Humanized Mice for Live-Attenuated Vaccine Research: From Unmet Potential to New Promises


Abstract
:1. Introduction
2. Live-Attenuated Vaccines
2.1. Using Closely Related Pathogens as LAVs: The Smallpox Vaccine
2.2. Generating LAV through Pathogen Attenuation
2.2.1. Attenuation of Related Pathogens: The Tuberculosis Vaccine
2.2.2. Attenuation of Virulent Pathogens
Yellow Fever
Measles
Polio
Influenza
2.3. Investigating the Molecular Mechanisms Governing LAV Attenuation
3. The Quest for Suitable and Cost-Effective in Vivo Systems to Investigate LAVs
3.1. Of Mice, Men, and Non-Human Primates
3.2. Human Immune System Mouse Models
3.2.1. Development of Mouse Strains for Human Hematopoietic Stem Cell Engraftment
3.2.2. Second Generation Humanized Mouse Models and Emerging Models
Human Microenvironment and Lymphocyte Education
3.2.3. Engraftment Protocols and Variables
4. Contributions of HIS Mice to LAV Research
4.1. Assessing LAV Replication Fitness and Safety in HIS Mice
4.2. Investigating LAV-Induced Immunity in HIS Mice
4.3. Emerging Models for LAV Research
5. HIS Mice and LAV: Limitations and Future Opportunities
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Commission for the Certification of Smallpox Eradication; WHO: Geneva, Switzerland; WHO Publications Centre: Albany, NY, USA, 1980. [Google Scholar]
- World Health Organization. Measles. Available online: https://www.who.int/news-room/fact-sheets/detail/measles (accessed on 16 December 2019).
- Baxter, D. Active and passive immunity, vaccine types, excipients and licensing. Occup. Med. 2007, 57, 552–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staples, J.E.; Bocchini, J.A., Jr.; Rubin, L.; Fischer, M. Yellow Fever Vaccine Booster Doses: Recommendations of the Advisory Committee on Immunization Practices, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 64, 647–650. [Google Scholar] [PubMed]
- Malvy, D.; McElroy, A.K.; De Clerck, H.; Günther, S.; Van Griensven, J. Ebola virus disease. Lancet 2019, 393, 936–948. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. ZIKA Epidemiology Update; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Weber, D.J.; Rutala, W.A.; Fischer, W.A.; Kanamori, H.; Sickbert-Bennett, E.E. Emerging infectious diseases: Focus on infection control issues for novel coronaviruses (Severe Acute Respiratory Syndrome-CoV and Middle East Respiratory Syndrome-CoV), hemorrhagic fever viruses (Lassa and Ebola), and highly pathogenic avian influenza viruses, A(H5N1) and A(H7N9). Am. J. Infect. Control. 2016, 44, e91–e100. [Google Scholar] [CrossRef] [Green Version]
- Gill, C.M.; Beckham, J.D.; Piquet, A.L.; Tyler, K.L.; Pastula, D.M. Five Emerging Neuroinvasive Arboviral Diseases: Cache Valley, Eastern Equine Encephalitis, Jamestown Canyon, Powassan, and Usutu. Semin. Neurol. 2019, 39, 419–427. [Google Scholar] [CrossRef]
- Minor, P.D. Live attenuated vaccines: Historical successes and current challenges. Virology 2015, 479, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Frierson, J.G. The yellow fever vaccine: A history. Yale J. Biol. Med. 2010, 83, 77–85. [Google Scholar]
- Theocharides, A.P.; Rongvaux, A.; Fritsch, K.; Flavell, R.A.; Manz, M.G. Humanized hemato-lymphoid system mice. Haematologica 2016, 101, 5–19. [Google Scholar] [CrossRef]
- Douam, F.; Ploss, A. The use of humanized mice for studies of viral pathogenesis and immunity. Curr. Opin. Virol. 2018, 29, 62–71. [Google Scholar] [CrossRef]
- Gaska, J.M.; Ploss, A. Study of viral pathogenesis in humanized mice. Curr. Opin. Virol. 2015, 11, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Rongvaux, A.; Willinger, T.; Martinek, J.; Strowig, T.; Gearty, S.V.; Teichmann, L.L.; Saito, Y.; Marches, F.; Halene, S.; Palucka, A.K.; et al. Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 2014, 32, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Cosgun, K.N.; Rahmig, S.; Mende, N.; Reinke, S.; Hauber, I.; Schafer, C.; Petzold, A.; Weisbach, H.; Heidkamp, G.; Purbojo, A.; et al. Kit regulates HSC engraftment across the human-mouse species barrier. Cell Stem Cell 2014, 15, 227–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, A.; De, C.; Abad Fernandez, M.; Lenarcic, E.M.; Xu, Y.; Cockrell, A.S.; Cleary, R.A.; Johnson, C.E.; Schramm, N.J.; Rank, L.M.; et al. Precision mouse models with expanded tropism for human pathogens. Nat. Biotechnol. 2019, 37, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Ziegler, C.G.K.; Hrebikova, G.; Fant, B.; Leach, R.; Parsons, L.; Wang, W.; Gaska, J.M.; Winer, B.Y.; Heller, B.; et al. Selective expansion of myeloid and NK cells in humanized mice yields human-like vaccine responses. Nat. Commun. 2018, 9, 5031. [Google Scholar] [CrossRef] [Green Version]
- Sippel, T.R.; Radtke, S.; Olsen, T.M.; Kiem, H.P.; Rongvaux, A. Human hematopoietic stem cell maintenance and myeloid cell development in next-generation humanized mouse models. Blood Adv. 2019, 3, 268–274. [Google Scholar] [CrossRef]
- Chang, C. Time Frame and Reasons of Kangxi Emperor Adopted Variolation. Chin. J. Med. Hist. 1996, 26, 30–32. [Google Scholar]
- Griffiths, J. Doctor Thomas Dimsdale, and Smallpox in Russia: The Variolation of the Empress Catherine the Great. Bristol Med. Chir. J. 1984, 99, 14. [Google Scholar]
- Housz, J.M.I.; Beale, N.; Beale, E. The life of Dr Jan Ingen Housz (1730–99), private counsellor and personal physician to Emperor Joseph II of Austria. J. Med Biogr. 2005, 13, 15–21. [Google Scholar] [CrossRef]
- Imperato, P.J.; Imperato, G.H. Smallpox Inoculation (Variolation) in East Africa with Special Reference to the Practice Among the Boran and Gabra of Northern Kenya. J. Community Health 2014, 39, 1053–1062. [Google Scholar] [CrossRef]
- Ma, B. Variolation, pioneer of modern immunology. Chin. J. Med. Hist. 1995, 25, 139–144. [Google Scholar]
- Weiss, R.A.; Esparza, J. The prevention and eradication of smallpox: A commentary on Sloane (1755)‘An account of inoculation’. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenner, E. The Three Original Publications on Vaccination Against Smallpox; Eliot, C.W., Ed.; P.F. Collier & Son: New York, NY, USA; Volume 38, pp. 1909–1914.
- Pasteur, L. De l’attenuation du virus du choléra des poules. C. R. Acad. Sci. Paris 1880, 91, 673–680. [Google Scholar]
- Pasteur, L.; Chamberland, C.E.; Roux, E. Sur la vaccination charbonneuse. C. R. Acad. Sci. Paris 1881, 92, 1378–1383. [Google Scholar]
- Pasteur, L. Mèthode pour prévenir la rage apres morsure. C. R. Acad. Sci. Paris 1885, 101, 765–772. [Google Scholar]
- Calmette, A.; Guérin, C.; Boquet, A.; Nègre, L. La vaccination préventive contre la tuberculose par le “BCG”; Masson et cie: Paris, France, 1927. [Google Scholar]
- Matson, D.O. The Pentavalent Rotavirus Vaccine, RotaTeq™. Semin. Pediatr. Infect. Dis. 2006, 17, 195–199. [Google Scholar] [CrossRef]
- Plotkin, S. History of vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 12283–12287. [Google Scholar] [CrossRef] [Green Version]
- Theiler, M.; Smith, H.H. The effect of prolonged cultivation in vitro upon the pathogenicty of yellow fever virus. J. Exp. Med. 1937, 65, 767–786. [Google Scholar] [CrossRef]
- Sabin, A.B.; Hennessen, W.A.; Winsser, J. Studies on variants of poliomyelitis virus: I. Experimental segregation and properties of avirulent variants of three immunologic types. J. Exp. Med. 1954, 99, 551–576. [Google Scholar] [CrossRef] [Green Version]
- Enders, J.F.; Peebles, T.C. Propagation in Tissue Cultures of Cytopathogenic Agents from Patients with Measles. Proc. Soc. Exp. Biol. Med. 1954, 86, 277–286. [Google Scholar] [CrossRef]
- Buynak, E.B.; Hilleman, M.R. Live attenuated mumps virus vaccine. 1. Vaccine development. Proc. Soc. Exp. Biol. Med. 1966, 123, 768–775. [Google Scholar] [CrossRef]
- World Health Organization. Rubella vaccines: WHO position paper. Wkly. Epidemiol. Rec. 2011, 86, 301–316. [Google Scholar]
- World Health Organization. Typhoid vaccines: WHO position paper, March 2018—Recommendations. Vaccine 2019, 37, 214–216. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M. Development and characterization of a live varicella vaccine (Oka strain). Biken J. 1984, 27, 31–36. [Google Scholar] [PubMed]
- Oxman, M.; Levin, M.; Johnson, G.; Schmader, K.; Straus, S.; Gelb, L.; Arbeit, R.; Simberkoff, M.; Gershon, A.; Davis, L. A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N. Engl. J. Med. 2005, 352, 2271–2284. [Google Scholar] [CrossRef] [Green Version]
- Ravel, G.; Mantel, N.; Silvano, J.; Rogue, A.; Guy, B.; Jackson, N.; Burdin, N. Biodistribution and safety of a live attenuated tetravalent dengue vaccine in the cynomolgus monkey. Vaccine 2017, 35, 5918–5923. [Google Scholar] [CrossRef]
- Ginsburg, A.S.; Meghani, A.; Halstead, S.B.; Yaich, M. Use of the live attenuated Japanese Encephalitis vaccine SA 14-14-2 in children: A review of safety and tolerability studies. Hum. Vaccin. Immunother. 2017, 13, 2222–2231. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.; Miller, C.; Catalan, J.; Myers, G.A.; Ratterree, M.S.; Trent, D.W.; Monath, T.P. ChimeriVax-West Nile virus live-attenuated vaccine: Preclinical evaluation of safety, immunogenicity, and efficacy. J. Virol. 2004, 78, 12497–12507. [Google Scholar] [CrossRef] [Green Version]
- Shan, C.; Muruato, A.E.; Jagger, B.W.; Richner, J.; Nunes, B.T.D.; Medeiros, D.B.A.; Xie, X.P.; Nunes, J.G.C.; Morabito, K.M.; Kong, W.P.; et al. A single-dose live-attenuated vaccine prevents Zika virus pregnancy transmission and testis damage. Nat. Commun. 2017, 8, 676. [Google Scholar] [CrossRef]
- Escriou, N.; Callendret, B.; Lorin, V.; Combredet, C.; Marianneau, P.; Fevrier, M.; Tangy, F. Protection from SARS coronavirus conferred by live measles vaccine expressing the spike glycoprotein. Virology 2014, 452, 32–41. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.E.; Tewari, K.; Lyke, K.E.; Sim, B.K.L.; Billingsley, P.F.; Laurens, M.B.; Gunasekera, A.; Chakravarty, S.; James, E.R.; Sedegah, M.; et al. Live Attenuated Malaria Vaccine Designed to Protect Through Hepatic CD8(+) T Cell Immunity. Science 2011, 334, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Coller, B.-A.G.; Blue, J.; Das, R.; Dubey, S.; Finelli, L.; Gupta, S.; Helmond, F.; Grant-Klein, R.J.; Liu, K.; Simon, J.; et al. Clinical development of a recombinant Ebola vaccine in the midst of an unprecedented epidemic. Vaccine 2017, 35, 4465–4469. [Google Scholar] [CrossRef] [PubMed]
