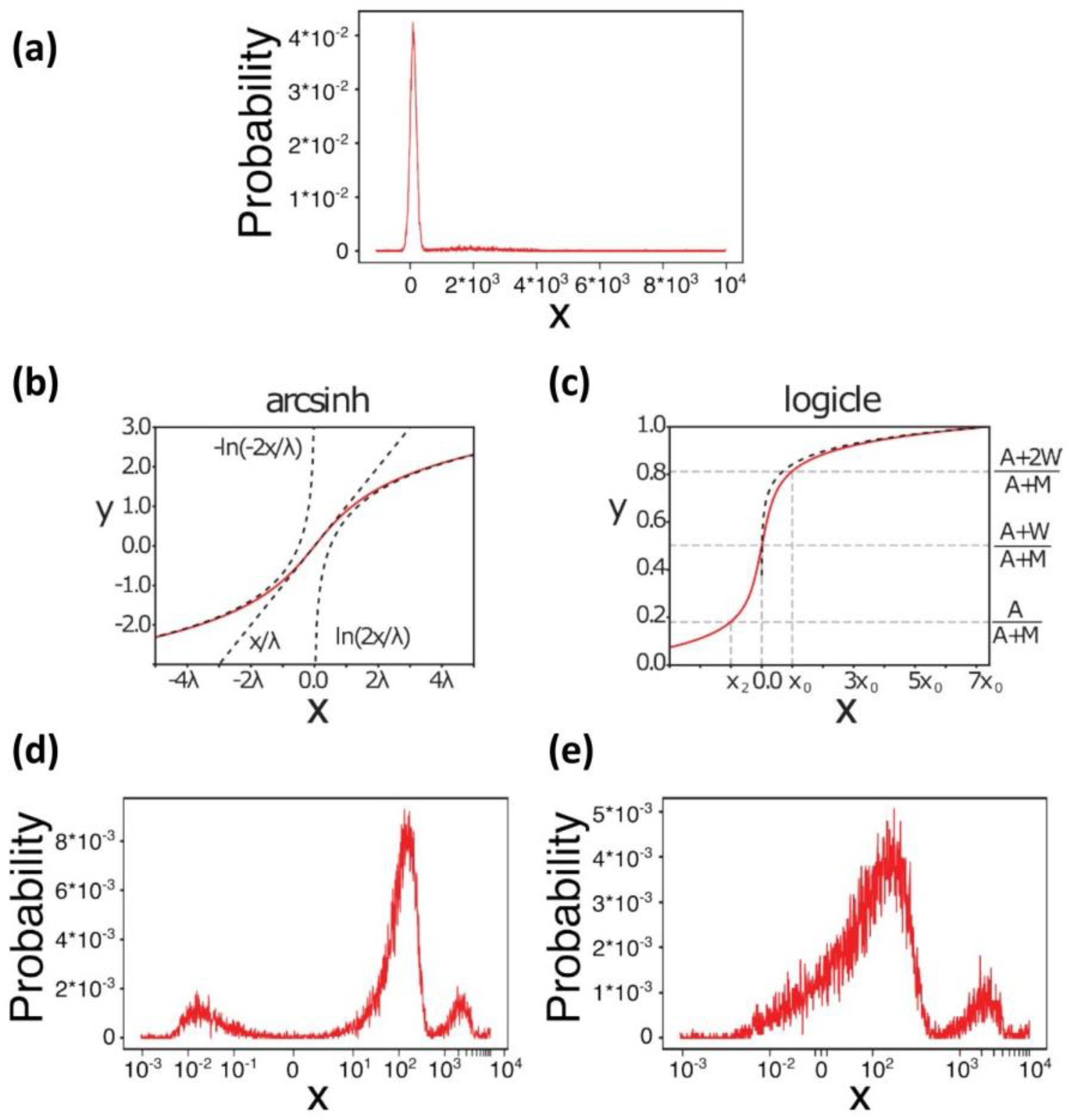
3. Data Pre-Processing
4. Automated Data Analysis
4.1. Automated Sequential Gating
4.2. Boolean Combination Gates
4.3. Multivariate Approach
4.3.1. Clustering
4.3.2. Dimensionality Reduction
4.3.3. Trajectory Inference
4.3.4. Multivariate Analysis Settings
5. Interpretation of the Results
6. Impact of Automated Analysis in the Knowledge of Biological Processes
7. Flow Cytometry in Vaccine Studies and the Advantages of Computational Analysis
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Adan, A.; Alizada, G.; Kiraz, Y.; Baran, Y.; Nalbant, A. Flow cytometry: Basic principles and applications. Crit. Rev. Biotechnol. 2017, 37, 163–176. [Google Scholar] [CrossRef]
- Nomura, L.; Maino, V.C.; Maecker, H.T. Standardization and optimization of multiparameter intracellular cytokine staining. Cytom. Part A 2008, 73, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.T.; McCoy, J.P.; Nussenblatt, R. Standardizing immunophenotyping for the Human Immunology Project. Nat. Rev. Immunol. 2012, 12, 191–200. [Google Scholar]
- Gouttefangeas, C.; Chan, C.; Attig, S.; Køllgaard, T.T.; Rammensee, H.-G.; Stevanović, S.; Wernet, D.; thor Straten, P.; Welters, M.J.P.; Ottensmeier, C.; et al. Data analysis as a source of variability of the HLA-peptide multimer assay: From manual gating to automated recognition of cell clusters. Cancer Immunol. Immunother. 2015, 64, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Irish, J.M. Beyond the age of cellular discovery. Nat. Immunol. 2014, 15, 1095–1097. [Google Scholar] [CrossRef]
- Aghaeepour, N.; Finak, G.; FlowCAP Consortium; DREAM Consortium; Hoos, H.; Mosmann, T.R.; Brinkman, R.; Gottardo, R.; Scheuermann, R.H. Critical assessment of automated flow cytometry data analysis techniques. Nat. Methods 2013, 10, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Conrad, V.K.; Dubay, C.J.; Malek, M.; Brinkman, R.R.; Koguchi, Y.; Redmond, W.L. Implementation and Validation of an Automated Flow Cytometry Analysis Pipeline for Human Immune Profiling. Cytom. Part A 2019, 95, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Huber, W.; Carey, V.J.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115. [Google Scholar] [CrossRef]
- Hahne, F.; LeMeur, N.; Brinkman, R.R.; Ellis, B.; Haaland, P.; Sarkar, D.; Spidlen, J.; Strain, E.; Gentleman, R. flowCore: A Bioconductor package for high throughput flow cytometry. BMC Bioinform. 2009, 10, 106. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Khodabakhshi, A.H.; Bashashati, A.; Wong, C.-J.; Gascoyne, R.D.; Weng, A.P.; Seyfert-Margolis, V.; Bourcier, K.; Asare, A.; Lumley, T.; et al. Per-channel basis normalization methods for flow cytometry data. Cytom. Part A 2010, 77, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Fletez-Brant, K.; Špidlen, J.; Brinkman, R.R.; Roederer, M.; Chattopadhyay, P.K. flowClean: Automated identification and removal of fluorescence anomalies in flow cytometry data. Cytom. Part A 2016, 89, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Monaco, G.; Chen, H.; Poidinger, M.; Chen, J.; de Magalhães, J.P.; Larbi, A. flowAI: Automatic and interactive anomaly discerning tools for flow cytometry data. Bioinformatics 2016, 32, 2473–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowell, H.L.; Zanotelli, V.R.T.; Chevrier, S.; Robinson, M.D.; Bodenmiller, B. CATALYST: Cytometry dATa anALYSis Tools. Available online: https://bioconductor.org/packages/release/bioc/html/CATALYST.html (accessed on 19 March 2020).
- Van Gassen, S.; Gaudilliere, B.; Angst, M.S.; Saeys, Y.; Aghaeepour, N. CytoNorm: A Normalization Algorithm for Cytometry Data. Cytom. Part A 2019, 97, 268–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, M.; Taghiyar, M.J.; Chong, L.; Finak, G.; Gottardo, R.; Brinkman, R.R. flowDensity: Reproducing manual gating of flow cytometry data by automated density-based cell population identification. Bioinformatics 2015, 31, 606–607. [Google Scholar] [CrossRef] [Green Version]
