Synthetic DNA Vaccines Adjuvanted with pIL-33 Drive Liver-Localized T Cells and Provide Protection from Plasmodium Challenge in a Mouse Model

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construct Design
2.2. Western Blot
2.3. Immunization and CELLECTRA Electroporation
2.4. Immune Cell Isolation
2.5. ELISPOT
2.6. Flow Cytometry
2.7. Immunofluorescence
2.8. Mosquito Feeding and Sporozoite Extraction
2.9. Challenge Study
3. Results
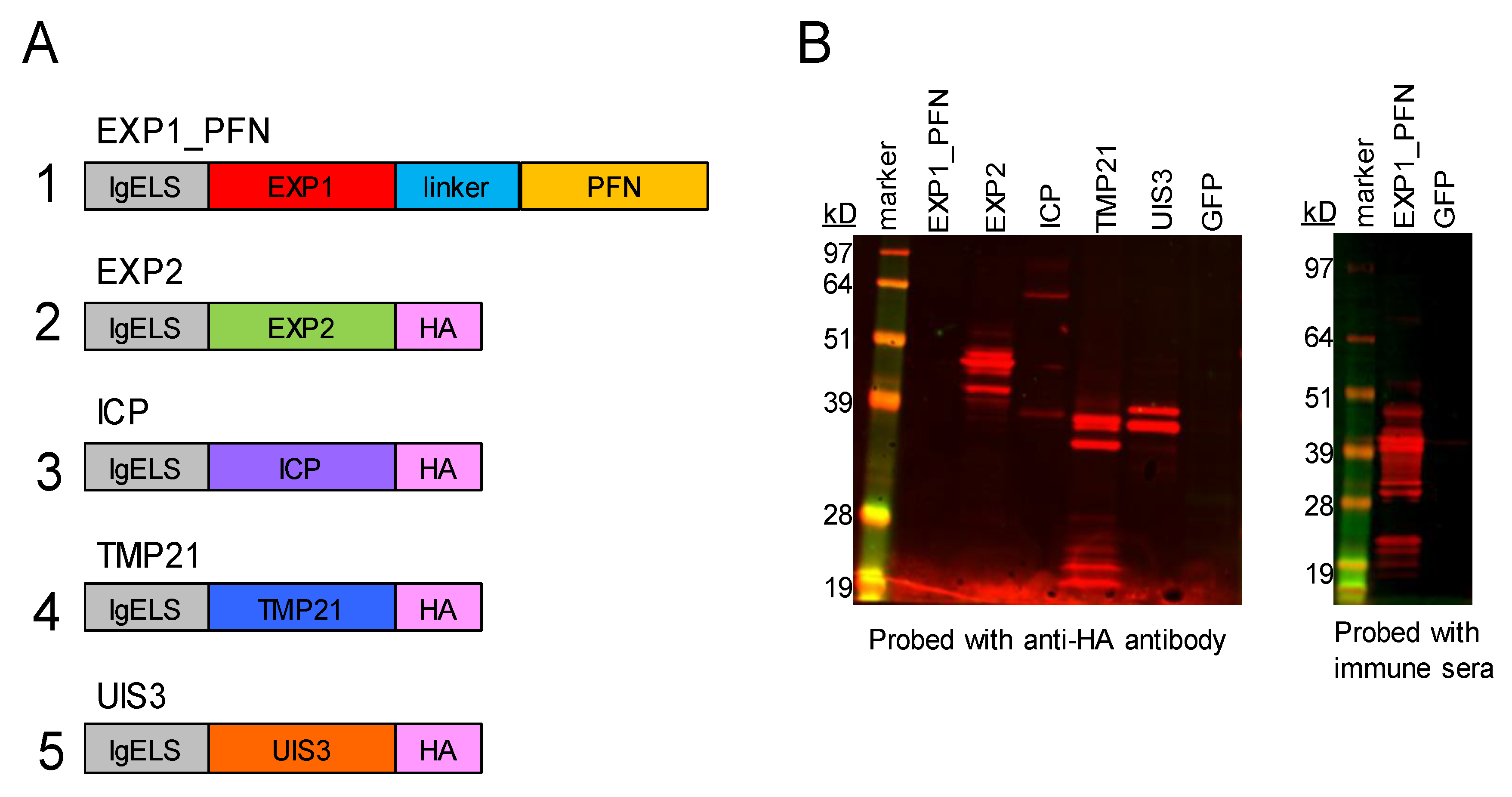
3.1. synDNA Vaccine Construct Design and In Vitro Expression
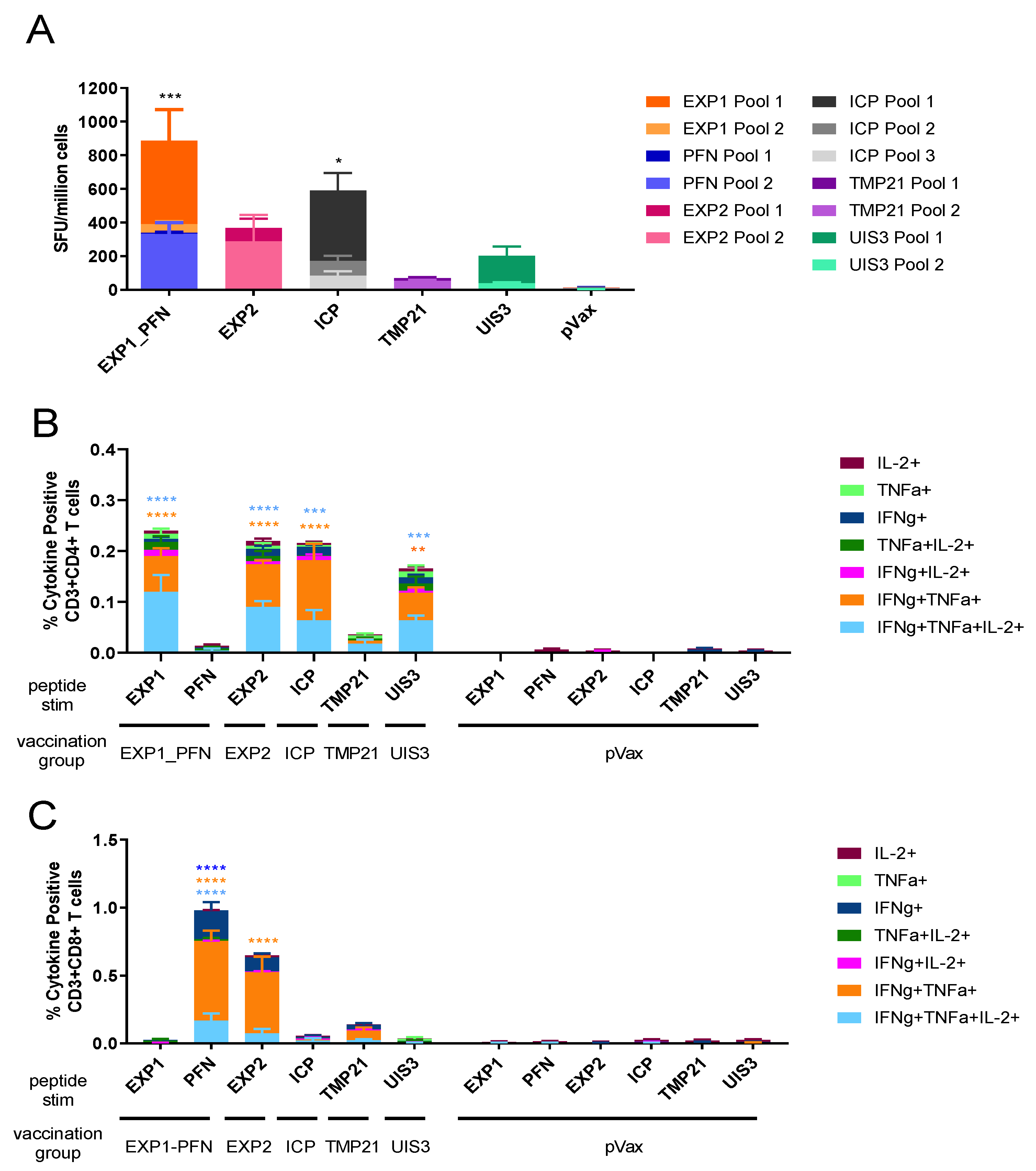
3.2. Py LS Vaccine Constructs Delivered Individually Elicit a Robust and Polyfunctional T Cell Response
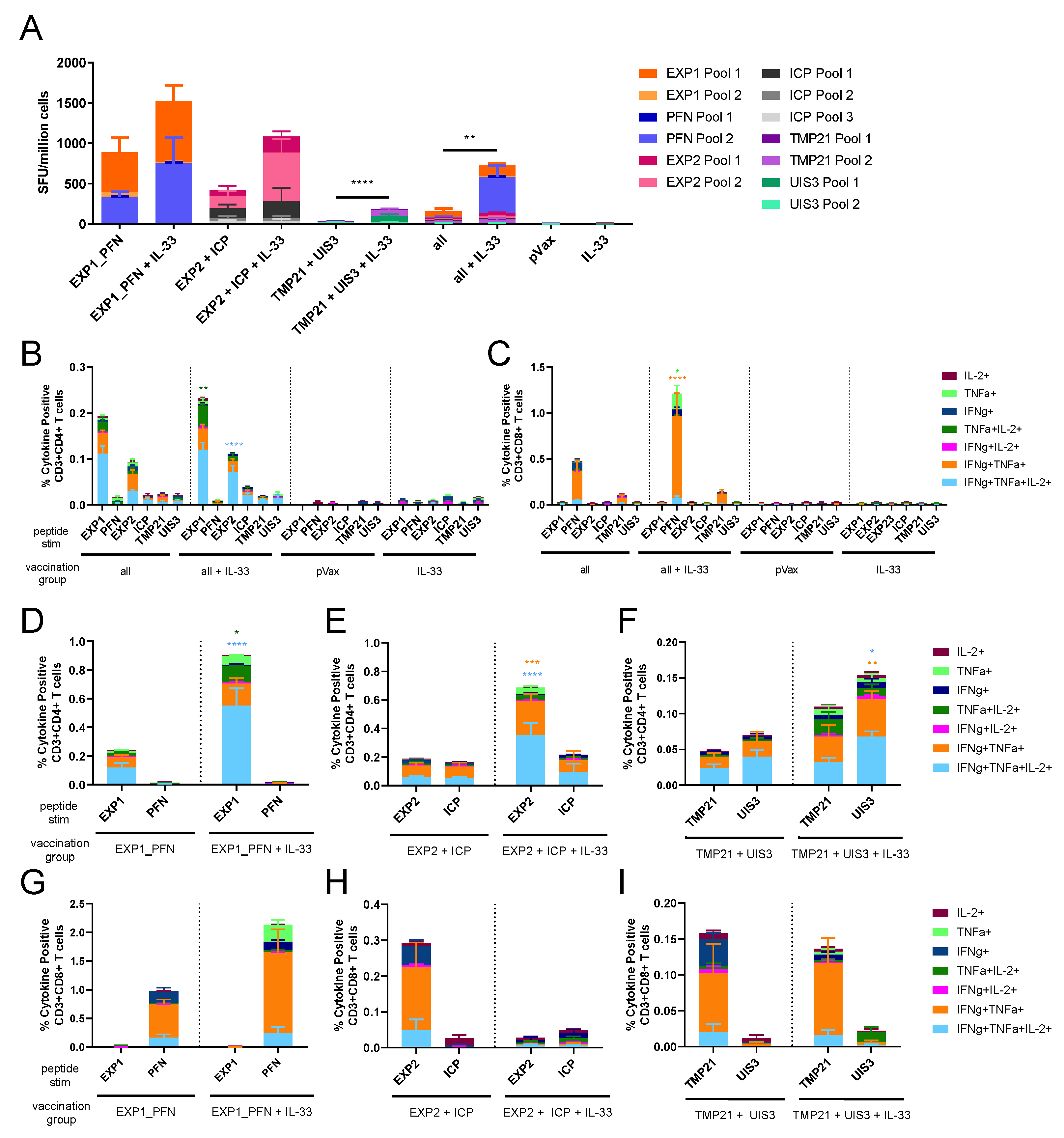
3.3. Co-Formulated Py LS Vaccines Delivered with and without Plasmid IL-33 Adjuvant Elicit a Robust and Polyfunctional T Cell Response
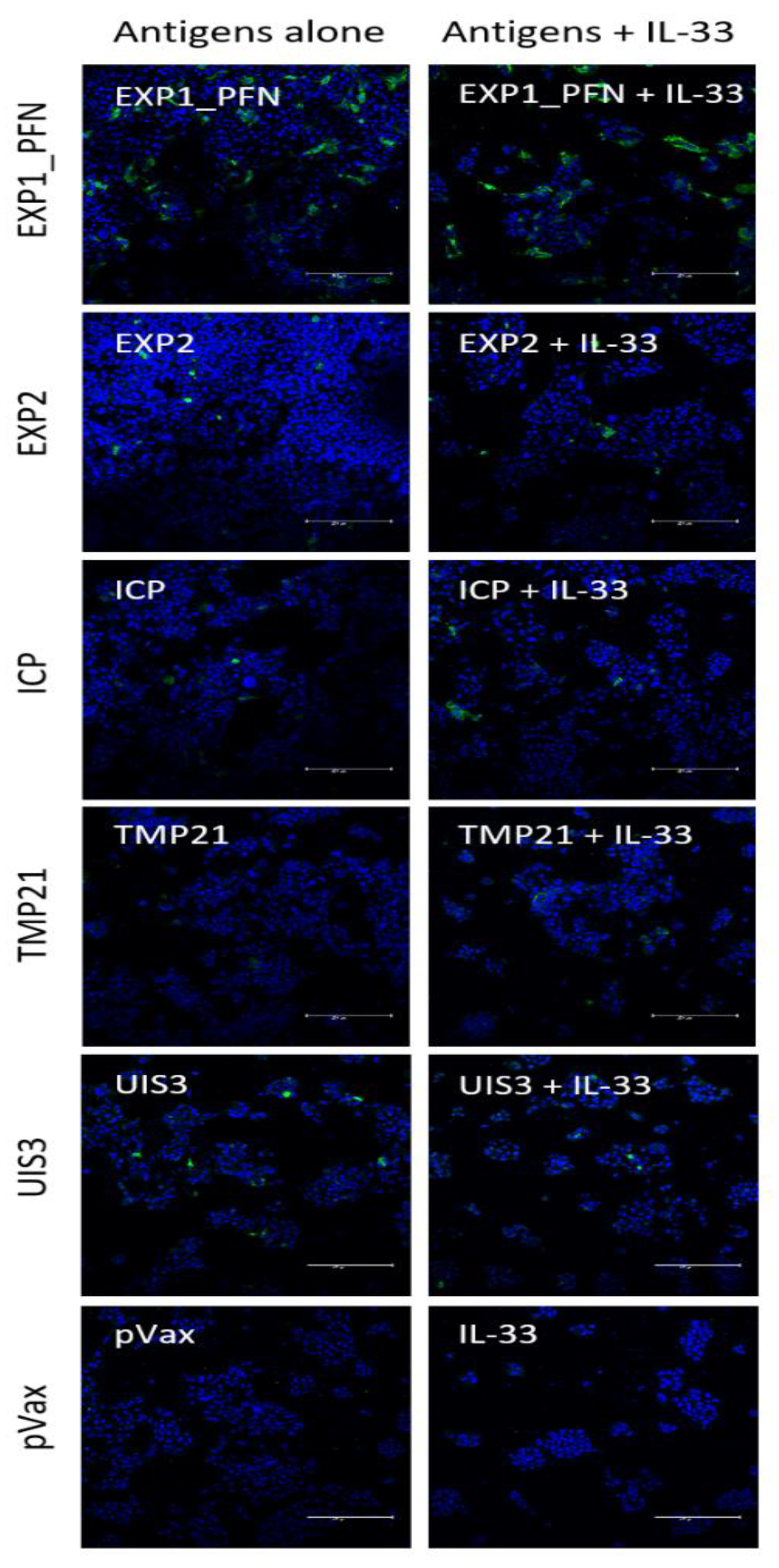
3.4. Py LS Antigen Vaccination Elicits a Robust and Polyfunctional Antigen-Specific T Cell Response in the Liver Which Is Enhanced with the Addition of Plasmid IL-33
3.5. Vaccine Delivered with and without Adjuvant Elicits Antigen-Specific Antibody Responses
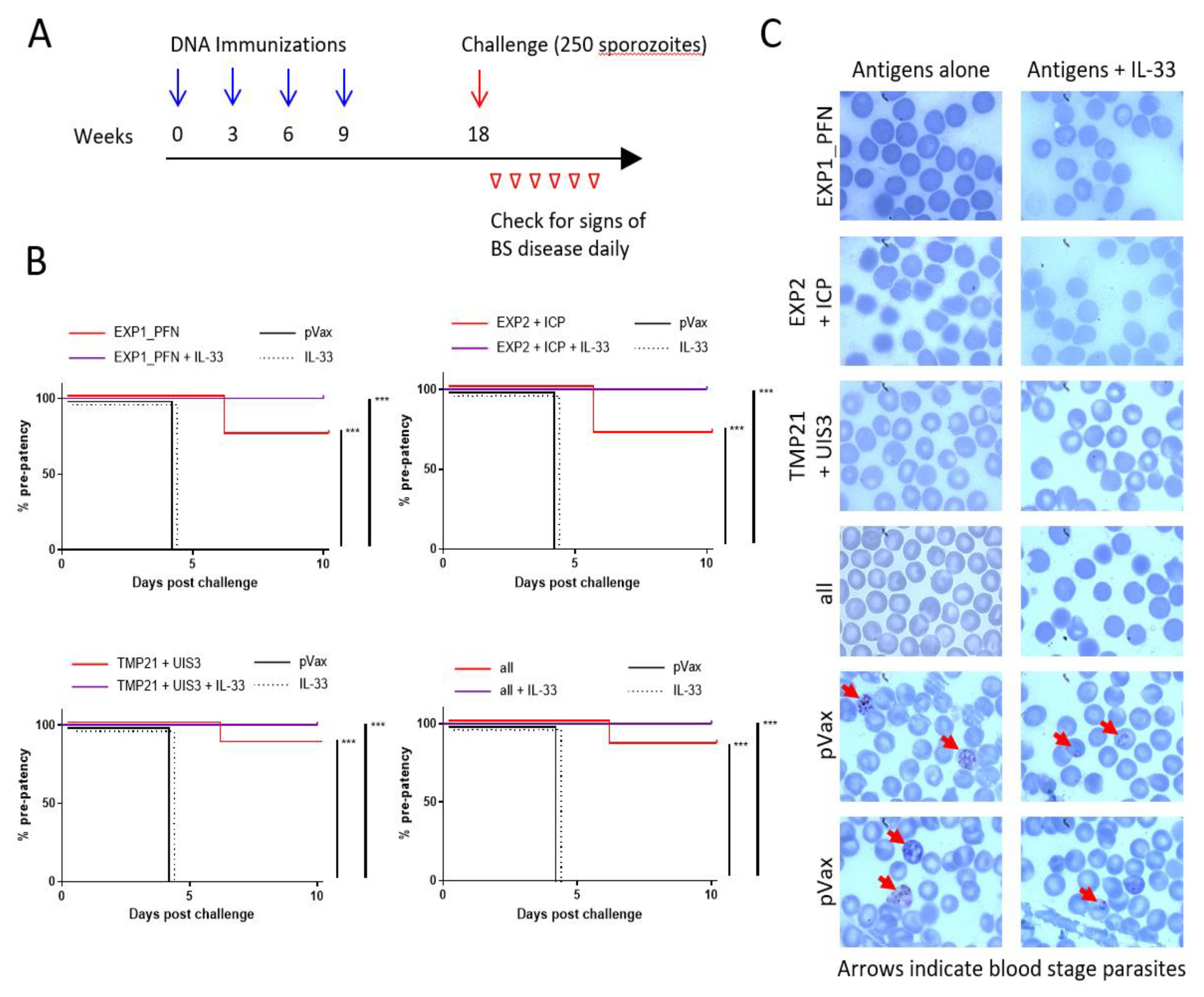
3.6. Plasmodium LS synDNA Vaccine Is Protective against Malaria Infection in a Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Price, R.N.; Tjitra, E.; Guerra, C.A.; Yeung, S.; White, N.J.; Anstey, N.M. Vivax malaria: Neglected and not benign. Am. J. Trop. Med. Hyg. 2007, 77, 79–87. [Google Scholar] [CrossRef]
- Aly, A.S.I.; Vaughan, A.M.; Kappe, S.H.I. Malaria Parasite Development in the Mosquito and Infection of the Mammalian Host. Annu. Rev. Microbiol. 2009, 63, 195–221. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, A.M.; Aly, A.S.I.; Kappe, S.H.I. Malaria Parasite Pre-Erythrocytic Stage Infection: Gliding and Hiding. Cell Host Microbe 2008, 4, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Kaushansky, A.; Kappe, S.H.I. The crucial role of hepatocyte growth factor receptor during liver-stage infection is not conserved among Plasmodium species. Nat. Med. 2011, 17, 1180. [Google Scholar] [CrossRef]
- Kaushansky, A.; Ye, A.S.; Austin, L.S.; Mikolajczak, S.A.; Vaughan, A.M.; Camargo, N.; Metzger, P.G.; Douglass, A.N.; MacBeath, G.; Kappe, S.H.I. Suppression of Host p53 Is Critical for Plasmodium Liver-Stage Infection. Cell Rep. 2013, 3, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Sturm, A.; Graewe, S.; Franke-Fayard, B.; Retzlaff, S.; Bolte, S.; Roppenser, B.; Aepfelbacher, M.; Janse, C.; Heussler, V. Alteration of the Parasite Plasma Membrane and the Parasitophorous Vacuole Membrane during Exo-Erythrocytic Development of Malaria Parasites. Protist 2009, 160, 51–63. [Google Scholar] [CrossRef]