- Regules, J.A.; Beigel, J.H.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Munoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A Recombinant Vesicular Stomatitis Virus Ebola Vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Carroll, D.S.; Gardner, S.N.; Walsh, M.C.; Vitalis, E.A.; Damon, I.K. On the origin of smallpox: Correlating variola phylogenics with historical smallpox records. Proc. Natl. Acad. Sci. USA 2007, 104, 15787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voigt, E.A.; Kennedy, R.B.; Poland, G.A. Defending against smallpox: A focus on vaccines. Expert Rev. Vaccines 2016, 15, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behbehani, A.M. The smallpox story: Life and death of an old disease. Microbiol. Rev. 1983, 47, 455–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenner, F.; Henderson, D.A.; Arita, I.; Jezek, Z.; Ladnyi, I.D.; World Health Organization. Smallpox and Its Eradication; WHO: Geneva, Switzerland, 1988. [Google Scholar]
- WHO. Requirements for Biological Substances: 5. Requirements for Smallpox Vaccine, Report of a Study Group [Meeting Held in Geneva From 3 to 8 November 1958]; WHO: Geneva, Switzerland, 1959. [Google Scholar]
- Shackell, L. An improved method of desiccation, with some applications to biological problems. Am. J. Physiol. Leg. Content 1909, 24, 325–340. [Google Scholar] [CrossRef]
- Rubin, B.A. A note on the development of the bifurcated needle for smallpox vaccination. WHO Chron. 1980, 34, 180–181. [Google Scholar]
- Cono, J.; Casey, C.G.; Bell, D.M. Smallpox vaccination and adverse reactions. Guidance for clinicians. MMWR Recomm. Rep. 2003, 52, 1–28. [Google Scholar]
- Petersen, B.W.; Damon, I.K.; Pertowski, C.A.; Meaney-Delman, D.; Guarnizo, J.T.; Beigi, R.H.; Edwards, K.M.; Fisher, M.C.; Frey, S.E.; Lynfield, R.; et al. Clinical guidance for smallpox vaccine use in a postevent vaccination program. MMWR Recomm. Rep. 2015, 64, 1–26. [Google Scholar] [PubMed]
- Springer, Y.P.; Hsu, C.H.; Werle, Z.R.; Olson, L.E.; Cooper, M.P.; Castrodale, L.J.; Fowler, N.; McCollum, A.M.; Goldsmith, C.S.; Emerson, G.L.; et al. Novel Orthopoxvirus Infection in an Alaska Resident. Clin. Infect. Dis. 2017, 64, 1737–1741. [Google Scholar] [CrossRef]
- Lanave, G.; Dowgier, G.; Decaro, N.; Albanese, F.; Brogi, E.; Parisi, A.; Losurdo, M.; Lavazza, A.; Martella, V.; Buonavoglia, C.; et al. Novel Orthopoxvirus and Lethal Disease in Cat, Italy. Emerg. Infect. Dis. 2018, 24, 1665–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balamurugan, V.; Venkatesan, G.; Bhanuprakash, V.; Singh, R.K. Camelpox, an emerging orthopox viral disease. Indian J. Virol. 2013, 24, 295–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durski, K.N.; McCollum, A.M.; Nakazawa, Y.; Petersen, B.W.; Reynolds, M.G.; Briand, S.; Djingarey, M.H.; Olson, V.; Damon, I.K.; Khalakdina, A. Emergence of monkeypox—West and central Africa, 1970–2017. Morb. Mortal. Wkly. Rep. 2018, 67, 306. [Google Scholar] [CrossRef] [PubMed]
- Nalca, A.; Zumbrun, E.E. ACAM2000: The new smallpox vaccine for United States Strategic National Stockpile. Drug. Des. Dev. Ther. 2010, 4, 71–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.H.; Kang, Y.M.; Kim, G.; Choe, P.G.; Song, J.S.; Lee, K.H.; Seong, B.L.; Park, W.B.; Kim, N.J.; Oh, M.D. An open-label, single arm, phase III clinical study to evaluate the efficacy and safety of CJ smallpox vaccine in previously vaccinated healthy adults. Vaccine 2013, 31, 5239–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, J.S.; Greenberg, R.N. IMVAMUNE®: Modified vaccinia Ankara strain as an attenuated smallpox vaccine. Expert Rev. Vaccines 2009, 8, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wilck, M.B.; Seaman, M.S.; Baden, L.R.; Walsh, S.R.; Grandpre, L.E.; Devoy, C.; Giri, A.; Kleinjan, J.A.; Noble, L.C.; Stevenson, K.E.; et al. Safety and immunogenicity of modified vaccinia Ankara (ACAM3000): Effect of dose and route of administration. J. Infect. Dis. 2010, 201, 1361–1370. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Tuberculosis Report 2018; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Houben, R.M.G.J.; Dodd, P.J. The Global Burden of Latent Tuberculosis Infection: A Re-estimation Using Mathematical Modelling. PLoS Med. 2016, 13, e1002152. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. BCG vaccine: WHO position paper, February 2018—Recommendations. Vaccine 2018, 36, 3408–3410. [Google Scholar] [CrossRef]
- Barberis, I.; Bragazzi, N.L.; Galluzzo, L.; Martini, M. The history of tuberculosis: From the first historical records to the isolation of Koch’s bacillus. J. Prev. Med. Hyg. 2017, 58, E9–E12. [Google Scholar]
- Smith, K.C.; Orme, I.M.; Starke, J.R. Tuberculosis vaccines. In Vaccines; Elsevier: Amsterdam, The Netherlands, 2013; pp. 789–811. [Google Scholar]
- Fine, P.E.; Carneiro, I.A.; Milstien, J.B.; Clements, C.J.; World Health Organization. Issues Relating to the Use of BCG in Immunization Programmes: A Discussion Document; World Health Organization: Geneva, Switzerland, 1999. [Google Scholar]
- Lange, B. Weitere Untersuchungen zur Klärung der Ursachen der Unglücksfälle in Lübeck. Tuberk 1931, 62, 335–351. [Google Scholar]
- Abubakar, I.; Pimpin, L.; Ariti, C.; Beynon, R.; Mangtani, P.; Sterne, J.A.; Fine, P.E.; Smith, P.G.; Lipman, M.; Elliman, D.; et al. Systematic review and meta-analysis of the current evidence on the duration of protection by bacillus Calmette-Guerin vaccination against tuberculosis. Health Technol. Assess. 2013, 17. [Google Scholar] [CrossRef] [PubMed]
- Mangtani, P.; Abubakar, I.; Ariti, C.; Beynon, R.; Pimpin, L.; Fine, P.E.; Rodrigues, L.C.; Smith, P.G.; Lipman, M.; Whiting, P.F.; et al. Protection by BCG vaccine against tuberculosis: A systematic review of randomized controlled trials. Clin. Infect. Dis. 2014, 58, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trunz, B.B.; Fine, P.; Dye, C. Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: A meta-analysis and assessment of cost-effectiveness. Lancet 2006, 367, 1173–1180. [Google Scholar] [CrossRef]
- Roy, A.; Eisenhut, M.; Harris, R.J.; Rodrigues, L.C.; Sridhar, S.; Habermann, S.; Snell, L.; Mangtani, P.; Adetifa, I.; Lalvani, A.; et al. Effect of BCG vaccination against Mycobacterium tuberculosis infection in children: Systematic review and meta-analysis. BMJ 2014, 349, g4643. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Vaccines against influenza WHO position paper—November 2012. Wkly. Epidemiol. Rec. Relev. Épidémiologique Hebd. 2012, 87, 461–476. [Google Scholar]
- Theiler, M. The Development of Vaccines Against Yellow Fever; Kungl. Boktryckeriet PA Norstedt: Stockholm, Sweden, 1952. [Google Scholar]
- Norrby, E. Yellow fever and Max Theiler: The only Nobel Prize for a virus vaccine. J. Exp. Med. 2007, 204, 2779–2784. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Yellow fever vaccine: WHO position paper. Wkly. Epidemiol. Rec. Geneva 2003, 78, 349. [Google Scholar]
- Patterson, K.D. Yellow fever epidemics and mortality in the United States, 1693–1905. Soc. Sci. Med. 1992, 34, 855–865. [Google Scholar] [CrossRef]
- Chippaux, J.P.; Chippaux, A. Yellow fever in Africa and the Americas: A historical and epidemiological perspective. J. Venom. Anim. Toxins. Incl. Trop. Dis. 2018, 24, 20. [Google Scholar] [CrossRef] [Green Version]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends. Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Stokes, A.; Bauer, J.H.; Hudson, N.P. The Transmission of Yellow Fever to Macacus Rhesus: Preliminary Note. JAMA 1928, 90, 253–254. [Google Scholar] [CrossRef]
- Mathis, C.; Sellards, A.W.; Laigret, J. Sensibilité du Macacus rhesus au virus de la fièvre jaune. C.R. Acad. Sci. 1928, 186, 604–606. [Google Scholar]
- Theiler, M.; Smith, H.H. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 1937, 65, 787–800. [Google Scholar] [CrossRef] [Green Version]
- Monath, T.P. 17D Yellow Fever Virus Vaccine. Am. J. Trop. Med. Hyg. 2013, 89, 1225. [Google Scholar] [CrossRef] [Green Version]
- Monath, T.P. Yellow fever: An update. Lancet Infect. Dis. 2001, 1, 11–20. [Google Scholar] [CrossRef]
- Poland, J.D.; Calisher, C.H.; Monath, T.P.; Downs, W.G.; Murphy, K. Persistence of neutralizing antibody 30-35 years after immunization with 17D yellow fever vaccine. Bull. World Health Organ. 1981, 59, 895–900. [Google Scholar]
- Casey, R.M.; Harris, J.B.; Ahuka-Mundeke, S.; Dixon, M.G.; Kizito, G.M.; Nsele, P.M.; Umutesi, G.; Laven, J.; Kosoy, O.; Paluku, G.; et al. Immunogenicity of Fractional-Dose Vaccine during a Yellow Fever Outbreak—Final Report. N. Engl. J. Med. 2018, 381, 444–454. [Google Scholar] [CrossRef]
- Griffin, D.E. Measles Vaccine. Viral Immunol. 2018, 31, 86–95. [Google Scholar] [CrossRef]
- World Health Organization. Global Measles and Rubella Strategic Plan; WHO: Geneva, Switzerland, 2012. [Google Scholar]
- Makino, S. Development and Characteristics of Live AIK-C Measles Virus Vaccine: A Brief Report. Clin. Infect. Dis. 1983, 5, 504–505. [Google Scholar] [CrossRef]
- Schwarz, A.J.F. Preliminary Tests of a Highly Attenuated Measles Vaccine. JAMA Pediatr. 1962, 103, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Ikic, D.; Beck, M.; Juzbasic, M. Characterization of Edmonston-Zagreb measles virus. In Proc. Symposium on Human Diploid Cells; Yugoslav Academy of Sciences and Arts: Zagreb, Yugoslavia, 1970; pp. 121–129. [Google Scholar]