- Finak, G.; Frelinger, J.; Jiang, W.; Newell, E.W.; Ramey, J.; Davis, M.M.; Kalams, S.A.; De Rosa, S.C.; Gottardo, R. OpenCyto: An open source infrastructure for scalable, robust, reproducible, and automated, end-to-end flow cytometry data analysis. PLoS Comput. Biol. 2014, 10, e1003806. [Google Scholar] [CrossRef] [Green Version]
- Meehan, S.; Walther, G.; Moore, W.; Orlova, D.; Meehan, C.; Parks, D.; Ghosn, E.; Philips, M.; Mitsunaga, E.; Waters, J.; et al. AutoGate: Automating analysis of flow cytometry data. Immunol. Res. 2014, 58, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Commenges, D.; Alkhassim, C.; Gottardo, R.; Hejblum, B.; Thiébaut, R. cytometree: A binary tree algorithm for automatic gating in cytometry analysis. Cytom. Part A 2018, 93, 1132–1140. [Google Scholar] [CrossRef] [Green Version]
- Meehan, S.; Kolyagin, G.A.; Parks, D.; Youngyunpipatkul, J.; Herzenberg, L.A.; Walther, G.; Ghosn, E.E.B.; Orlova, D.Y. Automated subset identification and characterization pipeline for multidimensional flow and mass cytometry data clustering and visualization. Commun. Biol. 2019, 2, 229. [Google Scholar] [CrossRef] [Green Version]
- Aghaeepour, N.; Chattopadhyay, P.K.; Ganesan, A.; O’Neill, K.; Zare, H.; Jalali, A.; Hoos, H.H.; Roederer, M.; Brinkman, R.R. Early immunologic correlates of HIV protection can be identified from computational analysis of complex multivariate T-cell flow cytometry assays. Bioinformatics 2012, 28, 1009–1016. [Google Scholar] [CrossRef]
- Van Gassen, S.; Vens, C.; Dhaene, T.; Lambrecht, B.N.; Saeys, Y. FloReMi: Flow density survival regression using minimal feature redundancy. Cytom. Part A 2016, 89, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Aghaeepour, N.; Jalali, A.; O’Neill, K.; Chattopadhyay, P.K.; Roederer, M.; Hoos, H.H.; Brinkman, R.R. RchyOptimyx: Cellular hierarchy optimization for flow cytometry. Cytom. Part A 2012, 81, 1022–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghaeepour, N.; Nikolic, R.; Hoos, H.H.; Brinkman, R.R. Rapid cell population identification in flow cytometry data. Cytom. Part A 2011, 79, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Qiu, P.; Simonds, E.F.; Bendall, S.C.; Gibbs, K.D.; Bruggner, R.V.; Linderman, M.D.; Sachs, K.; Nolan, G.P.; Plevritis, S.K. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat. Biotechnol. 2011, 29, 886–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cron, A.; Gouttefangeas, C.; Frelinger, J.; Lin, L.; Singh, S.K.; Britten, C.M.; Welters, M.J.P.; van der Burg, S.H.; West, M.; Chan, C. Hierarchical Modeling for Rare Event Detection and Cell Subset Alignment across Flow Cytometry Samples. PLOS Comput. Biol. 2013, 9, e1003130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruggner, R.V.; Bodenmiller, B.; Dill, D.L.; Tibshirani, R.J.; Nolan, G.P. Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E2770–E2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gassen, S.; Callebaut, B.; Van Helden, M.J.; Lambrecht, B.N.; Demeester, P.; Dhaene, T.; Saeys, Y. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytom. Part A 2015, 87, 636–645. [Google Scholar] [CrossRef]
- Samusik, N.; Good, Z.; Spitzer, M.H.; Davis, K.L.; Nolan, G.P. Automated mapping of phenotype space with single-cell data. Nat. Methods 2016, 13, 493–496. [Google Scholar] [CrossRef] [Green Version]
- Lo, K.; Hahne, F.; Brinkman, R.R.; Gottardo, R. flowClust: A Bioconductor package for automated gating of flow cytometry data. BMC Bioinform. 2009, 10, 145. [Google Scholar] [CrossRef] [Green Version]
- Sörensen, T.; Baumgart, S.; Durek, P.; Grützkau, A.; Häupl, T. immunoClust--An automated analysis pipeline for the identification of immunophenotypic signatures in high-dimensional cytometric datasets. Cytom. Part A 2015, 87, 603–615. [Google Scholar] [CrossRef]