- Sturm, A.; Heussler, V. Live and let die: Manipulation of host hepatocytes by exoerythrocytic Plasmodium parasites. Med Microbiol. Immunol. 2007, 196, 127–133. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Kappe, S.H. Malaria vaccine development: Persistent challenges. Curr. Opin. Immunol. 2012, 24, 324–331. [Google Scholar] [CrossRef]
- Sinnis, P.; Nardin, E. Sporozoite Antigens: Biology and Immunology of the Circumsporozoite Protein and Thrombospondin-Related Anonymous Protein. In Malaria Immunology; KARGER: Basel, Switzerland, 2002; Volume 80, pp. 70–96. [Google Scholar]
- RTSS Clinical Trials Partnership. Efficacy and Safety of the RTS,S/AS01 Malaria Vaccine during 18 Months after Vaccination: A Phase 3 Randomized, Controlled Trial in Children and Young Infants at 11 African Sites. PLoS Med. 2014, 11. [Google Scholar] [CrossRef]
- Rts, S.C.T.P. Efficacy and safety of RTS,S/AS01 malaria vaccine with or without a booster dose in infants and children in Africa: Final results of a phase 3, individually randomised, controlled trial. Lancet 2015, 386, 31–45. [Google Scholar] [CrossRef] [Green Version]
- RTS, S Clinical Trials Partnership. First Results of Phase 3 Trial of RTS,S/AS01 Malaria Vaccine in African Children. N. Engl. J. Med. 2011, 365, 1863–1875. [Google Scholar] [CrossRef] [Green Version]
- Phase, A. Trial of RTS,S/AS01 malaria vaccine in African infants. N. Engl. J. Med. 2012, 367, 2284–2295. [Google Scholar] [CrossRef] [Green Version]
- White, N.J. A Vaccine for Malaria. N. Engl. J. Med. 2011. [Google Scholar] [CrossRef]
- Vaughan, A.M.; Kappe, S.H.I. Vaccination using radiation- or genetically attenuated live sporozoites. Methods Mol. Biol. 2013, 549–566. [Google Scholar] [CrossRef]
- National Institute of Health Home. Available online: https://clinicaltrials.gov/ (accessed on 21 November 2019).
- Butler, N.S.; Schmidt, N.W.; Vaughan, A.M.; Aly, A.S.; Kappe, S.H.I.; Harty, J.T. Superior Antimalarial Immunity after Vaccination with Late Liver Stage-Arresting Genetically Attenuated Parasites. Cell Host Microbe 2011, 9, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Doll, K.L.; Butler, N.S.; Harty, J.T. CD8 T cell independent immunity after single dose infection-treatment-vaccination (ITV) against Plasmodium yoelii. Vaccine 2014, 32, 483–491. [Google Scholar] [CrossRef] [Green Version]