- Hilleman, M.R.; Buynak, E.B.; Weibel, R.E.; Stokes, J., Jr.; Whitman, J.E., Jr.; Leagus, M.B. Development and Evaluation of the Moraten Measles Virus Vaccine. JAMA 1968, 206, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Takahashi, M.; Minekawa, Y.; Ogino, T.; Suzuki, N. Studies on further attenuated live measles vaccine. I. Adaptation of measles virus to the chorioallantoic membrane of chick embryo and clinical tests on the strain. Biken J. 1970, 13, 111–116. [Google Scholar] [PubMed]
- Smorodintsev, A.; Boichuk, L.; Shikina, E.; Batanova, T.; Bystryakova, L.; Peradze, T. Clinical and immunological response to live tissue culture vaccine against measles. Acta Virol. 1960, 4, 201–214. [Google Scholar] [PubMed]
- Bankamp, B.; Takeda, M.; Zhang, Y.; Xu, W.; Rota, P.A. Genetic Characterization of Measles Vaccine Strains. J. Infect. Dis. 2011, 204, S533–S548. [Google Scholar] [CrossRef] [Green Version]
- Dabbagh, A.; Laws, R.L.; Steulet, C.; Dumolard, L.; Mulders, M.N.; Kretsinger, K.; Alexander, J.P.; Rota, P.A.; Goodson, J.L. Progress Toward Regional Measles Elimination—Worldwide, 2000–2017. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1323–1329. [Google Scholar] [CrossRef]
- World Health Organization. Global Vaccine Action Plan 2011–2020; WHO: Geneva, Switzerland, 2013. [Google Scholar]
- Minor, P.D. The polio-eradication programme and issues of the end game. J. Gen. Virol. 2012, 93, 457–474. [Google Scholar] [CrossRef] [Green Version]
- Sabin, A.B. Pathogenesis of Poliomyelitis Reappraisal in the Light of New Data. Science 1956, 123, 1151. [Google Scholar] [CrossRef]
- Bodian, D. Emerging concept of poliomyelitis infection. Science 1955, 122, 105–108. [Google Scholar] [CrossRef]
- Sabin, A.B.; Ward, R. The natural history of human poliomyelitis: I. Distribution of virus in nervous and non-nervous tissues. J. Exp. Med. 1941, 73, 771–793. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A.; Orenstein, W.A.; Offit, P.A. Vaccines; Elsevier/Saunders: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Nathanson, N.; Kew, O.M. From emergence to eradication: The epidemiology of poliomyelitis deconstructed. Am. J. Epidemiol. 2010, 172, 1213–1229. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Biellik, R. Inactivated polio vaccine: Its proposed role in the final stages of polio eradication. Pan. Afr. Med. J. 2013, 14, 102. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global eradication of poliomyelitis by the year 2000. Wkly. Epidemiol. Rec. Relev. épidémiologique Hebd. 1988, 63, 161–162. [Google Scholar]
- World Health Organization. Polio Endgame Strategy 2019–2023: Eradication, Integration, Certification and Containment; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Global Polio Eradication Initiative. Global eradication of wild poliovirus type 2 declared. Retrieved Novemb. 2015, 30, 2015. [Google Scholar]
- Dyer, O. Polio: WHO declares type 3 poliovirus eradicated after 31 year campaign. BMJ 2019, 367, l6201. [Google Scholar] [CrossRef]
- Cox, R.; Brokstad, K.; Ogra, P. Influenza virus: Immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand. J. Immunol. 2004, 59, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shrestha, S.; Foxman, B.; Berus, J.; Van Panhuis, W.G.; Steiner, C.; Viboud, C.; Rohani, P. The role of influenza in the epidemiology of pneumonia. Sci. Rep. 2015, 5, 15314. [Google Scholar] [CrossRef]
- Arriola, C.; Garg, S.; Anderson, E.J.; Ryan, P.A.; George, A.; Zansky, S.M.; Bennett, N.; Reingold, A.; Bargsten, M.; Miller, L.; et al. Influenza Vaccination Modifies Disease Severity Among Community-dwelling Adults Hospitalized With Influenza. Clin. Infect. Dis. 2017, 65, 1289–1297. [Google Scholar] [CrossRef]
- Smith, W.; Andrewes, C.H.; Laidlaw, P.P. A Virus Obtained from Influenza Patients. Lancet 1933, 222, 66–68. [Google Scholar] [CrossRef] [Green Version]
- Burnet, F. Influenza virus infections of the chick embryo lung. Br. J. Exp. Pathol. 1940, 21, 147. [Google Scholar]
- Stanley, W. The preparation and properties of influenza virus vaccines concentrated and purified by differential centrifugation. J. Exp. Med. 1945, 81, 193–218. [Google Scholar] [CrossRef] [PubMed]
- Barberis, I.; Myles, P.; Ault, S.K.; Bragazzi, N.L.; Martini, M. History and evolution of influenza control through vaccination: From the first monovalent vaccine to universal vaccines. J. Prev. Med. Hyg. 2016, 57, E115–E120. [Google Scholar] [PubMed]
- Wareing, M.D.; Tannock, G.A. Live attenuated vaccines against influenza; an historical review. Vaccine 2001, 19, 3320–3330. [Google Scholar] [CrossRef]
- Alexandrova, G.; Smorodintsev, A. Obtaining of an Additionally Attenuated Vaccinating Cryophilic Influenza Strain. Rev. Roum. Inframicrobiol. 1965, 2, 179–186. [Google Scholar]
- Maassab, H. Adaptation and growth characteristics of influenza virus at 25 C. Nature 1967, 213, 612. [Google Scholar] [CrossRef]
- Justewicz, D.M.; Morin, M.J.; Robinson, H.L.; Webster, R.G. Antibody-forming cell response to virus challenge in mice immunized with DNA encoding the influenza virus hemagglutinin. J. Virol. 1995, 69, 7712–7717. [Google Scholar] [CrossRef] [Green Version]
- Rudenko, L.G.; Lonskaya, N.I.; Klimov, A.I.; Vasilieva, R.I.; Ramirez, A. Clinical and epidemiological evaluation of a live, cold-adapted influenza vaccine for 3–14-year-olds. Bull. World Health Organ. 1996, 74, 77–84. [Google Scholar]
- Herrlich, A.; Mayr, A.; Mahnel, H.; Munz, E. Experimental studies on transformation of the variola virus into the vaccinia virus. Arch. Für Die Gesamte Virusforsch. 1963, 12, 579–599. [Google Scholar] [CrossRef]
- Evans, D.M.A.; Dunn, G.; Minor, P.D.; Schild, G.C.; Cann, A.J.; Stanway, G.; Almond, J.W.; Currey, K.; Maizel, J.V. Increased neurovirulence associated with a single nucleotide change in a noncoding region of the Sabin type 3 poliovaccine genome. Nature 1985, 314, 548–550. [Google Scholar] [CrossRef]
- Guillot, S.; Otelea, D.; Delpeyroux, F.; Crainic, R. Point mutations involved in the attenuation/neurovirulence alternation in type 1 and 2 oral polio vaccine strains detected by site-specific polymerase chain reaction. Vaccine 1994, 12, 503–507. [Google Scholar] [CrossRef]
- Hahn, C.S.; Dalrymple, J.M.; Strauss, J.H.; Rice, C.M. Comparison of the virulent Asibi strain of yellow fever virus with the 17D vaccine strain derived from it. Proc. Natl. Acad. Sci. USA 1987, 84, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, C.L.; Lerch, R.A.; Walpita, P.; Wang, H.-P.; Sidhu, M.S.; Udem, S.A. Comparison of Predicted Amino Acid Sequences of Measles Virus Strains in the Edmonston Vaccine Lineage. J. Virol. 2001, 75, 910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahairas, G.G.; Sabo, P.J.; Hickey, M.J.; Singh, D.C.; Stover, C.K. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 1996, 178, 1274. [Google Scholar] [CrossRef] [Green Version]
- Garnier, T.; Eiglmeier, K.; Camus, J.C.; Medina, N.; Mansoor, H.; Pryor, M.; Duthoy, S.; Grondin, S.; Lacroix, C.; Monsempe, C.; et al. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 2003, 100, 7877–7882. [Google Scholar] [CrossRef] [Green Version]
- Bartz, R.; Brinckmann, U.; Dunster, L.M.; Rima, B.; Ter Meulen, V.; Schneider-Schaulies, J. Mapping amino acids of the measles virus hemagglutinin responsible for receptor (CD46) downregulation. Virology 1996, 224, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.B.; Ohgimoto, S.; Kato, S.; Kurazono, S.; Ayata, M.; Takeuchi, K.; Ihara, T.; Ogura, H. Contribution of matrix, fusion, hemagglutinin, and large protein genes of the CAM-70 measles virus vaccine strain to efficient growth in chicken embryonic fibroblasts. J. Virol. 2009, 83, 11645–11654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Garcia, M.D.; Meertens, L.; Chazal, M.; Hafirassou, M.L.; Dejarnac, O.; Zamborlini, A.; Despres, P.; Sauvonnet, N.; Arenzana-Seisdedos, F.; Jouvenet, N.; et al. Vaccine and Wild-Type Strains of Yellow Fever Virus Engage Distinct Entry Mechanisms and Differentially Stimulate Antiviral Immune Responses. MBio 2016, 7, e01956-15. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.; Lobigs, M. E protein domain III determinants of yellow fever virus 17D vaccine strain enhance binding to glycosaminoglycans, impede virus spread, and attenuate virulence. J. Virol. 2008, 82, 6024–6033. [Google Scholar] [CrossRef] [Green Version]
- Lefeuvre, A.; Contamin, H.; Decelle, T.; Fournier, C.; Lang, J.; Deubel, V.; Marianneau, P. Host-cell interaction of attenuated and wild-type strains of yellow fever virus can be differentiated at early stages of hepatocyte infection. Microbes Infect. 2006, 8, 1530–1538. [Google Scholar] [CrossRef]
- Woodson, S.E.; Holbrook, M.R. Infection of hepatocytes with 17-D vaccine-strain yellow fever virus induces a strong pro-inflammatory host response. J. Gen. Virol. 2011, 92, 2262–2271. [Google Scholar] [CrossRef]
- Woodson, S.E.; Freiberg, A.N.; Holbrook, M.R. Coagulation factors, fibrinogen and plasminogen activator inhibitor-1, are differentially regulated by yellow fever virus infection of hepatocytes. Virus Res. 2013, 175, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Woodson, S.E.; Freiberg, A.N.; Holbrook, M.R. Differential cytokine responses from primary human Kupffer cells following infection with wild-type or vaccine strain yellow fever virus. Virology 2011, 412, 188–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaiboullina, S.F.; Rizvanov, A.A.; Holbrook, M.R.; St Jeor, S. Yellow fever virus strains Asibi and 17D-204 infect human umbilical cord endothelial cells and induce novel changes in gene expression. Virology 2005, 342, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, Y.; McArthur, M.A.; Cohen, M.; Jahrling, P.B.; Janosko, K.B.; Josleyn, N.; Kang, K.; Zhang, T.F.; Holbrook, M.R. Characterization of Yellow Fever Virus Infection of Human and Non-human Primate Antigen Presenting Cells and Their Interaction with CD4(+) T Cells. PLoS Negl. Trop. Dis. 2016, 10. [Google Scholar] [CrossRef]
- Guest, S.; Pilipenko, E.; Sharma, K.; Chumakov, K.; Roos, R.P. Molecular Mechanisms of Attenuation of the Sabin Strain of Poliovirus Type 3. J. Virol. 2004, 78, 11097. [Google Scholar] [CrossRef] [Green Version]