- Mosmann, T.R.; Naim, I.; Rebhahn, J.; Datta, S.; Cavenaugh, J.S.; Weaver, J.M.; Sharma, G. SWIFT-scalable clustering for automated identification of rare cell populations in large, high-dimensional flow cytometry datasets, part 2: Biological evaluation. Cytom. Part A 2014, 85, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Wei, C.; Eun-Hyung Lee, F.; Campbell, J.; Halliley, J.; Lee, J.A.; Cai, J.; Kong, Y.M.; Sadat, E.; Thomson, E.; et al. Elucidation of seventeen human peripheral blood B-cell subsets and quantification of the tetanus response using a density-based method for the automated identification of cell populations in multidimensional flow cytometry data. Cytom. B Clin. Cytom. 2010, 78 (Suppl. 1), S69–S82. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Sealfon, S.C. flowPeaks: A fast unsupervised clustering for flow cytometry data via K-means and density peak finding. Bioinformatics 2012, 28, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Laio, A. Clustering by fast search and find of density peaks. Science 2014, 344, 1492–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, J.H.; Simonds, E.F.; Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Litvin, O.; Fienberg, H.G.; Jager, A.; Zunder, E.R.; et al. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 2015, 162, 184–197. [Google Scholar] [CrossRef] [Green Version]
- van der Maaten, L.; Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Shekhar, K.; Brodin, P.; Davis, M.M.; Chakraborty, A.K. Automatic Classification of Cellular Expression by Nonlinear Stochastic Embedding (ACCENSE). Proc. Natl. Acad. Sci. USA 2014, 111, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Amir, E.D.; Davis, K.L.; Tadmor, M.D.; Simonds, E.F.; Levine, J.H.; Bendall, S.C.; Shenfeld, D.K.; Krishnaswamy, S.; Nolan, G.P.; Pe’er, D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013, 31, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Kratochvíl, M.; Koladiya, A.; Balounova, J.; Novosadova, V.; Fišer, K.; Sedlacek, R.; Vondrášek, J.; Drbal, K. Rapid single-cell cytometry data visualization with EmbedSOM. bioRxiv 2018, 496869. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Saul, N.; Großberger, L. UMAP: Uniform Manifold Approximation and Projection. JOSS 2018, 3, 861. [Google Scholar] [CrossRef]
- Angerer, P.; Haghverdi, L.; Büttner, M.; Theis, F.J.; Marr, C.; Buettner, F. destiny: Diffusion maps for large-scale single-cell data in R. Bioinformatics 2016, 32, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- Linderman, G.C.; Rachh, M.; Hoskins, J.G.; Steinerberger, S.; Kluger, Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat. Methods 2019, 16, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Davis, K.L.; Amir, E.D.; Tadmor, M.D.; Simonds, E.F.; Chen, T.J.; Shenfeld, D.K.; Nolan, G.P.; Pe’er, D. Single-Cell Trajectory Detection Uncovers Progression and Regulatory Coordination in Human B Cell Development. Cell 2014, 157, 714–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Setty, M.; Tadmor, M.D.; Reich-Zeliger, S.; Angel, O.; Salame, T.M.; Kathail, P.; Choi, K.; Bendall, S.; Friedman, N.; Pe’er, D. Wishbone identifies bifurcating developmental trajectories from single-cell data. Nat. Biotechnol. 2016, 34, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Moon, K.R.; van Dijk, D.; Wang, Z.; Gigante, S.; Burkhardt, D.B.; Chen, W.S.; Yim, K.; van den Elzen, A.; Hirn, M.J.; Coifman, R.R.; et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 2019, 37, 1482–1492. [Google Scholar] [CrossRef]
- Bioconductor-Home. Available online: https://bioconductor.org/ (accessed on 19 March 2020).
- FlowJo Exchange. Available online: https://www.flowjo.com/exchange/#/ (accessed on 19 March 2020).
- Cytobank. Available online: https://www.cytobank.org/ (accessed on 19 March 2020).
- ImmPort Shared Data. Available online: https://www.immport.org/shared/home (accessed on 19 March 2020).