- Douradinha, B.; Van Dijk, M.; Van Gemert, G.J.; Khan, S.M.; Janse, C.J.; Waters, A.P.; Sauerwein, R.W.; Luty, A.J.F.; Silva-Santos, B.; Mota, M.M.; et al. Immunization with genetically attenuated P52-deficient Plasmodium berghei sporozoites induces a long-lasting effector memory CD8+T cell response in the liver. J. Immune Based Ther. Vaccines 2011, 9, 6. [Google Scholar] [CrossRef] [Green Version]
- Spring, M.; Murphy, J.; Nielsen, R.; Dowler, M.; Bennett, J.W.; Zarling, S.; Williams, J.; de la Vega, P.; Ware, L.; Komisar, J.; et al. First-in-human evaluation of genetically attenuated Plasmodium falciparum sporozoites administered by bite of Anopheles mosquitoes to adult volunteers. Vaccine 2013, 31. [Google Scholar] [CrossRef]
- Tarun, A.S.; Dumpit, R.F.; Camargo, N.; Labaied, M.; Liu, P.; Takagi, A.; Wang, R.; Kappe, S.H.I. Protracted Sterile Protection with Plasmodium yoelii Pre-erythrocytic Genetically Attenuated Parasite Malaria Vaccines Is Independent of Significant Liver-Stage Persistence and Is Mediated by CD8+ T Cells. J. Infect. Dis. 2007, 196, 608–616. [Google Scholar] [CrossRef] [Green Version]
- Tse, S.W.; Radtke, A.J.; Espinosa, D.A.; Cockburn, I.A.; Zavala, F. The chemokine receptor CXCR6 is required for the maintenance of liver memory CD8+ T cells specific for infectious pathogens. J. Infect. Dis. 2014, 210, 1508–1516. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, D.; Ng, W.Y.; Holz, L.E.; Ma, J.Z.; Zaid, A.; Wong, Y.C.; Lau, L.S.; Mollard, V.; Cozijnsen, A.; Collins, N.; et al. Liver-Resident Memory CD8+ T Cells Form a Front-Line Defense against Malaria Liver-Stage Infection. Immunity 2016, 45, 889–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gola, A.; Silman, D.; Walters, A.A.; Sridhar, S.; Uderhardt, S.; Salman, A.M.; Halbroth, B.R.; Bellamy, D.; Bowyer, G.; Powlson, J.; et al. Prime and target immunization protects against liver-stage malaria in mice. Sci. Transl. Med. 2018, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halbroth, B.R.; Sebastian, S.; Salman, A.M.; Ulaszewska, M.; Gola, A.; Longley, R.J.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Pre-clinical development and assessment of viral vectors expressing a fusion antigen of P. falciparum LSA1 and LSAP2 for efficacy against liver-stage malaria. Infect. Immun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Lisewski, A.M.; Quiros, J.P.; Ng, C.L.; Adikesavan, A.K.; Miura, K.; Putluri, N.; Eastman, R.T.; Scanfeld, D.; Regenbogen, S.J.; Altenhofen, L.; et al. Supergenomic network compression and the discovery of exp1 as a glutathione transferase inhibited by artesunate. Cell 2014, 158, 916–928. [Google Scholar] [CrossRef] [Green Version]