- Barba-Spaeth, G.; Longman, R.S.; Albert, M.L.; Rice, C.M. Live attenuated yellow fever 17D infects human DCs and allows for presentation of endogenous and recombinant T cell epitopes. J. Exp. Med. 2005, 202, 1179–1184. [Google Scholar] [CrossRef]
- Palmer, D.R.; Fernandez, S.; Bisbing, J.; Peachman, K.K.; Rao, M.; Barvir, D.; Gunther, V.; Burgess, T.; Kohno, Y.; Padmanabhan, R.; et al. Restricted replication and lysosomal trafficking of yellow fever 17D vaccine virus in human dendritic cells. J. Gen. Virol. 2007, 88, 148–156. [Google Scholar] [CrossRef]
- Beck, A.; Tesh, R.B.; Wood, T.G.; Widen, S.G.; Ryman, K.D.; Barrett, A.D. Comparison of the live attenuated yellow fever vaccine 17D-204 strain to its virulent parental strain Asibi by deep sequencing. J. Infect. Dis. 2014, 209, 334–344. [Google Scholar] [CrossRef]
- Davis, E.H.; Beck, A.S.; Strother, A.E.; Thompson, J.K.; Widen, S.G.; Higgs, S.; Wood, T.G.; Barrett, A.D.T. Attenuation of Live-Attenuated Yellow Fever 17D Vaccine Virus Is Localized to a High-Fidelity Replication Complex. Mbio 2019, 10, e02294-19. [Google Scholar] [CrossRef] [Green Version]
- Hsu, T.; Hingley-Wilson, S.M.; Chen, B.; Chen, M.; Dai, A.Z.; Morin, P.M.; Marks, C.B.; Padiyar, J.; Goulding, C.; Gingery, M.; et al. The primary mechanism of attenuation of bacillus Calmette–Guérin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc. Natl. Acad. Sci. USA 2003, 100, 12420. [Google Scholar] [CrossRef] [Green Version]
- Watson, A.M.; Lam, L.K.; Klimstra, W.B.; Ryman, K.D. The 17D-204 Vaccine Strain-Induced Protection against Virulent Yellow Fever Virus Is Mediated by Humoral Immunity and CD4+ but not CD8+ T Cells. PLoS Pathog. 2016, 12, e1005786. [Google Scholar] [CrossRef] [PubMed]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, L.K.M.; Watson, A.M.; Ryman, K.D.; Klimstra, W.B. Gamma-interferon exerts a critical early restriction on replication and dissemination of yellow fever virus vaccine strain 17D-204. Npj Vaccines 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankamp, B.; Hodge, G.; McChesney, M.B.; Bellini, W.J.; Rota, P.A. Genetic changes that affect the virulence of measles virus in a rhesus macaque model. Virology 2008, 373, 39–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelmann, F.; Josset, L.; Girke, T.; Park, B.; Barron, A.; Dewane, J.; Hammarlund, E.; Lewis, A.; Axthelm, M.K.; Slifka, M.K.; et al. Pathophysiologic and transcriptomic analyses of viscerotropic yellow fever in a rhesus macaque model. PLoS Negl. Trop. Dis. 2014, 8, e3295. [Google Scholar] [CrossRef] [Green Version]
- McGoldrick, A.; Macadam, A.J.; Dunn, G.; Rowe, A.; Burlison, J.; Minor, P.D.; Meredith, J.; Evans, D.J.; Almond, J.W. Role of mutations G-480 and C-6203 in the attenuation phenotype of Sabin type 1 poliovirus. J. Virol. 1995, 69, 7601. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, M.J.; Lam, D.H.; Racaniello, V.R. Determinants of attenuation and temperature sensitivity in the type 1 poliovirus Sabin vaccine. J. Virol. 1995, 69, 4972. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.S.; Roos, J.M.; McGuigan, L.C.; Smith, K.A.; Cormier, N.; Cohen, L.K.; Roberts, B.E.; Payne, L.G. Molecular attenuation of vaccinia virus: Mutant generation and animal characterization. J. Virol. 1992, 66, 2617. [Google Scholar] [CrossRef] [Green Version]
- Tscharke, D.C.; Reading, P.C.; Smith, G.L. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J. Gen. Virol. 2002, 83, 1977–1986. [Google Scholar] [CrossRef]
- Darrah, P.A.; Zeppa, J.J.; Maiello, P.; Hackney, J.A.; Wadsworth, M.H., 2nd; Hughes, T.K.; Pokkali, S.; Swanson, P.A., 2nd; Grant, N.L.; Rodgers, M.A.; et al. Prevention of tuberculosis in macaques after intravenous BCG immunization. Nature 2020, 577, 95–102. [Google Scholar] [CrossRef]
- Naniche, D.; Yeh, A.; Eto, D.; Manchester, M.; Friedman, R.M.; Oldstone, M.B. Evasion of host defenses by measles virus: Wild-type measles virus infection interferes with induction of alpha/beta interferon production. J. Virol. 2000, 74, 7478–7484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatsu, Y.; Takeda, M.; Ohno, S.; Koga, R.; Yanagi, Y. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J. Virol. 2006, 80, 11861–11867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, S.; Ono, N.; Takeda, M.; Takeuchi, K.; Yanagi, Y. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J. Gen. Virol. 2004, 85, 2991–2999. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Kadota, S.-i.; Takeda, M.; Miyajima, N.; Nagata, K. Measles virus V protein blocks interferon (IFN)-α/β but not IFN-γ signaling by inhibiting STAT1 and STAT2 phosphorylation. FEBS Lett. 2003, 545, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Yokota, S.-i.; Okabayashi, T.; Yokosawa, N.; Fujii, N. Growth arrest of epithelial cells during measles virus infection is caused by upregulation of interferon regulatory factor 1. J. Virol. 2004, 78, 4591–4598. [Google Scholar] [CrossRef] [Green Version]
- Shivakoti, R.; Siwek, M.; Hauer, D.; Schultz, K.L.W.; Griffin, D.E. Induction of Dendritic Cell Production of Type I and Type III Interferons by Wild-Type and Vaccine Strains of Measles Virus: Role of Defective Interfering RNAs. J. Virol. 2013, 87, 7816. [Google Scholar] [CrossRef] [Green Version]
- Querec, T.; Bennouna, S.; Alkan, S.; Laouar, Y.; Gorden, K.; Flavell, R.; Akira, S.; Ahmed, R.; Pulendran, B. Yellow fever vaccine YF-17D activates multiple dendritic cell subsets via TLR2, 7, 8, and 9 to stimulate polyvalent immunity. J. Exp. Med. 2006, 203, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Akondy, R.S.; Fitch, M.; Edupuganti, S.; Yang, S.; Kissick, H.T.; Li, K.W.; Youngblood, B.A.; Abdelsamed, H.A.; McGuire, D.J.; Cohen, K.W.; et al. Origin and differentiation of human memory CD8 T cells after vaccination. Nature 2017, 552, 362–367. [Google Scholar] [CrossRef]
- Akondy, R.S.; Johnson, P.L.; Nakaya, H.I.; Edupuganti, S.; Mulligan, M.J.; Lawson, B.; Miller, J.D.; Pulendran, B.; Antia, R.; Ahmed, R. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. USA 2015, 112, 3050–3055. [Google Scholar] [CrossRef] [Green Version]
- Akondy, R.S.; Monson, N.D.; Miller, J.D.; Edupuganti, S.; Teuwen, D.; Wu, H.; Quyyumi, F.; Garg, S.; Altman, J.D.; Del Rio, C.; et al. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 2009, 183, 7919–7930. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.D.; Van der Most, R.G.; Akondy, R.S.; Glidewell, J.T.; Albott, S.; Masopust, D.; Murali-Krishna, K.; Mahar, P.L.; Edupuganti, S.; Lalor, S.; et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 2008, 28, 710–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomoto, A.; Omata, T.; Toyoda, H.; Kuge, S.; Horie, H.; Kataoka, Y.; Genba, Y.; Nakano, Y.; Imura, N. Complete nucleotide sequence of the attenuated poliovirus Sabin 1 strain genome. Proc. Natl. Acad. Sci. USA 1982, 79, 5793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, S.R.; Dunn, G.; Cammack, N.; Minor, P.D.; Almond, J.W. Nucleotide sequence of a neurovirulent variant of the type 2 oral poliovirus vaccine. J. Virol. 1989, 63, 4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanway, G.; Hughes, P.J.; Mountford, R.C.; Reeve, P.; Minor, P.D.; Schild, G.C.; Almond, J.W. Comparison of the complete nucleotide sequences of the genomes of the neurovirulent poliovirus P3/Leon/37 and its attenuated Sabin vaccine derivative P3/Leon 12a1b. Proc. Natl. Acad. Sci. USA 1984, 81, 1539–1543. [Google Scholar] [CrossRef] [Green Version]
- Alcamí, A.; Symons, J.A.; Collins, P.D.; Williams, T.J.; Smith, G.L. Blockade of Chemokine Activity by a Soluble Chemokine Binding Protein from Vaccinia Virus. J. Immunol. 1998, 160, 624. [Google Scholar]
- He, Y.; Manischewitz, J.; Meseda, C.A.; Merchlinsky, M.; Vassell, R.A.; Sirota, L.; Berkower, I.; Golding, H.; Weiss, C.D. Antibodies to the A27 protein of vaccinia virus neutralize and protect against infection but represent a minor component of Dryvax vaccine--induced immunity. J. Infect. Dis. 2007, 196, 1026–1032. [Google Scholar] [CrossRef]
- Kennedy, R.B.; Poland, G.A. The identification of HLA class II-restricted T cell epitopes to vaccinia virus membrane proteins. Virology 2010, 408, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Parkinson, J.E.; Smith, G.L. Vaccinia Virus Gene A36R Encodes a Mr 43-50 K Protein on the Surface of Extracellular Enveloped Virus. Virology 1994, 204, 376–390. [Google Scholar] [CrossRef]
- Leung, A.S.; Tran, V.; Wu, Z.; Yu, X.; Alexander, D.C.; Gao, G.F.; Zhu, B.; Liu, J. Novel genome polymorphisms in BCG vaccine strains and impact on efficacy. BMC Genom. 2008, 9, 413. [Google Scholar] [CrossRef] [Green Version]
- Lewis, K.N.; Liao, R.; Guinn, K.M.; Hickey, M.J.; Smith, S.; Behr, M.A.; Sherman, D.R. Deletion of RD1 from Mycobacterium tuberculosis Mimics Bacille Calmette-Guérin Attenuation. J. Infect. Dis. 2003, 187, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Mattow, J.; Jungblut, P.R.; Schaible, U.E.; Mollenkopf, H.-J.; Lamer, S.; Zimny-Arndt, U.; Hagens, K.; Müller, E.-C.; Kaufmann, S.H.E. Identification of proteins from Mycobacterium tuberculosis missing in attenuated Mycobacterium bovis BCG strains. Electrophoresis 2001, 22, 2936–2946. [Google Scholar] [CrossRef]
- Sil, B.K.; Dunster, L.M.; Ledger, T.N.; Wills, M.R.; Minor, P.D.; Barrett, A.D. Identification of envelope protein epitopes that are important in the attenuation process of wild-type yellow fever virus. J. Virol. 1992, 66, 4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyajima, N.; Takeda, M.; Tashiro, M.; Hashimoto, K.; Yanagi, Y.; Nagata, K.; Takeuchi, K. Cell tropism of wild-type measles virus is affected by amino acid substitutions in the P, V and M proteins, or by a truncation in the C protein. J. Gen. Virol. 2004, 85, 3001–3006. [Google Scholar] [CrossRef] [PubMed]
- Xin, J.Y.; Ihara, T.; Komase, K.; Nakayama, T. Amino acid substitutions in matrix, fusion and hemagglutinin proteins of wild measles virus for adaptation to vero cells. Intervirology 2011, 54, 217–228. [Google Scholar] [CrossRef]