- Finak, G.; Perez, J.-M.; Weng, A.; Gottardo, R. Optimizing transformations for automated, high throughput analysis of flow cytometry data. BMC Bioinform. 2010, 11, 546. [Google Scholar] [CrossRef] [Green Version]
- Finck, R.; Simonds, E.F.; Jager, A.; Krishnaswamy, S.; Sachs, K.; Fantl, W.; Pe’er, D.; Nolan, G.P.; Bendall, S.C. Normalization of mass cytometry data with bead standards. Cytom. Part A 2013, 83, 483–494. [Google Scholar] [CrossRef]
- Zunder, E.R.; Finck, R.; Behbehani, G.K.; Amir, E.-A.D.; Krishnaswamy, S.; Gonzalez, V.D.; Lorang, C.G.; Bjornson, Z.; Spitzer, M.H.; Bodenmiller, B.; et al. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015, 10, 316–333. [Google Scholar] [CrossRef]
- Anchang, B.; Hart, T.D.P.; Bendall, S.C.; Qiu, P.; Bjornson, Z.; Linderman, M.; Nolan, G.P.; Plevritis, S.K. Visualization and cellular hierarchy inference of single-cell data using SPADE. Nat. Protoc. 2016, 11, 1264–1279. [Google Scholar] [CrossRef]
- Weber, L.M.; Robinson, M.D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytom. Part A 2016, 89, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, C.; Liu, M.; Stanton, R.; McGee, M.; Qian, Y.; Scheuermann, R.H. Mapping cell populations in flow cytometry data for cross-sample comparison using the Friedman-Rafsky test statistic as a distance measure. Cytom. Part A 2016, 89, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, S.; Nolfi, E.; Pettini, E.; Pastore, G.; Fiorino, F.; Pozzi, G.; Medaglini, D.; Ciabattini, A. Computational Analysis of Multiparametric Flow Cytometric Data to Dissect B Cell Subsets in Vaccine Studies. Cytom. Part A 2019, 97, 259–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coifman, R.R.; Lafon, S.; Lee, A.B.; Maggioni, M.; Nadler, B.; Warner, F.; Zucker, S.W. Geometric diffusions as a tool for harmonic analysis and structure definition of data: Diffusion maps. PNAS 2005, 102, 7426–7431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghverdi, L.; Buettner, F.; Theis, F.J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics 2015, 31, 2989–2998. [Google Scholar] [CrossRef]
- Saeys, Y.; Van Gassen, S.; Lambrecht, B.N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016, 16, 449–462. [Google Scholar] [CrossRef]
- Cannoodt, R.; Saelens, W.; Saeys, Y. Computational methods for trajectory inference from single-cell transcriptomics. Eur. J. Immunol. 2016, 46, 2496–2506. [Google Scholar] [CrossRef]
- Moon, K.R.; Stanley, J.S.; Burkhardt, D.; van Dijk, D.; Wolf, G.; Krishnaswamy, S. Manifold learning-based methods for analyzing single-cell RNA-sequencing data. Curr. Opin. Syst. Opin. 2018, 7, 36–46. [Google Scholar] [CrossRef]
- Saelens, W.; Cannoodt, R.; Todorov, H.; Saeys, Y. A comparison of single-cell trajectory inference methods. Nat. Biotechnol. 2019, 37, 547–554. [Google Scholar] [CrossRef]
- Jin, S.; MacLean, A.L.; Peng, T.; Nie, Q. scEpath: Energy landscape-based inference of transition probabilities and cellular trajectories from single-cell transcriptomic data. Bioinformatics 2018, 34, 2077–2086. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Teschendorff, A.E.; Chen, W.; Chen, L.; Li, T. Quantifying Waddington’s epigenetic landscape: A comparison of single-cell potency measures. Brief. Bioinform. 2018, 21, 248–261. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, C.B.; Olsen, L.R. Algorithmic Clustering Of Single-Cell Cytometry Data-How Unsupervised Are These Analyses Really? Cytom. Part A 2019, 97, 219–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlova, D.Y.; Herzenberg, L.A.; Walther, G. Science not art: Statistically sound methods for identifying subsets in multi-dimensional flow and mass cytometry data sets. Nat. Rev. Immunol. 2018, 18, 77. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Van Gassen, S.; Lambrecht, B. Response to Orlova et al. “Science not art: Statistically sound methods for identifying subsets in multi-dimensional flow and mass cytometry data sets”. Nat. Rev. Immunol. 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- FlowCAP—Flow Cytometry: Critical Assessment of Population Identification Methods. Available online: http://flowcap.flowsite.org/ (accessed on 19 March 2020).