- Spielmann, T.; Gardiner, D.L.; Beck, H.P.; Trenholme, K.R.; Kemp, D.J. Organization of ETRAMPs and EXP-1 at the parasite-host cell interface of malaria parasites. Mol. Microbiol. 2006, 59. [Google Scholar] [CrossRef]
- Doolan, D.L.; Hoffman, S.L.; Southwood, S.; Wentworth, P.A.; Sidney, J.; Chesnut, R.W.; Keogh, E.; Appella, E.; Nutman, T.B.; Lal, A.; et al. Degenerate cytotoxic T cell epitopes from P. falciparum restricted by multiple HLA-A and HLA-B supertype alleles. Immunity 1997, 7, 97–112. [Google Scholar] [CrossRef] [Green Version]
- Dobaño, C.; Widera, G.; Rabussay, D.; Doolan, D.L. Enhancement of antibody and cellular immune responses to malaria DNA vaccines by in vivo electroporation. Vaccine 2007, 25, 6635–6645. [Google Scholar] [CrossRef]
- Meraldi, V.; Nebié, I.; Tiono, A.B.; Diallo, D.; Sanogo, E.; Theisen, M.; Druilhe, P.; Corradin, G.; Moret, R.; Sirima, B.S. Natural antibody response to Plasmodium falciparum Exp-1, MSP-3 and GLURP long synthetic peptides and association with protection. Parasite Immunol. 2004, 26, 265–272. [Google Scholar] [CrossRef]
- Moreau, C.A.; Bhargav, S.P.; Kumar, H.; Quadt, K.A.; Piirainen, H.; Strauss, L.; Kehrer, J.; Streichfuss, M.; Spatz, J.P.; Wade, R.C.; et al. A unique profilin-actin interface is important for malaria parasite motility. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef]
- Kursula, I.; Kursula, P.; Ganter, M.; Panjikar, S.; Matuschewski, K.; Schüler, H. Structural Basis for Parasite-Specific Functions of the Divergent Profilin of Plasmodium falciparum. Structure 2008, 16, 1638–1648. [Google Scholar] [CrossRef] [Green Version]
- Sattler, J.M.; Ganter, M.; Hliscs, M.; Matuschewski, K.; Schüler, H. Actin regulation in the malaria parasite. Eur. J. Cell Biol. 2011, 90, 966–971. [Google Scholar] [CrossRef] [PubMed]
- Alam, A.; Bhatnagar, R.K.; Relan, U.; Mukherjee, P.; Chauhan, V.S. Proteolytic activity of Plasmodium falciparum subtilisin-like protease 3 on parasite profilin, a multifunctional protein. Mol. Biochem. Parasitol. 2013, 191, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Hliscs, M.; Dunst, J.; Goosmann, C.; Brinkmann, V.; Montagna, G.N.; Matuschewski, K. Comparative Plasmodium gene overexpression reveals distinct perturbation of sporozoite transmission by profilin. Mol. Biol. Cell 2016, 27, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Marti, T.; Rick, B.; Johnson, D.; Benting, J.; Baumeister, S.; Helmbrecht, C.; Lanzer, M.; Lingelbach, K. Characterization and cloning of the gene encoding the vacuolar membrane protein EXP-2 from Plasmodium falciparum. Mol. Biochem. Parasitol. 1998, 92, 47–57. [Google Scholar] [CrossRef]