- Condack, C.; Grivel, J.-C.; Devaux, P.; Margolis, L.; Cattaneo, R. Measles Virus Vaccine Attenuation: Suboptimal Infection of Lymphatic Tissue and Tropism Alteration. J. Infect. Dis. 2007, 196, 541–549. [Google Scholar] [CrossRef]
- Druelle, J.; Sellin, C.I.; Waku-Kouomou, D.; Horvat, B.; Wild, F.T. Wild type measles virus attenuation independent of type I IFN. Virol. J. 2008, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Kato, A.; Kobune, F.; Sakata, H.; Li, Y.; Shioda, T.; Sakai, Y.; Asakawa, M.; Nagai, Y. Measles Virus Attenuation Associated with Transcriptional Impediment and a Few Amino Acid Changes in the Polymerase and Accessory Proteins. J. Virol. 1998, 72, 8690. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Ohno, S.; Tahara, M.; Takeuchi, H.; Shirogane, Y.; Ohmura, H.; Nakamura, T.; Yanagi, Y. Measles viruses possessing the polymerase protein genes of the Edmonston vaccine strain exhibit attenuated gene expression and growth in cultured cells and SLAM knock-in mice. J. Virol. 2008, 82, 11979–11984. [Google Scholar] [CrossRef] [Green Version]
- Leonard, V.H.; Hodge, G.; Reyes-Del Valle, J.; McChesney, M.B.; Cattaneo, R. Measles virus selectively blind to signaling lymphocytic activation molecule (SLAM, CD150) is attenuated and induces strong adaptive immune responses in rhesus monkeys. J. Virol. 2010, 84, 3413–3420. [Google Scholar] [CrossRef] [Green Version]
- Rowe, A.; Ferguson, G.L.; Minor, P.D.; Macadam, A.J. Coding Changes in the Poliovirus Protease 2A Compensate for 5′NCR Domain V Disruptions in a Cell-Specific Manner. Virology 2000, 269, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Westrop, G.D.; Wareham, K.A.; Evans, D.M.; Dunn, G.; Minor, P.D.; Magrath, D.I.; Taffs, F.; Marsden, S.; Skinner, M.A.; Schild, G.C. Genetic basis of attenuation of the Sabin type 3 oral poliovirus vaccine. J. Virol. 1989, 63, 1338–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, R.; Minor, P.; Dunn, G.; Modlin, J.F.; Ogra, P.L. Shedding of Virulent Poliovirus Revertants during Immunization with Oral Poliovirus Vaccine after Prior Immunization with Inactivated Polio Vaccine. J. Infect. Dis. 1993, 168, 1105–1109. [Google Scholar] [CrossRef] [PubMed]
- Omata, T.; Kohara, M.; Kuge, S.; Komatsu, T.; Abe, S.; Semler, B.L.; Kameda, A.; Itoh, H.; Arita, M.; Wimmer, E. Genetic analysis of the attenuation phenotype of poliovirus type 1. J. Virol. 1986, 58, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macadam, A.J.; Pollard, S.R.; Ferguson, G.; Skuce, R.; Wood, D.; Almond, J.W.; Minor, P.D. Genetic Basis of Attenuation of the Sabin Type 2 Vaccine Strain of Poliovirus in Primates. Virology 1993, 192, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Bergmann, M.; Garcia-Sastre, A.; Palese, P. Mechanism of attenuation of a chimeric influenza A/B transfectant virus. J. Virol. 1992, 66, 4679–4685. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, N.; Ye, Y.; Fenstermacher, K.J.; Liu, H.; Lane, A.P.; Pekosz, A. The M2 protein of live, attenuated influenza vaccine encodes a mutation that reduces replication in human nasal epithelial cells. Vaccine 2017, 35, 6691–6699. [Google Scholar] [CrossRef]
- Lagranderie, M.; Balazuc, A.-M.; Deriaud, E.; Leclerc, C.D.; Gheorghiu, M. Comparison of immune responses of mice immunized with five different Mycobacterium bovis BCG vaccine strains. Infect. Immun. 1996, 64, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Lanthier, P.A.; Huston, G.E.; Moquin, A.; Eaton, S.M.; Szaba, F.M.; Kummer, L.W.; Tighe, M.P.; Kohlmeier, J.E.; Blair, P.J.; Broderick, M.; et al. Live attenuated influenza vaccine (LAIV) impacts innate and adaptive immune responses. Vaccine 2011, 29, 7849–7856. [Google Scholar] [CrossRef] [Green Version]
- Jegaskanda, S.; Mason, R.D.; Andrews, S.F.; Wheatley, A.K.; Zhang, R.; Reynoso, G.V.; Ambrozak, D.R.; Santos, C.P.; Luke, C.J.; Matsuoka, Y.; et al. Intranasal Live Influenza Vaccine Priming Elicits Localized B Cell Responses in Mediastinal Lymph Nodes. J. Virol. 2018, 92, e01970-17. [Google Scholar] [CrossRef] [Green Version]
- Pyo, H.-M.; Zhou, Y. Protective efficacy of intranasally administered bivalent live influenza vaccine and immunological mechanisms underlying the protection. Vaccine 2014, 32, 3835–3842. [Google Scholar] [CrossRef]
- Rennick, L.J.; De Vries, R.D.; Carsillo, T.J.; Lemon, K.; Van Amerongen, G.; Ludlow, M.; Nguyen, D.T.; Yueksel, S.; Verburgh, R.J.; Haddock, P.; et al. Live-Attenuated Measles Virus Vaccine Targets Dendritic Cells and Macrophages in Muscle of Nonhuman Primates. J. Virol. 2015, 89, 2192–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masopust, D.; Sivula, C.P.; Jameson, S.C. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J. Immunol. 2017, 199, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horie, H.; Koike, S.; Kurata, T.; Sato-Yoshida, Y.; Ise, I.; Ota, Y.; Abe, S.; Hioki, K.; Kato, H.; Taya, C.; et al. Transgenic mice carrying the human poliovirus receptor: New animal models for study of poliovirus neurovirulence. J. Virol. 1994, 68, 681–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arroyo, J.; Guirakhoo, F.; Fenner, S.; Zhang, Z.X.; Monath, T.P.; Chambers, T.J. Molecular basis for attenuation of neurovirulence of a yellow fever Virus/Japanese encephalitis virus chimera vaccine (ChimeriVax-JE). J. Virol. 2001, 75, 934–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, R.R.; Knuchel, M.C.; Morin, T.N.; Naim, H.Y. An immune competent mouse model for the characterization of recombinant measles vaccines. Hum. Vaccin. Immunother. 2015, 11, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Xu, P.; Glazko, G. Estimation of divergence times from multiprotein sequences for a few mammalian species and several distantly related organisms. Proc. Natl. Acad. Sci. USA 2001, 98, 2497–2502. [Google Scholar] [CrossRef] [Green Version]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [Green Version]
- Baba, T.W.; Jeong, Y.S.; Penninck, D.; Bronson, R.; Greene, M.F.; Ruprecht, R.M. Pathogenicity of Live, Attenuated Siv after Mucosal Infection of Neonatal Macaques. Science 1995, 267, 1820–1825. [Google Scholar] [CrossRef]
- StahlHennig, C.; Dittmer, U.; Nisslein, T.; Petry, H.; Jurkiewicz, E.; Fuchs, D.; Wachter, H.; MatzRensing, K.; Kuhn, E.M.; Kaup, F.J.; et al. Rapid development of vaccine protection in macaques by live-attenuated simian immunodeficiency virus. J. Gen. Virol. 1996, 77, 2969–2981. [Google Scholar] [CrossRef]
- Reynolds, M.R.; Weiler, A.M.; Weisgrau, K.L.; Piaskowski, S.M.; Furlott, J.R.; Weinfurter, J.T.; Kaizu, M.; Soma, T.; Leon, E.J.; MacNair, C.; et al. Macaques vaccinated with live-attenuated SIV control replication of heterologous virus. J. Exp. Med. 2008, 205, 2537–2550. [Google Scholar] [CrossRef] [Green Version]
- Gundlach, B.R.; Lewis, M.G.; Sopper, S.; Schnell, T.; Sodroski, J.; Stahl-Hennig, C.; Uberla, K. Evidence for recombination of live, attenuated immunodeficiency virus vaccine with challenge virus to a more virulent strain. J. Virol. 2000, 74, 3537–3542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, T.W.; Liska, V.; Khimani, A.H.; Ray, N.B.; Dailey, P.J.; Penninck, D.; Bronson, R.; Greene, M.F.; McClure, H.M.; Martin, L.N.; et al. Live attenuated, multiply deleted simian immunodeficiency virus causes AIDS in infant and adult macaques (vol 5, pg 194, 1999). Nat. Med. 1999, 5, 590. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Liu, Z.; Sheffer, D.; Smith, M.; Singh, D.K.; Buch, S.; Narayan, O. Protection of macaques against AIDS with a live attenuated SHIV vaccine is of finite duration. Virology 2008, 371, 238–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putkonen, P.; Walther, L.; Zhang, Y.J.; Li, S.L.; Nilsson, C.; Albert, J.; Biberfeld, P.; Thorstensson, R.; Biberfeld, G. Long-Term Protection against Siv-Induced Disease in Macaques Vaccinated with a Live Attenuated Hiv-2 Vaccine. Nat. Med. 1995, 1, 914–918. [Google Scholar] [CrossRef]
- Hodara, V.L.; Parodi, L.M.; Keckler, M.S.; Giavedoni, L.D. Increases in NKG2C Expression on T Cells and Higher Levels of Circulating CD8(+) B Cells Are Associated with Sterilizing Immunity Provided by a Live Attenuated SIV Vaccine. Aids. Res. Hum. Retrov. 2016, 32, 1125–1134. [Google Scholar] [CrossRef]
- Virnik, K.; Hockenbury, M.; Ni, Y.S.; Beren, J.; Pavlakis, G.N.; Felber, B.K.; Berkower, I. Live attenuated rubella vectors expressing SIV and HIV vaccine antigens replicate and elicit durable immune responses in rhesus macaques. Retrovirology 2013, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Rose, N.F.; Marx, P.A.; Luckay, A.; Nixon, D.F.; Moretto, W.J.; Donahoe, S.M.; Montefiori, D.; Roberts, A.; Buonocore, L.; Rose, J.K. An effective AIDS vaccine based on live attenuated vesicular stomatitis virus recombinants. Cell 2001, 106, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, H.; Jones, S.M.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Stroher, U.; Grolla, A.; Bray, M.; Fritz, E.A.; Fernando, L.; Feldmann, F.; et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog. 2007, 3, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Matassov, D.; Mire, C.E.; Latham, T.; Geisbert, J.B.; Xu, R.; Ota-Setlik, A.; Agans, K.N.; Kobs, D.J.; Wendling, M.Q.S.; Burnaugh, A.; et al. Single-Dose Trivalent VesiculoVax Vaccine Protects Macaques from Lethal Ebolavirus and Marburgvirus Challenge. J. Virol. 2018, 92, e01190-17. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Hanley, P.W.; Haddock, E.; Martellaro, C.; Kobinger, G.; Feldmann, H. Efficacy of Vesicular Stomatitis Virus-Ebola Virus Postexposure Treatment in Rhesus Macaques Infected With Ebola Virus Makona. J. Infect. Dis. 2016, 214, S360–S366. [Google Scholar] [CrossRef]