- Brinkman, R.R.; Aghaeepour, N.; Finak, G.; Gottardo, R.; Mosmann, T.; Scheuermann, R.H. Automated analysis of flow cytometry data comes of age. Cytom. Part A 2016, 89, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, R.R.; Aghaeepour, N.; Finak, G.; Gottardo, R.; Mosmann, T.; Scheuermann, R.H. State-of-the-Art in the Computational Analysis of Cytometry Data. Cytom. Part A 2015, 87, 591–593. [Google Scholar] [CrossRef]
- Mittag, A.; Tarnok, A. Recent advances in cytometry applications: Preclinical, clinical, and cell biology. Methods Cell Biol. 2011, 103, 1–20. [Google Scholar]
- Coustan-Smith, E.; Song, G.; Shurtleff, S.; Yeoh, A.E.-J.; Chng, W.J.; Chen, S.P.; Rubnitz, J.E.; Pui, C.-H.; Downing, J.R.; Campana, D. Universal monitoring of minimal residual disease in acute myeloid leukemia. JCI Insight 2018, 3, 98561. [Google Scholar] [CrossRef]
- DiGiuseppe, J.A.; Tadmor, M.D.; Pe’er, D. Detection of minimal residual disease in B lymphoblastic leukemia using viSNE. Cytom. B Clin. Cytom. 2015, 88, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Good, Z.; Sarno, J.; Jager, A.; Samusik, N.; Aghaeepour, N.; Simonds, E.F.; White, L.; Lacayo, N.J.; Fantl, W.J.; Fazio, G.; et al. Single-cell developmental classification of B cell precursor acute lymphoblastic leukemia at diagnosis reveals predictors of relapse. Nat. Med. 2018, 24, 474–483. [Google Scholar] [CrossRef]
- Reiter, M.; Diem, M.; Schumich, A.; Maurer-Granofszky, M.; Karawajew, L.; Rossi, J.G.; Ratei, R.; Groeneveld-Krentz, S.; Sajaroff, E.O.; Suhendra, S.; et al. Automated Flow Cytometric MRD Assessment in Childhood Acute B-Lymphoblastic Leukemia Using Supervised Machine Learning. Cytom. Part A 2019, 95, 966–975. [Google Scholar] [CrossRef]
- Ko, B.-S.; Wang, Y.-F.; Li, J.-L.; Li, C.-C.; Weng, P.-F.; Hsu, S.-C.; Hou, H.-A.; Huang, H.-H.; Yao, M.; Lin, C.-T.; et al. Clinically validated machine learning algorithm for detecting residual diseases with multicolor flow cytometry analysis in acute myeloid leukemia and myelodysplastic syndrome. EBioMedicine 2018, 37, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajwa, B.; Wallace, P.K.; Griffiths, E.A.; Dundar, M. Automated Assessment of Disease Progression in Acute Myeloid Leukemia by Probabilistic Analysis of Flow Cytometry Data. IEEE Trans. Biomed. Eng. 2017, 64, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Chretien, A.-S.; Granjeaud, S.; Gondois-Rey, F.; Harbi, S.; Orlanducci, F.; Blaise, D.; Vey, N.; Arnoulet, C.; Fauriat, C.; Olive, D. Increased NK Cell Maturation in Patients with Acute Myeloid Leukemia. Front. Immunol. 2015, 6, 564. [Google Scholar] [CrossRef] [Green Version]
- Zare, H.; Bashashati, A.; Kridel, R.; Aghaeepour, N.; Haffari, G.; Connors, J.M.; Gascoyne, R.D.; Gupta, A.; Brinkman, R.R.; Weng, A.P. Automated analysis of multidimensional flow cytometry data improves diagnostic accuracy between mantle cell lymphoma and small lymphocytic lymphoma. Am. J. Clin. Pathol. 2012, 137, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Lakoumentas, J.; Drakos, J.; Karakantza, M.; Nikiforidis, G.C.; Sakellaropoulos, G.C. Bayesian clustering of flow cytometry data for the diagnosis of B-chronic lymphocytic leukemia. J. Biomed. Inform. 2009, 42, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Schlitzer, A.; Chen, J.; Mair, F.; Sumatoh, H.R.; Teng, K.W.W.; Low, D.; Ruedl, C.; Riccardi-Castagnoli, P.; Poidinger, M.; et al. High-dimensional analysis of the murine myeloid cell system. Nat. Immunol. 2014, 15, 1181–1189. [Google Scholar] [CrossRef]
- Wong, M.T.; Chen, J.; Narayanan, S.; Lin, W.; Anicete, R.; Kiaang, H.T.K.; De Lafaille, M.A.C.; Poidinger, M.; Newell, E.W. Mapping the Diversity of Follicular Helper T Cells in Human Blood and Tonsils Using High-Dimensional Mass Cytometry Analysis. Cell Rep. 2015, 11, 1822–1833. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Kim, H.; Brennan, P.J.; Han, B.; Baecher-Allan, C.M.; De Jager, P.L.; Brenner, M.B.; Raychaudhuri, S. Application of user-guided automated cytometric data analysis to large-scale immunoprofiling of invariant natural killer T cells. Proc. Natl. Acad. Sci. USA 2013, 110, 19030–19035. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Kim, D.; Tse, H.T.; Tseng, P.; Peng, L.; Dhar, M.; Karumbayaram, S.; Di Carlo, D. High-throughput physical phenotyping of cell differentiation. Microsyst. Nanoeng. 2017, 3, 17013. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; van Unen, V.; Abdelaal, T.; Guo, N.; Kasatskaya, S.A.; Ladell, K.; McLaren, J.E.; Egorov, E.S.; Izraelson, M.; de Sousa Lopes, S.M.C.; et al. Memory CD4+ T cells are generated in the human fetal intestine. Nat. Immunol. 2019, 20, 301–312. [Google Scholar] [CrossRef]