- De Koning-Ward, T.F.; Gilson, P.R.; Boddey, J.A.; Rug, M.; Smith, B.J.; Papenfuss, A.T.; Sanders, P.R.; Lundie, R.J.; Maier, A.G.; Cowman, A.F.; et al. A newly discovered protein export machine in malaria parasites. Nature 2009, 459. [Google Scholar] [CrossRef] [Green Version]
- Boysen, K.E.; Matuschewski, K. Inhibitor of Cysteine Proteases Is Critical for Motility and Infectivity of Plasmodium Sporozoites. MBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, C.; Heitmann, A.; Mishra, S.; Burda, P.C.; Singer, M.; Prado, M.; Niklaus, L.; Lacroix, C.; Ménard, R.; Frischknecht, F.; et al. A Cysteine Protease Inhibitor of Plasmodium berghei Is Essential for Exo-erythrocytic Development. PLoS Pathog. 2014, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Keitany, G.J.; Peng, X.; Gibson, C.; Mohar, I.; Vignali, M.; Crispe, I.N.; Huang, F.; Wang, R. Identification of pre-erythrocytic malaria antigens that target hepatocytes for killing in vivo and contribute to protection elicited by whole-parasite vaccination. PLoS ONE 2014, 9, e102225. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Yogavel, M.; Akhouri, R.R.; Gill, J.; Sharma, A. Crystal structure of soluble domain of malaria sporozoite protein UIS3 in complex with lipid. J. Biol. Chem. 2008, 283. [Google Scholar] [CrossRef] [Green Version]
- Mikolajczak, S.A.; Jacobs-Lorena, V.; MacKellar, D.C.; Camargo, N.; Kappe, S.H. L-FABP is a critical host factor for successful malaria liver stage development. Int. J. Parasitol. 2007, 37, 483–489. [Google Scholar] [CrossRef]
- Mueller, A.K.; Labaled, M.; Kappe, S.H.I.; Matuschewski, K. Genetically modified Plasmodium parasites as a protective experimental malaria vaccine. Nature 2005, 433, 164. [Google Scholar] [CrossRef] [PubMed]
- Longley, R.J.; Halbroth, B.R.; Salman, A.M.; Ewer, K.J.; Hodgson, S.H.; Janse, C.J.; Khan, S.M.; Hill, A.V.S.; Spencer, A.J. Assessment of the Plasmodium falciparum preerythrocytic antigen UIS3 as a potential candidate for a malaria vaccine. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudchodkar, S.B.; Choi, H.; Reuschel, E.L.; Esquivel, R.; Jin-Ah Kwon, J.; Jeong, M.; Maslow, J.N.; Reed, C.C.; White, S.; Kim, J.J.; et al. Rapid response to an emerging infectious disease—Lessons learned from development of a synthetic DNA vaccine targeting Zika virus. Microbes Infect. 