- Qiu, X.G.; Fernando, L.; Alimonti, J.B.; Melito, P.L.; Feldmann, F.; Dick, D.; Stroher, U.; Feldmann, H.; Jones, S.M. Mucosal Immunization of Cynomolgus Macaques with the VSV Delta G/ZEBOVGP Vaccine Stimulates Strong Ebola GP-Specific Immune Responses. PLoS ONE 2009, 4, e5547. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Lewis, M.G.; Geisbert, J.B.; Grolla, A.; Leung, A.; Paragas, J.; Matthias, L.; Smith, M.A.; Jones, S.M.; et al. Vesicular Stomatitis Virus-Based Ebola Vaccine Is Well-Tolerated and Protects Immunocompromised Nonhuman Primates. PLoS Pathog. 2008, 4, e1000225. [Google Scholar] [CrossRef] [PubMed]
- Yee, J.L.; McChesney, M.B.; Christe, K.L.; Management, N.B.C. Multicenter Safety and Immunogenicity Trial of an Attenuated Measles Vaccine for NHP. Comp. Med. 2015, 65, 448–454. [Google Scholar] [PubMed]
- Van Binnendijk, R.S.; Poelen, M.C.M.; Van Amerongen, G.; De Vries, P.; Osterhaus, A.D.M.E. Protective immunity in macaques vaccinated with live attenuated, recombinant, and subunit measles vaccines in the presence of passively acquired antibodies. J. Infect. Dis. 1997, 175, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.H.; Griffin, D.E.; Rota, P.A.; Papania, M.; Cape, S.P.; Bennett, D.; Quinn, B.; Sievers, R.E.; Shermer, C.; Powell, K.; et al. Successful respiratory immunization with dry powder live-attenuated measles virus vaccine in rhesus macaques. Proc. Natl. Acad. Sci. USA 2011, 108, 2987–2992. [Google Scholar] [CrossRef] [Green Version]
- Reed, D.S.; Lind, C.M.; Lackemeyer, M.G.; Sullivan, L.J.; Pratt, W.D.; Parker, M.D. Genetically engineered, live, attenuated vaccines protect nonhuman primates against aerosol challenge with a virulent IE strain of Venezuelan equine encephalitis virus. Vaccine 2005, 23, 3139–3147. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Marioni, J.C.; Blekhman, R.; Stephens, M.; Gilad, Y. Functional comparison of innate immune signaling pathways in primates. PLoS Genet. 2010, 6, e1001249. [Google Scholar] [CrossRef] [Green Version]
- Gaska, J.M.; Parsons, L.; Balev, M.; Cirincione, A.; Wang, W.; Schwartz, R.E.; Ploss, A. Conservation of cell-intrinsic immune responses in diverse nonhuman primate species. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Querec, T.D.; Akondy, R.S.; Lee, E.K.; Cao, W.; Nakaya, H.I.; Teuwen, D.; Pirani, A.; Gernert, K.; Deng, J.; Marzolf, B.; et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat. Immunol. 2009, 10, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Gaucher, D.; Therrien, R.; Kettaf, N.; Angermann, B.R.; Boucher, G.; Filali-Mouhim, A.; Moser, J.M.; Mehta, R.S.; Drake, D.R., 3rd; Castro, E.; et al. Yellow fever vaccine induces integrated multilineage and polyfunctional immune responses. J. Exp. Med. 2008, 205, 3119–3131. [Google Scholar] [CrossRef]
- Wahid, R.; Cannon, M.J.; Chow, M. Virus-specific CD4+ and CD8+ cytotoxic T-cell responses and long-term T-cell memory in individuals vaccinated against polio. J. Virol. 2005, 79, 5988–5995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovsyannikova, I.G.; Dhiman, N.; Jacobson, R.M.; Vierkant, R.A.; Poland, G.A. Frequency of measles virus-specific CD4+ and CD8+ T cells in subjects seronegative or highly seropositive for measles vaccine. Clin. Diagn. Lab. Immunol. 2003, 10, 411–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Shinkai, Y.; Rathbun, G.; Lam, K.P.; Oltz, E.M.; Stewart, V.; Mendelsohn, M.; Charron, J.; Datta, M.; Young, F.; Stall, A.M.; et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 1992, 68, 855–867. [Google Scholar] [CrossRef]
- McCune, J.M.; Namikawa, R.; Kaneshima, H.; Shultz, L.D.; Lieberman, M.; Weissman, I.L. The SCID-hu mouse: Murine model for the analysis of human hematolymphoid differentiation and function. Science 1988, 241, 1632–1639. [Google Scholar] [CrossRef]
- Mosier, D.E.; Gulizia, R.J.; Baird, S.M.; Wilson, D.B. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988, 335, 256–259. [Google Scholar] [CrossRef]
- Kamel-Reid, S.; Dick, J.E. Engraftment of immune-deficient mice with human hematopoietic stem cells. Science 1988, 242, 1706–1709. [Google Scholar] [CrossRef]
- Lapidot, T.; Pflumio, F.; Doedens, M.; Murdoch, B.; Williams, D.E.; Dick, J.E. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science 1992, 255, 1137–1141. [Google Scholar] [CrossRef]
- Hesselton, R.M.; Greiner, D.L.; Mordes, J.P.; Rajan, T.V.; Sullivan, J.L.; Shultz, L.D. High levels of human peripheral blood mononuclear cell engraftment and enhanced susceptibility to human immunodeficiency virus type 1 infection in NOD/LtSz-scid/scid mice. J. Infect. Dis. 1995, 172, 974–982. [Google Scholar] [CrossRef]
- Lowry, P.A.; Shultz, L.D.; Greiner, D.L.; Hesselton, R.M.; Kittler, E.L.; Tiarks, C.Y.; Rao, S.S.; Reilly, J.; Leif, J.H.; Ramshaw, H.; et al. Improved engraftment of human cord blood stem cells in NOD/LtSz-scid/scid mice after irradiation or multiple-day injections into unirradiated recipients. Biol. Blood Marrow Transplant. 1996, 2, 15–23. [Google Scholar] [PubMed]
- Pflumio, F.; Izac, B.; Katz, A.; Shultz, L.D.; Vainchenker, W.; Coulombel, L. Phenotype and function of human hematopoietic cells engrafting immune-deficient CB17-severe combined immunodeficiency mice and nonobese diabetic-severe combined immunodeficiency mice after transplantation of human cord blood mononuclear cells. Blood 1996, 88, 3731–3740. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 8, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- DiSanto, J.P.; Muller, W.; Guy-Grand, D.; Fischer, A.; Rajewsky, K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor gamma chain. Proc. Natl. Acad. Sci. USA 1995, 92, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Mazurier, F.; Fontanellas, A.; Salesse, S.; Taine, L.; Landriau, S.; Moreau-Gaudry, F.; Reiffers, J.; Peault, B.; Di Santo, J.P.; De Verneuil, H. A novel immunodeficient mouse model--RAG2 x common cytokine receptor gamma chain double mutants--requiring exogenous cytokine administration for human hematopoietic stem cell engraftment. J. Interferon Cytokine Res. 1999, 19, 533–541. [Google Scholar] [CrossRef]
- Goldman, J.P.; Blundell, M.P.; Lopes, L.; Kinnon, C.; Di Santo, J.P.; Thrasher, A.J. Enhanced human cell engraftment in mice deficient in RAG2 and the common cytokine receptor gamma chain. Br. J. Haematol. 1998, 103, 335–342. [Google Scholar] [CrossRef]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Traggiai, E.; Chicha, L.; Mazzucchelli, L.; Bronz, L.; Piffaretti, J.C.; Lanzavecchia, A.; Manz, M.G. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004, 304, 104–107. [Google Scholar] [CrossRef]
- Okada, S.; Harada, H.; Ito, T.; Saito, T.; Suzu, S. Early development of human hematopoietic and acquired immune systems in new born NOD/Scid/Jak3null mice intrahepatic engrafted with cord blood-derived CD34 + cells. Int. J. Hematol. 2008, 88, 476–482. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. 2005, 174, 6477–6489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, D.T.; Badowski, M.; Balamurugan, A.; Yang, O.O. Long-term human immune system reconstitution in non-obese diabetic (NOD)-Rag (-)-gamma chain (-) (NRG) mice is similar but not identical to the original stem cell donor. Clin. Exp. Immunol. 2013, 174, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Hrebikova, G.; Albrecht, Y.E.; Sellau, J.; Sharon, Y.; Ding, Q.; Ploss, A. Single-cell tracking of flavivirus RNA uncovers species-specific interactions with the immune system dictating disease outcome. Nat. Commun. 2017, 8, 14781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rongvaux, A.; Willinger, T.; Takizawa, H.; Rathinam, C.; Auerbach, W.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Eynon, E.E.; Stevens, S.; et al. Human thrombopoietin knockin mice efficiently support human hematopoiesis in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 2378–2383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willinger, T.; Rongvaux, A.; Takizawa, H.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Auerbach, W.; Eynon, E.E.; Stevens, S.; Manz, M.G.; et al. Human IL-3/GM-CSF knock-in mice support human alveolar macrophage development and human immune responses in the lung. Proc. Natl. Acad. Sci. USA 2011, 108, 2390–2395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehm, M.A.; Racki, W.J.; Leif, J.; Burzenski, L.; Hosur, V.; Wetmore, A.; Gott, B.; Herlihy, M.; Ignotz, R.; Dunn, R.; et al. Engraftment of human HSCs in nonirradiated newborn NOD-scid IL2rgamma null mice is enhanced by transgenic expression of membrane-bound human SCF. Blood 2012, 119, 2778–2788. [Google Scholar] [CrossRef]
- Takagi, S.; Saito, Y.; Hijikata, A.; Tanaka, S.; Watanabe, T.; Hasegawa, T.; Mochizuki, S.; Kunisawa, J.; Kiyono, H.; Koseki, H.; et al. Membrane-bound human SCF/KL promotes in vivo human hematopoietic engraftment and myeloid differentiation. Blood 2012, 119, 2768–2777. [Google Scholar] [CrossRef] [Green Version]
- Ito, R.; Takahashi, T.; Katano, I.; Kawai, K.; Kamisako, T.; Ogura, T.; Ida-Tanaka, M.; Suemizu, H.; Nunomura, S.; Ra, C.; et al. Establishment of a human allergy model using human IL-3/GM-CSF-transgenic NOG mice. J. Immunol. 2013, 191, 2890–2899. [Google Scholar] [CrossRef] [Green Version]
- Nicolini, F.E.; Cashman, J.D.; Hogge, D.E.; Humphries, R.K.; Eaves, C.J. NOD/SCID mice engineered to express human IL-3, GM-CSF and Steel factor constitutively mobilize engrafted human progenitors and compromise human stem cell regeneration. Leukemia 2004, 18, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Rathinam, C.; Poueymirou, W.T.; Rojas, J.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Rongvaux, A.; Eynon, E.E.; Manz, M.G.; Flavell, R.A. Efficient differentiation and function of human macrophages in humanized CSF-1 mice. Blood 2011, 118, 3119–3128. [Google Scholar] [CrossRef] [Green Version]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Ploss, A. Development of human CD4+FoxP3+ regulatory T cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing NOD-SCID IL2Rgamma(null) humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Borsotti, C.; Schickel, J.N.; Zhu, S.; Strowig, T.; Eynon, E.E.; Frleta, D.; Gurer, C.; Murphy, A.J.; Yancopoulos, G.D.; et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood 2017, 129, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Herndler-Brandstetter, D.; Shan, L.; Yao, Y.; Stecher, C.; Plajer, V.; Lietzenmayer, M.; Strowig, T.; De Zoete, M.R.; Palm, N.W.; Chen, J.; et al. Humanized mouse model supports development, function, and tissue residency of human natural killer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E9626–E9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, M.; Chou, F.S.; Link, K.A.; Mizukawa, B.; Perry, R.L.; Carroll, M.; Mulloy, J.C. AML xenograft efficiency is significantly improved in NOD/SCID-IL2RG mice constitutively expressing human SCF, GM-CSF and IL-3. Leukemia 2010, 24, 1785–1788. [Google Scholar] [CrossRef] [Green Version]