- Liu, M.; Barton, E.S.; Jennings, R.N.; Oldenburg, D.G.; Whirry, J.M.; White, D.W.; Grayson, J.M. Unsupervised learning techniques reveal heterogeneity in memory CD8+ T cell differentiation following acute, chronic and latent viral infections. Virology 2017, 509, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Barcenilla, H.; Åkerman, L.; Pihl, M.; Ludvigsson, J.; Casas, R. Mass Cytometry Identifies Distinct Subsets of Regulatory T Cells and Natural Killer Cells Associated With High Risk for Type 1 Diabetes. Front. Immunol. 2019, 10, 982. [Google Scholar] [CrossRef] [PubMed]
- Emmaneel, A.; Bogaert, D.J.; Van Gassen, S.; Tavernier, S.J.; Dullaers, M.; Haerynck, F.; Saeys, Y. A Computational Pipeline for the Diagnosis of CVID Patients. Front. Immunol. 2019, 10, 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, R.; Barman, P.K.; Thatoi, P.K.; Tripathy, R.; Kumar Das, B.; Ravindran, B. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep. 2015, 5, 13886. [Google Scholar] [CrossRef] [Green Version]
- Lacombe, F.; Lechevalier, N.; Vial, J.P.; Béné, M.C. An R-Derived FlowSOM Process to Analyze Unsupervised Clustering of Normal and Malignant Human Bone Marrow Classical Flow Cytometry Data. Cytom. Part A 2019, 95, 1191–1197. [Google Scholar] [CrossRef]
- Coindre, S.; Tchitchek, N.; Alaoui, L.; Vaslin, B.; Bourgeois, C.; Goujard, C.; Lecuroux, C.; Bruhns, P.; Le Grand, R.; Beignon, A.-S.; et al. Mass Cytometry Analysis Reveals Complex Cell-State Modifications of Blood Myeloid Cells During HIV Infection. Front. Immunol. 2019, 10, 2677. [Google Scholar] [CrossRef] [Green Version]
- Leite Pereira, A.; Bitoun, S.; Paoletti, A.; Nocturne, G.; Marcos Lopez, E.; Cosma, A.; Le Grand, R.; Mariette, X.; Tchitchek, N. Characterization of Phenotypes and Functional Activities of Leukocytes From Rheumatoid Arthritis Patients by Mass Cytometry. Front. Immunol. 2019, 10, 2384. [Google Scholar] [CrossRef]
- Duetz, C.; Bachas, C.; Westers, T.M.; van de Loosdrecht, A.A. Computational analysis of flow cytometry data in hematological malignancies: Future clinical practice? Curr. Opin. Oncol. 2020, 32, 162–169. [Google Scholar] [CrossRef]
- Ciabattini, A.; Pettini, E.; Medaglini, D. CD4(+) T Cell Priming as Biomarker to Study Immune Response to Preventive Vaccines. Front. Immunol. 2013, 4, 421. [Google Scholar] [CrossRef] [Green Version]
- Jelley-Gibbs, D.M.; Strutt, T.M.; McKinstry, K.K.; Swain, S.L. Influencing the fates of CD4 T cells on the path to memory: Lessons from influenza. Immunol. Cell Biol. 2008, 86, 343–352. [Google Scholar] [CrossRef]
- Altman, J.D.; Moss, P.A.; Goulder, P.J.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.J.; Chu, H.H.; Pepper, M.; McSorley, S.J.; Jameson, S.C.; Kedl, R.M.; Jenkins, M.K. Naive CD4(+) T cell frequency varies for different epitopes and predicts repertoire diversity and response magnitude. Immunity 2007, 27, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prota, G.; Christensen, D.; Andersen, P.; Medaglini, D.; Ciabattini, A. Peptide-specific T helper cells identified by MHC class II tetramers differentiate into several subtypes upon immunization with CAF01 adjuvanted H56 tuberculosis vaccine formulation. Vaccine 2015, 33, 6823–6830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciabattini, A.; Pettini, E.; Fiorino, F.; Pastore, G.; Andersen, P.; Pozzi, G.; Medaglini, D. Modulation of Primary Immune Response by Different Vaccine Adjuvants. Front. Immunol. 2016, 7, 427. [Google Scholar] [CrossRef] [Green Version]
- Uchtenhagen, H.; Rims, C.; Blahnik, G.; Chow, I.-T.; Kwok, W.W.; Buckner, J.H.; James, E.A. Efficient ex vivo analysis of CD4+ T-cell responses using combinatorial HLA class II tetramer staining. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pastore, G.; Carraro, M.; Pettini, E.; Nolfi, E.; Medaglini, D.; Ciabattini, A. Optimized Protocol for the Detection of Multifunctional Epitope-Specific CD4+ T Cells Combining MHC-II Tetramer and Intracellular Cytokine Staining Technologies. Front. Immunol. 2019, 10, 2304. [Google Scholar] [CrossRef]