2018, 20, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, D.O.; Weiner, D.B. IL-33 isoforms: Their future as vaccine adjuvants? Expert Rev. Vaccines 2015, 14, 489–492. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Edwards, L.; Parker, R.L.; Denny, L.; Giffear, M.; et al. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, D.O.; Wise, M.C.; Walters, J.N.; Reuschel, E.L.; Choi, M.J.; Obeng-Adjei, N.; Yan, J.; Morrow, M.P.; Weiner, D.B. Alarmin IL-33 Acts as an Immunoadjuvant to Enhance Antigen-Specific Tumor Immunity. Cancer Res. 2014, 74, 1789–1800. [Google Scholar] [CrossRef] [Green Version]
- Villarreal, D.O.; Weiner, D.B. Interleukin 33: A switch-hitting cytokine. Curr. Opin. Immunol. 2014, 28, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Morrow, M.P.; Kraynyak, K.A.; Sylvester, A.J.; Dallas, M.; Knoblock, D.; Boyer, J.D.; Yan, J.; Vang, R.; Khan, A.S.; Humeau, L.; et al. Clinical and immunologic biomarkers for histologic regression of high-grade cervical dysplasia and clearance of HPV16 and HPV18 after immunotherapy. Clin. Cancer Res. 2018, 24, 276–294. [Google Scholar] [CrossRef] [Green Version]
- Obeng-Adjei, N.; Hutnick, N.A.; Yan, J.; Chu, J.S.; Myles, D.J.F.; Morrow, M.P.; Sardesai, N.Y.; Weiner, D.B. DNA vaccine cocktail expressing genotype A and C HBV surface and consensus core antigens generates robust cytotoxic and antibody responses in mice and Rhesus macaques. Cancer Gene Ther. 2013, 20, 652. [Google Scholar] [CrossRef]
- Choi, H.; Kudchodkar, S.B.; Reuschel, E.L.; Asijaid, K.; Borole, P.; Ho, M.; Wojtak, K.; Reed, C.; Ramos, S.; Bopp, N.E.; et al. Protective immunity by an engineered DNA vaccine for mayaro virus. PLoS Negl. Trop. Dis. 2019, 13. [Google Scholar] [CrossRef]
- Aly, A.S.I.; Deveci, G.; Yilmaz, I.; Abraham, A.; Golshan, A.; Hart, R.J. Phenotypic Analysis of Rodent Malaria Parasite Asexual and Sexual Blood Stages and Mosquito Stages. JoVE J. Vis. Exp. 2019, e55688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, R.J.; Ghaffar, A.; Abdalal, S.; Perrin, B.; Aly, A.S.I. Plasmodium AdoMetDC/ODC bifunctional enzyme is essential for male sexual stage development and mosquito transmission. Biol. Open 2016, 5, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, R.J.; Cornillot, E.; Abraham, A.; Molina, E.; Nation, C.S.; Ben Mamoun, C.; Aly, A.S.I. Genetic characterization of plasmodium putative pantothenate kinase genes reveals their essential