- Feuring-Buske, M.; Gerhard, B.; Cashman, J.; Humphries, R.K.; Eaves, C.J.; Hogge, D.E. Improved engraftment of human acute myeloid leukemia progenitor cells in beta 2-microglobulin-deficient NOD/SCID mice and in NOD/SCID mice transgenic for human growth factors. Leukemia 2003, 17, 760–763. [Google Scholar] [CrossRef] [Green Version]
- McIntosh, B.E.; Brown, M.E.; Duffin, B.M.; Maufort, J.P.; Vereide, D.T.; Slukvin, I.I.; Thomson, J.A. Nonirradiated NOD, B6.SCID Il2rgamma-/- Kit(W41/W41) (NBSGW) mice support multilineage engraftment of human hematopoietic cells. Stem Cell Rep. 2015, 4, 171–180. [Google Scholar] [CrossRef] [Green Version]
- Hassan, H.T. c-Kit expression in human normal and malignant stem cells prognostic and therapeutic implications. Leuk Res. 2009, 33, 5–10. [Google Scholar] [CrossRef]
- Waskow, C.; Liu, K.; Darrasse-Jeze, G.; Guermonprez, P.; Ginhoux, F.; Merad, M.; Shengelia, T.; Yao, K.; Nussenzweig, M. The receptor tyrosine kinase Flt3 is required for dendritic cell development in peripheral lymphoid tissues. Nat. Immunol. 2008, 9, 676–683. [Google Scholar] [CrossRef]
- Mackarehtschian, K.; Hardin, J.D.; Moore, K.A.; Boast, S.; Goff, S.P.; Lemischka, I.R. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity 1995, 3, 147–161. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Wilkinson, A.; Idris, A.; Fancke, B.; O’Keeffe, M.; Khalil, D.; Ju, X.; Lahoud, M.H.; Caminschi, I.; Shortman, K.; et al. FLT3-ligand treatment of humanized mice results in the generation of large numbers of CD141+ and CD1c+ dendritic cells in vivo. J. Immunol. 2014, 192, 1982–1989. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Mention, J.J.; Court, N.; Masse-Ranson, G.; Toubert, A.; Spits, H.; Legrand, N.; Corcuff, E.; Strick-Marchand, H.; Di Santo, J.P. A novel Flt3-deficient HIS mouse model with selective enhancement of human DC development. Eur. J. Immunol. 2016, 46, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Lastra, S.; Masse-Ranson, G.; Fiquet, O.; Darche, S.; Serafini, N.; Li, Y.; Dusseaux, M.; Strick-Marchand, H.; Di Santo, J.P. A functional DC cross talk promotes human ILC homeostasis in humanized mice. Blood Adv. 2017, 1, 601–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonboehmer, H. T-Cell Development and Selection in the Thymus. Bone Marrow Transpl. 1992, 9, 46–48. [Google Scholar]
- Billips, L.G.; Lassoued, K.; Nunez, C.; Wang, J.Y.; Kubagawa, H.; Gartland, G.L.; Burrows, P.D.; Cooper, M.D. Human B-cell development. Immunoglobulin Gene Expr. Dev. Dis. 1995, 764, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Gurer, C.; Ploss, A.; Liu, Y.F.; Arrey, F.; Sashihara, J.; Koo, G.; Rice, C.M.; Young, J.W.; Chadburn, A.; et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J. Exp. Med. 2009, 206, 1423–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billerbeck, E.; Horwitz, J.A.; Labitt, R.N.; Donovan, B.M.; Vega, K.; Budell, W.C.; Koo, G.C.; Rice, C.M.; Ploss, A. Characterization of human antiviral adaptive immune responses during hepatotropic virus infection in HLA-transgenic human immune system mice. J. Immunol. 2013, 191, 1753–1764. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Pearson, T.; Friberg, H.; Shultz, L.D.; Greiner, D.L.; Rothman, A.L.; Mathew, A. Dengue virus infection and virus-specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rgammanull mice. PLoS ONE 2009, 4, e7251. [Google Scholar] [CrossRef]
- Namikawa, R.; Weilbaecher, K.N.; Kaneshima, H.; Yee, E.J.; McCune, J.M. Long-term human hematopoiesis in the SCID-hu mouse. J. Exp. Med. 1990, 172, 1055–1063. [Google Scholar] [CrossRef]
- Lan, P.; Tonomura, N.; Shimizu, A.; Wang, S.; Yang, Y.G. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 2006, 108, 487–492. [Google Scholar] [CrossRef] [Green Version]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Gatlin, J.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Covassin, L.; Jangalwe, S.; Jouvet, N.; Laning, J.; Burzenski, L.; Shultz, L.D.; Brehm, M.A. Human immune system development and survival of non-obese diabetic (NOD)-scid IL2rgamma(null) (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin. Exp. Immunol. 2013, 174, 372–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brainard, D.M.; Seung, E.; Frahm, N.; Cariappa, A.; Bailey, C.C.; Hart, W.K.; Shin, H.S.; Brooks, S.F.; Knight, H.L.; Eichbaum, Q.; et al. Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol. 2009, 83, 7305–7321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, P.W.; Estes, J.D.; Sun, Z.; Othieno, F.A.; Wei, B.L.; Wege, A.K.; Powell, D.A.; Payne, D.; Haase, A.T.; Garcia, J.V. Antiretroviral pre-exposure prophylaxis prevents vaginal transmission of HIV-1 in humanized BLT mice. PLoS Med. 2008, 5, e16. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Denton, P.W.; Estes, J.D.; Othieno, F.A.; Wei, B.L.; Wege, A.K.; Melkus, M.W.; Padgett-Thomas, A.; Zupancic, M.; Haase, A.T.; et al. Intrarectal transmission, systemic infection, and CD4+ T cell depletion in humanized mice infected with HIV-1. J. Exp. Med. 2007, 204, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Lavender, K.J.; Pang, W.W.; Messer, R.J.; Duley, A.K.; Race, B.; Phillips, K.; Scott, D.; Peterson, K.E.; Chan, C.K.; Dittmer, U.; et al. BLT-humanized C57BL/6 Rag2-/-gammac-/-CD47-/- mice are resistant to GVHD and develop B- and T-cell immunity to HIV infection. Blood 2013, 122, 4013–4020. [Google Scholar] [CrossRef]
- Marsden, M.D.; Kovochich, M.; Suree, N.; Shimizu, S.; Mehta, R.; Cortado, R.; Bristol, G.; An, D.S.; Zack, J.A. HIV latency in the humanized BLT mouse. J. Virol. 2012, 86, 339–347. [Google Scholar] [CrossRef] [Green Version]
- Stoddart, C.A.; Maidji, E.; Galkina, S.A.; Kosikova, G.; Rivera, J.M.; Moreno, M.E.; Sloan, B.; Joshi, P.; Long, B.R. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rgamma(−/−) (NSG) BLT mice. Virology 2011, 417, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.M.; Patel, S.N.; Shimizu, S.; Rao, D.S.; Gnanapragasam, P.N.; An, D.S.; Yang, L.; Baltimore, D. Inhibitory effect of HIV-specific neutralizing IgA on mucosal transmission of HIV in humanized mice. Blood 2012, 120, 4571–4582. [Google Scholar] [CrossRef] [Green Version]
- Wahl, A.; Swanson, M.D.; Nochi, T.; Olesen, R.; Denton, P.W.; Chateau, M.; Garcia, J.V. Human Breast Milk and Antiretrovirals Dramatically Reduce Oral HIV-1 Transmission in BLT Humanized Mice. PLoS Pathog. 2012, 8, e1002732. [Google Scholar] [CrossRef] [Green Version]
- Zou, W.; Denton, P.W.; Watkins, R.L.; Krisko, J.F.; Nochi, T.; Foster, J.L.; Garcia, J.V. Nef functions in BLT mice to enhance HIV-1 replication and deplete CD4(+)CD8(+) thymocytes. Retrovirology 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Frias-Staheli, N.; Dorner, M.; Marukian, S.; Billerbeck, E.; Labitt, R.N.; Rice, C.M.; Ploss, A. Utility of humanized BLT mice for analysis of dengue virus infection and antiviral drug testing. J. Virol. 2014, 88, 2205–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Pazoles, P.; Woda, M.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A.; Mathew, A. Enhanced humoral and HLA-A2-restricted dengue virus-specific T-cell responses in humanized BLT NSG mice. Immunology 2012, 136, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Smith, K.; Ramirez, A.; Woda, M.; Pazoles, P.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A.; Mathew, A. Dengue virus infection induces broadly cross-reactive human IgM antibodies that recognize intact virions in humanized BLT-NSG mice. Exp. Biol. Med. 2015, 240, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.V.; Ye, W.; Chen, Q.; Teixeira, M.M.; Preiser, P.; Ooi, E.E.; Chen, J. Dengue Virus-Infected Dendritic Cells, but Not Monocytes, Activate Natural Killer Cells through a Contact-Dependent Mechanism Involving Adhesion Molecules. Mbio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Robinson, L.N.; Tharakaraman, K.; Rowley, K.J.; Costa, V.V.; Chan, K.R.; Wong, Y.H.; Ong, L.C.; Tan, H.C.; Koch, T.; Cain, D.; et al. Structure-Guided Design of an Anti-dengue Antibody Directed to a Non-immunodominant Epitope. Cell 2015, 162, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.V.; Russ, H.A.; Khan, I.S.; LaFlam, T.N.; Metzger, T.C.; Anderson, M.S.; Hebrok, M. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem Cell 2013, 13, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.E.; Zhou, Y.; McIntosh, B.E.; Norman, I.G.; Lou, H.E.; Biermann, M.; Sullivan, J.A.; Kamp, T.J.; Thomson, J.A.; Anagnostopoulos, P.V.; et al. A Humanized Mouse Model Generated Using Surplus Neonatal Tissue. Stem Cell Rep. 2018, 10, 1175–1183. [Google Scholar] [CrossRef] [Green Version]
- Reinisch, A.; Etchart, N.; Thomas, D.; Hofmann, N.A.; Fruehwirth, M.; Sinha, S.; Chan, C.K.; Senarath-Yapa, K.; Seo, E.Y.; Wearda, T.; et al. Epigenetic and in vivo comparison of diverse MSC sources reveals an endochondral signature for human hematopoietic niche formation. Blood 2015, 125, 249–260. [Google Scholar] [CrossRef]
- Holzapfel, B.M.; Hutmacher, D.W.; Nowlan, B.; Barbier, V.; Thibaudeau, L.; Theodoropoulos, C.; Hooper, J.D.; Loessner, D.; Clements, J.A.; Russell, P.J.; et al. Tissue engineered humanized bone supports human hematopoiesis in vivo. Biomaterials 2015, 61, 103–114. [Google Scholar] [CrossRef]