- Tesfa, L.; Volk, H.D.; Kern, F. A protocol for combining proliferation, tetramer staining and intracellular cytokine detection for the flow-cytometric analysis of antigen specific T-cells. J. Biol. Regul. Homeost. Agents 2003, 17, 366–370. [Google Scholar]
- Chung, A.W.; Kumar, M.P.; Arnold, K.B.; Yu, W.H.; Schoen, M.K.; Dunphy, L.J.; Suscovich, T.J.; Frahm, N.; Linde, C.; Mahan, A.E.; et al. Dissecting Polyclonal Vaccine-Induced Humoral Immunity against HIV Using Systems Serology. Cell 2015, 163, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Kimble, J.B.; Malherbe, D.C.; Meyer, M.; Gunn, B.M.; Karim, M.M.; Ilinykh, P.A.; Iampietro, M.; Mohamed, K.S.; Negi, S.; Gilchuk, P.; et al. Antibody-Mediated Protective Mechanisms Induced by a Trivalent Parainfluenza Virus-Vectored Ebolavirus Vaccine. J. Virol. 2019, 93, e01845-18. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Frelinger, J.; Jiang, W.; Finak, G.; Seshadri, C.; Bart, P.-A.; Pantaleo, G.; McElrath, J.; DeRosa, S.; Gottardo, R. Identification and visualization of multidimensional antigen-specific T-cell populations in polychromatic cytometry data. Cytom. Part A 2015, 87, 675–682. [Google Scholar] [CrossRef]
- Ciabattini, A.; Pettini, E.; Fiorino, F.; Lucchesi, S.; Pastore, G.; Brunetti, J.; Santoro, F.; Andersen, P.; Bracci, L.; Pozzi, G.; et al. Heterologous Prime-Boost Combinations Highlight the Crucial Role of Adjuvant in Priming the Immune System. Front. Immunol. 2018, 9, 380. [Google Scholar] [CrossRef] [PubMed]
- Billeskov, R.; Wang, Y.; Solaymani-Mohammadi, S.; Frey, B.; Kulkarni, S.; Andersen, P.; Agger, E.M.; Sui, Y.; Berzofsky, J.A. Low Antigen Dose in Adjuvant-Based Vaccination Selectively Induces CD4 T Cells with Enhanced Functional Avidity and Protective Efficacy. J. Immunol. 2017, 198, 3494–3506. [Google Scholar] [CrossRef] [PubMed]
- Kvistborg, P.; Gouttefangeas, C.; Aghaeepour, N.; Cazaly, A.; Chattopadhyay, P.K.; Chan, C.; Eckl, J.; Finak, G.; Hadrup, S.R.; Maecker, H.T.; et al. Thinking outside the gate: Single-cell assessments in multiple dimensions. Immunity 2015, 42, 591–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
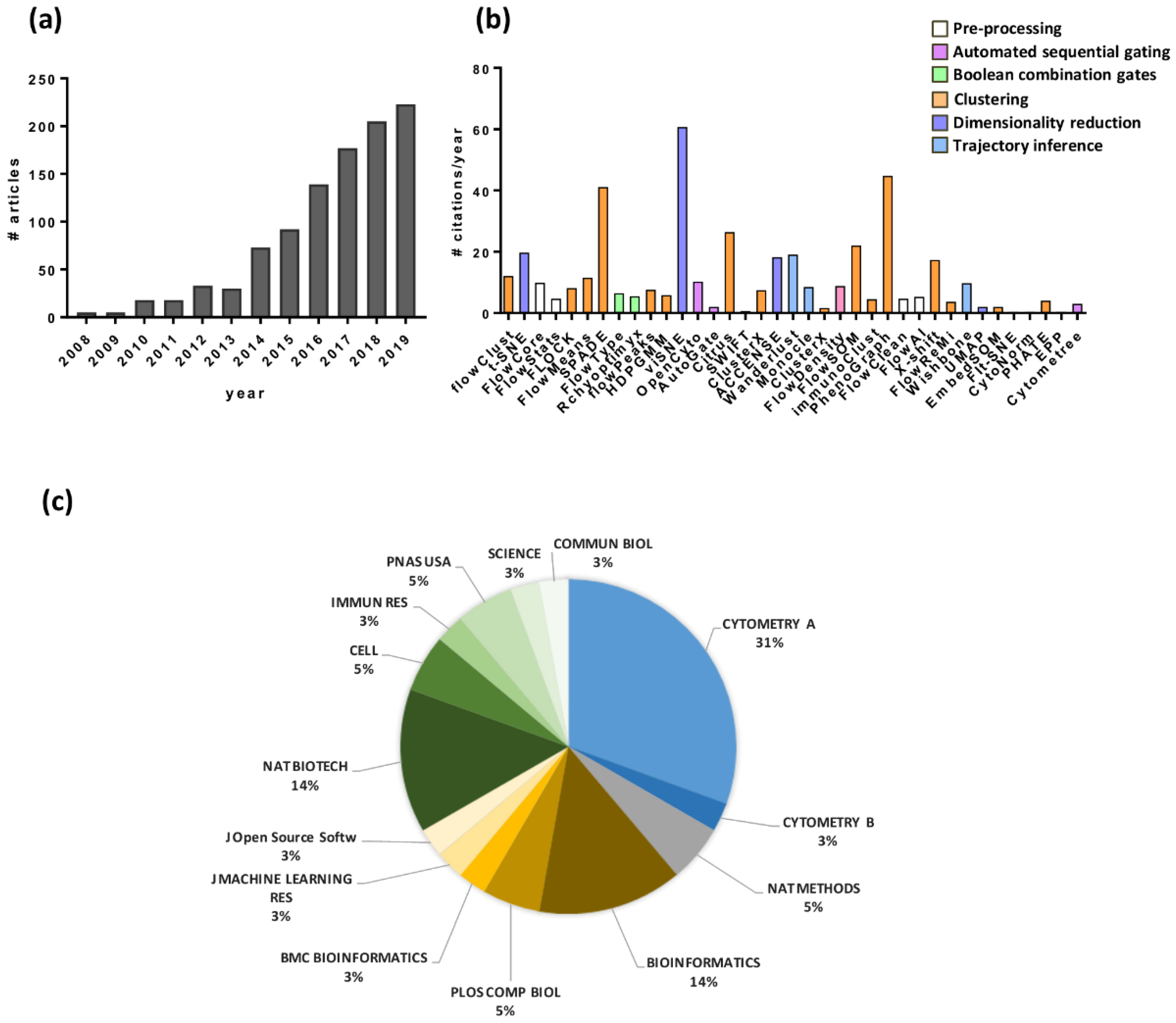{kind=link}
| Function | Software | Availability | Description | Reference |
|---|---|---|---|---|
| Pre-processing | FlowCore | R, Bioconductor | Import, compensate and transform FCS files in R environment | [9] |
| FlowStats | R, Bioconductor | Collection of algorithms to analyze flow cytometry data, including correction of batch effect | [10] | |
| FlowClean | R, Bioconductor FlowJo plugin | Quality control of data set based on compositional analysis | [11] | |
| FlowAI | R, Bioconductor FlowJo plugin | Quality control of data set based on flow rate, signal acquisition and dynamic range | [12] | |
| CATALYST | R, Bioconductor | Collection of algorithms to pre-process cytometric data and to perform data analysis (with FlowSOM clustering and dimensionality reduction) | [13] | |
| CytoNorm | R | Normalized batch effect using control sample and clustering algorithm | [14] | |
| Automated sequential gating | FlowDensity | R, Bioconductor | Provides tools for automated 1-D and 2-D sequential gating | [15] |
| OpenCyto | R, Bioconductor | Facilitates automated 1-D and 2-D gating methods in sequential way to mimic the manual gating | [16] | |
| AutoGate | Standalone software | Performs 2-D sequential gating to obviate the need to draw arbitrary gates to define the subsets in a gating | [17] | |
| cytometree | R | The algorithm relies on the construction of a binary tree, the nodes of which represents cellular populations | [18] | |
| EPP | Standalone software | AutoGate extension. Algorithm that detects the best 2-D gating strategy to identify cellular populations | [19] | |
| Boolean combination gates | flowType | R, Bioconductor | Phenotyping cytometric using multi-dimensional expansion of 1-D partitions | [20] |
| FloReMi | R | Starting from flowType results identifies the populations that best correlates with an external outcome | [21] | |