role in malaria parasite transmission to the mosquito. Sci. Rep. 2016, 6, 33518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aly, A.S.I.; Mikolajczak, S.A.; Rivera, H.S.; Camargo, N.; Jacobs-Lorena, V.; Labaied, M.; Coppens, I.; Kappe, S.H.I. Targeted deletion of SAP1 abolishes the expression of infectivity factors necessary for successful malaria parasite liver infection. Mol. Microbiol. 2008, 69, 152–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aly, A.S.I.; Lindner, S.E.; MacKellar, D.C.; Peng, X.; Kappe, S.H.I. SAP1 is a critical post-transcriptional regulator of infectivity in malaria parasite sporozoite stages. Mol. Microbiol. 2011, 79, 929–939. [Google Scholar] [CrossRef]
- Xu, Z.; Wise, M.C.; Choi, H.; Perales-Puchalt, A.; Patel, A.; Tello-Ruiz, E.; Chu, J.D.; Muthumani, K.; Weiner, D.B. Synthetic DNA delivery by electroporation promotes robust in vivo sulfation of broadly neutralizing anti-HIV immunoadhesin eCD4-Ig. EBioMedicine 2018, 35, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Wise, M.C.; Hutnick, N.A.; Pollara, J.; Myles, D.J.F.; Williams, C.; Yan, J.; LaBranche, C.C.; Khan, A.S.; Sardesai, N.Y.; Montefiori, D.; et al. An Enhanced Synthetic Multiclade DNA Prime Induces Improved Cross-Clade-Reactive Functional Antibodies when Combined with an Adjuvanted Protein Boost in Nonhuman Primates. J. Virol. 2015, 89, 9154–9166. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Reuschel, E.L.; Kraynyak, K.A.; Racine, T.; Park, D.H.; Scott, V.L.; Audet, J.; Amante, D.; Wise, M.C.; Keaton, A.A.; et al. Protective Efficacy and Long-Term Immunogenicity in Cynomolgus Macaques by Ebola Virus Glycoprotein Synthetic DNA Vaccines. J. Infect. Dis. 2019, 219, 544–555. [Google Scholar] [CrossRef]
- Modjarrad, K.; Roberts, C.C.; Mills, K.T.; Castellano, A.R.; Paolino, K.; Muthumani, K.; Reuschel, E.L.; Robb, M.L.; Racine, T.; Oh, M.; et al. Safety and immunogenicity of an anti-Middle East respiratory syndrome coronavirus DNA vaccine: A phase 1, open-label, single-arm, dose-escalation trial. Lancet Infect. Dis. 2019, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kalams, S.A.; Parker, S.D.; Elizaga, M.; Metch, B.; Edupuganti, S.; Hural, J.; De Rosa, S.; Carter, D.K.; Rybczyk, K.; Frank, I.; et al. Safety and comparative immunogenicity of an HIV-1 DNA vaccine in combination with plasmid interleukin 12 and impact of intramuscular electroporation for delivery. J. Infect. Dis. 2013, 208, 818–829. [Google Scholar] [CrossRef]
- Tebas, P.; Kraynyak, K.A.; Patel, A.; Maslow, J.N.; Morrow, M.P.; Sylvester, A.J.; Knoblock, D.; Gillespie, E.; Amante, D.; Racine, T.; et al. Intradermal SynCon® Ebola GP DNA Vaccine Is Temperature Stable and Safely Demonstrates Cellular and Humoral Immunogenicity Advantages in Healthy Volunteers. J. Infect. Dis. 2019, 220, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.A.; Snaith, R.; Cottingham, M.G.; Gilbert, S.C.; Hill, A.V.S. Enhancing protective immunity to malaria with a highly immunogenic virus-like particle vaccine. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villafana, T.; Lemnge, M.M.; Marsh, K.; Vansadia, P.; Bejon, P.; Demoitié, M.-A.; Carter, T.; Mturi, N.; Lang, T.; Njuguna, P.; et al. Efficacy of RTS,S/AS01E Vaccine against Malaria in Children 5 to 17 Months of Age. N. Engl. J. Med. 2008, 359, 2521–2532. [Google Scholar] [CrossRef] [Green Version]
- Moris, P.; Jongert, E.; van der Most, R.G. Characterization of T-cell immune responses in clinical trials of the candidate RTS,S malaria vaccine. Hum. Vaccines Immunother. 2018, 14, 17–27. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reeder, S.M.; Reuschel, E.L.; Bah, M.A.; Yun, K.; Tursi, N.J.; Kim, K.Y.; Chu, J.; Zaidi, F.I.; Yilmaz, I.; Hart, R.J.; et al. Synthetic DNA Vaccines Adjuvanted with pIL-33 Drive Liver-Localized T Cells and Provide Protection from Plasmodium Challenge in a Mouse Model. Vaccines 2020, 8, 21. https://doi.org/10.3390/vaccines8010021
Reeder SM, Reuschel EL, Bah MA, Yun K, Tursi NJ, Kim KY, Chu J, Zaidi FI, Yilmaz I, Hart RJ, et al. Synthetic DNA Vaccines Adjuvanted with pIL-33 Drive Liver-Localized T Cells and Provide Protection from Plasmodium Challenge in a Mouse Model. Vaccines. 2020; 8(1):21. https://doi.org/10.3390/vaccines8010021
Chicago/Turabian StyleReeder, Sophia M., Emma L. Reuschel, Mamadou A. Bah, Kun Yun, Nicholas J. Tursi, Kevin Y. Kim, Jacqueline Chu, Faraz I. Zaidi, Ilknur Yilmaz, Robert J. Hart, and et al. 2020. "Synthetic DNA Vaccines Adjuvanted with pIL-33 Drive Liver-Localized T Cells and Provide Protection from Plasmodium Challenge in a Mouse Model" Vaccines 8, no. 1: 21. https://doi.org/10.3390/vaccines8010021
APA StyleReeder, S. M., Reuschel, E. L., Bah, M. A., Yun, K., Tursi, N. J., Kim, K. Y., Chu, J., Zaidi, F. I., Yilmaz, I., Hart, R. J., Perrin, B., Xu, Z., Humeau, L., Weiner, D. B., & Aly, A. S. I. (2020). Synthetic DNA Vaccines Adjuvanted with pIL-33 Drive Liver-Localized T Cells and Provide Protection from Plasmodium Challenge in a Mouse Model. Vaccines, 8(1), 21. https://doi.org/10.3390/vaccines8010021