| RchyOptimyx | R, Bioconductor | Starting from flowType results, constructs a hierarchy of cells selecting the most informative phenotypes for biomarker detection | [22] | |
| Clustering | FlowMeans | R, Bioconductor FlowJo plugin | Automated gating tool based on K-means algorithm | [23] |
| SPADE | R, Matlab, Cytobank, FlowJo plugin | Clustering method based combining density-based sampling with hierarchical clustering | [24] | |
| HDPGMM | Python | Clustering based on hierarchical modeling extensions to the Dirichlet Process Gaussian Mixture Model | [25] | |
| Citrus | Cytobank, R | Identifies cell populations with hierarchical clustering and make prediction with regression model | [26] | |
| FlowSOM | R, Bioconductor FlowJo plugin, Cytobank | Clustering method combining SOM and hierarchical clustering | [27] | |
| X-shift | Standalone software, FlowJo plugin | Clustering based on kNN density estimation and cluster merging according Mahalanobis distances | [28] | |
| flowClust | R, Bioconductor | Model-based clustering using a t-mixture model | [29] | |
| immunoClust | R, Bioconductor | Model-based clustering on individual samples. Includes an additional step to map cluster between samples | [30] | |
| SWIFT | Matlab | Clustering method based on splitting and merging of Gaussian mixture models | [31] | |
| FLOCK | C, Immport | Automated method partitioning of each dimension into bins, followed by merging of dense regions, and density-based clustering | [32] | |
| flowPeaks | R, Bioconductor | Clustering method combining density-based clustering and K-means | [33] | |
| ClusterX | R | Fast clustering by automatic search and find of density peaks | [34] | |
| PhenoGraph | Matlab, Python | Cells are visualized in a graph structure and connected with weighted edge based on neighbor shared by cell. Graph is then partitioned in group of cells sharing similar phenotypes | [35] | |
| Dimensionality reduction | t-SNE | FlowJo plugin | Performs t-SNE in FlowJo, allowing to manually gate region in dimensionality reduced space to compare cell frequency across samples | [36] |
| ACCENSE | Standalone software | Performs dimensionality reduction with t-SNE algorithm, followed by clustering of dimensionality reduced events with K-means or DBSCAN algorithms | [37] | |
| Rtsne | R | Performs t-SNE dimensionality reduction in R environment | [36] | |
| viSNE | Cytobank, Matlab | Visualization tool based on implementation of t-SNE algorithm | [38] | |
| EmbedSOM | R, Bioconductor FlowJo plugin | Dimensionality reduction technique based on SOM | [39] | |
| UMAP | R, Python, FlowJo plugin | Dimensionality reduction technique based on Uniform Manifold Approximation and Projection (UMAP) | [40] | |
| Destiny | R, Bioconductor | Performs dimensionality reduction with diffusion map | [41] | |
| Fit-SNE | R, Matlab, Python, FlowJo plugin | Tool to perform dimensionality reduction using Fast Fourier Transform-accelerated Interpolation-based t-SNE | [42] | |
| Trajectory inference | Wanderlust | Matlab | Trajectory inference method based on kNN graph: Developed to identify linear transitions | [43] |
| Wishbone | Matlab, Python | Evolution of Wanderlust, it can identify bifurcation in the trajectories | [44] | |
| Monocle | R, Bioconductor | Identification of bifurcated trajectory based on MST | [45] | |
| PHATE | Matlab, Python | Identification of trajectory preserving continual progressions, branches and clusters | [46] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucchesi, S.; Furini, S.; Medaglini, D.; Ciabattini, A. From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies. Vaccines 2020, 8, 138. https://doi.org/10.3390/vaccines8010138
Lucchesi S, Furini S, Medaglini D, Ciabattini A. From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies. Vaccines. 2020; 8(1):138. https://doi.org/10.3390/vaccines8010138
Chicago/Turabian StyleLucchesi, Simone, Simone Furini, Donata Medaglini, and Annalisa Ciabattini. 2020. "From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies" Vaccines 8, no. 1: 138. https://doi.org/10.3390/vaccines8010138
APA StyleLucchesi, S., Furini, S., Medaglini, D., & Ciabattini, A. (2020). From Bivariate to Multivariate Analysis of Cytometric Data: Overview of Computational Methods and Their Application in Vaccination Studies. Vaccines, 8(1), 138. https://doi.org/10.3390/vaccines8010138