- Chen, Y.; Jacamo, R.; Shi, Y.X.; Wang, R.Y.; Battula, V.L.; Konoplev, S.; Strunk, D.; Hofmann, N.A.; Reinisch, A.; Konopleva, M.; et al. Human extramedullary bone marrow in mice: A novel in vivo model of genetically controlled hematopoietic microenvironment. Blood 2012, 119, 4971–4980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groen, R.W.; Noort, W.A.; Raymakers, R.A.; Prins, H.J.; Aalders, L.; Hofhuis, F.M.; Moerer, P.; Van Velzen, J.F.; Bloem, A.C.; Van Kessel, B.; et al. Reconstructing the human hematopoietic niche in immunodeficient mice: Opportunities for studying primary multiple myeloma. Blood 2012, 120, e9–e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinisch, A.; Hernandez, D.C.; Schallmoser, K.; Majeti, R. Generation and use of a humanized bone-marrow-ossicle niche for hematopoietic xenotransplantation into mice. Nat. Protoc. 2017, 12, 2169–2188. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Noort, W.A.; Jaques, J.; De Boer, B.; De Jong-Korlaar, R.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; Van Velzen, J.F.; Bloem, A.C.; Yuan, H.; et al. Establishing human leukemia xenograft mouse models by implanting human bone marrow-like scaffold-based niches. Blood 2016, 128, 2949–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abarrategi, A.; Foster, K.; Hamilton, A.; Mian, S.A.; Passaro, D.; Gribben, J.; Mufti, G.; Bonnet, D. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J. Clin. Investig. 2017, 127, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, M.; Sacchetti, B.; Pievani, A.; Redaelli, D.; Remoli, C.; Biondi, A.; Riminucci, M.; Bianco, P. Establishment of bone marrow and hematopoietic niches in vivo by reversion of chondrocyte differentiation of human bone marrow stromal cells. Stem Cell Res. 2014, 12, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontakke, P.; Carretta, M.; Jaques, J.; Brouwers-Vos, A.Z.; Lubbers-Aalders, L.; Yuan, H.; De Bruijn, J.D.; Martens, A.C.; Vellenga, E.; Groen, R.W.; et al. Modeling BCR-ABL and MLL-AF9 leukemia in a human bone marrow-like scaffold-based xenograft model. Leukemia 2016, 30, 2064–2073. [Google Scholar] [CrossRef]
- Martine, L.C.; Holzapfel, B.M.; McGovern, J.A.; Wagner, F.; Quent, V.M.; Hesami, P.; Wunner, F.M.; Vaquette, C.; De-Juan-Pardo, E.M.; Brown, T.D.; et al. Engineering a humanized bone organ model in mice to study bone metastases. Nat. Protoc. 2017, 12, 639–663. [Google Scholar] [CrossRef]
- Chappaz, S.; Finke, D. The IL-7 signaling pathway regulates lymph node development independent of peripheral lymphocytes. J. Immunol. 2010, 184, 3562–3569. [Google Scholar] [CrossRef]
- Chappaz, S.; Flueck, L.; Farr, A.G.; Rolink, A.G.; Finke, D. Increased TSLP availability restores T- and B-cell compartments in adult IL-7 deficient mice. Blood 2007, 110, 3862–3870. [Google Scholar] [CrossRef]
- Li, Y.; Masse-Ranson, G.; Garcia, Z.; Bruel, T.; Kok, A.; Strick-Marchand, H.; Jouvion, G.; Serafini, N.; Lim, A.I.; Dusseaux, M.; et al. A human immune system mouse model with robust lymph node development. Nat. Methods 2018, 15, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Katano, I.; Ito, R.; Goto, M.; Abe, H.; Mizuno, S.; Kawai, K.; Sugiyama, F.; Ito, M. Enhanced Antibody Responses in a Novel NOG Transgenic Mouse with Restored Lymph Node Organogenesis. Front. Immunol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- King, M.A.; Covassin, L.; Brehm, M.A.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Racki, W.J.; Covassin, L.; Brehm, M.; Pino, S.; Ignotz, R.; Dunn, R.; Laning, J.; Graves, S.K.; Rossini, A.A.; Shultz, L.D.; et al. NOD-scid IL2rgamma(null) mouse model of human skin transplantation and allograft rejection. Transplantation 2010, 89, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Nadig, S.N.; Wieckiewicz, J.; Wu, D.C.; Warnecke, G.; Zhang, W.; Luo, S.; Schiopu, A.; Taggart, D.P.; Wood, K.J. In vivo prevention of transplant arteriosclerosis by ex vivo-expanded human regulatory T cells. Nat. Med. 2010, 16, 809–813. [Google Scholar] [CrossRef]
- McDermott, S.P.; Eppert, K.; Lechman, E.R.; Doedens, M.; Dick, J.E. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood 2010, 116, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turgeon, N.A.; Banuelos, S.J.; Shultz, L.D.; Lyons, B.L.; Iwakoshi, N.; Greiner, D.L.; Mordes, J.P.; Rossini, A.A.; Appel, M.C. Alloimmune injury and rejection of human skin grafts on human peripheral blood lymphocyte-reconstituted non-obese diabetic severe combined immunodeficient beta2-microglobulin-null mice. Exp. Biol. Med. 2003, 228, 1096–1104. [Google Scholar] [CrossRef]
- Misharin, A.V.; Haines, G.K., 3rd; Rose, S.; Gierut, A.K.; Hotchkiss, R.S.; Perlman, H. Development of a new humanized mouse model to study acute inflammatory arthritis. J. Transl. Med. 2012, 10, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, M.; Brooks, R.A.; Panchal, R.; Rhyasen, G.W.; Danet-Desnoyers, G.; Mulloy, J.C. OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood 2014, 123, e134–e144. [Google Scholar] [CrossRef] [Green Version]
- Brehm, M.A.; Shultz, L.D.; Luban, J.; Greiner, D.L. Overcoming current limitations in humanized mouse research. J. Infect. Dis. 2013, 208, S125–S130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor Garcia, J. Humanized mice for HIV and AIDS research. Curr. Opin. Virol. 2016, 19, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legrand, N.; Van der Velden, G.J.; Ho Tsong Fang, R.; Douaisi, M.; Weijer, K.; Das, A.T.; Blom, B.; Uittenbogaart, C.H.; Berkhout, B.; Centlivre, M. A doxycycline-dependent human immunodeficiency virus type 1 replicates in vivo without inducing CD4+ T-cell depletion. J. Gen. Virol. 2012, 93, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, Y.U.; Kleibeuker, W.; Harwig, A.; Klaver, B.; Siteur-van Rijnstra, E.; Frankin, E.; Berkhout, B.; Das, A.T. Construction of Nef-positive doxycycline-dependent HIV-1 variants using bicistronic expression elements. Virology 2016, 488, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, S.; Suzuki, M.O.; Muhsen, M.; Ishige, M.; Kobayashi-Ishihara, M.; Ohno, S.; Takeda, M.; Nakayama, T.; Morikawa, Y.; Terahara, K.; et al. Sensitive detection of measles virus infection in the blood and tissues of humanized mouse by one-step quantitative RT-PCR. Front. Microbiol. 2013, 4, 298. [Google Scholar] [CrossRef] [Green Version]
- Valsamakis, A.; Auwaerter, P.G.; Rima, B.K.; Kaneshima, H.; Griffin, D.E. Altered virulence of vaccine strains of measles virus after prolonged replication in human tissue. J. Virol. 1999, 73, 8791–8797. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.I.; Gallegos, M.; Marches, F.; Zurawski, G.; Ramilo, O.; Garcia-Sastre, A.; Banchereau, J.; Palucka, A.K. Broad influenza-specific CD8+ T-cell responses in humanized mice vaccinated with influenza virus vaccines. Blood 2008, 112, 3671–3678. [Google Scholar] [CrossRef]
- Yu, C.I.; Becker, C.; Wang, Y.; Marches, F.; Helft, J.; Leboeuf, M.; Anguiano, E.; Pourpe, S.; Goller, K.; Pascual, V.; et al. Human CD1c+ dendritic cells drive the differentiation of CD103+ CD8+ mucosal effector T cells via the cytokine TGF-beta. Immunity 2013, 38, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.I.; Becker, C.; Metang, P.; Marches, F.; Wang, Y.; Toshiyuki, H.; Banchereau, J.; Merad, M.; Palucka, A.K. Human CD141+ dendritic cells induce CD4+ T cells to produce type 2 cytokines. J. Immunol. 2014, 193, 4335–4343. [Google Scholar] [CrossRef] [Green Version]
- Arrey, F.; Lowe, D.; Kuhlmann, S.; Kaiser, P.; Moura-Alves, P.; Krishnamoorthy, G.; Lozza, L.; Maertzdorf, J.; Skrahina, T.; Skrahina, A.; et al. Humanized Mouse Model Mimicking Pathology of Human Tuberculosis for in vivo Evaluation of Drug Regimens. Front. Immunol. 2019, 10, 89. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Lai, R.; Afkhami, S.; Haddadi, S.; Zganiacz, A.; Vahedi, F.; Ashkar, A.A.; Kaushic, C.; Jeyanathan, M.; Xing, Z. Enhancement of Antituberculosis Immunity in a Humanized Model System by a Novel Virus-Vectored Respiratory Mucosal Vaccine. J. Infect. Dis. 2017, 216, 135–145. [Google Scholar] [CrossRef]
- Grover, A.; Troy, A.; Rowe, J.; Troudt, J.M.; Creissen, E.; McLean, J.; Banerjee, P.; Feuer, G.; Izzo, A.A. Humanized NOG mice as a model for tuberculosis vaccine-induced immunity: A comparative analysis with the mouse and guinea pig models of tuberculosis. Immunology 2017, 152, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Douam, F.; Albrecht, Y.E.S.; Hrebikova, G.; Sadimin, E.; Davidson, C.; Kotenko, S.V.; Ploss, A. Type III Interferon-Mediated Signaling Is Critical for Controlling Live Attenuated Yellow Fever Virus Infection In Vivo. Mbio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibodeaux, B.A.; Garbino, N.C.; Liss, N.M.; Piper, J.; Blair, C.D.; Roehrig, J.T. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine 2012, 30, 3180–3187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shultz, L.D.; Brehm, M.A.; Bavari, S.; Greiner, D.L. Humanized mice as a preclinical tool for infectious disease and biomedical research. Ann. N. Y. Acad. Sci. 2011, 1245, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.A.; Jouvet, N.; Greiner, D.L.; Shultz, L.D. Humanized mice for the study of infectious diseases. Curr. Opin. Immunol. 2013, 25, 428–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jangalwe, S.; Shultz, L.D.; Mathew, A.; Brehm, M.A. Improved B cell development in humanized NOD-scid IL2Rgamma(null) mice transgenically expressing human stem cell factor, granulocyte-macrophage colony-stimulating factor and interleukin-3. Immun. Inflamm. Dis. 2016, 4, 427–440. [Google Scholar] [CrossRef]
- Okamoto, N.; Chihara, R.; Shimizu, C.; Nishimoto, S.; Watanabe, T. Artificial lymph nodes induce potent secondary immune responses in naive and immunodeficient mice. J. Clin. Investig. 2007, 117, 997–1007. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.C.; Hu, J.; Arber, D.; LeBon, T.; Forman, S.J. Transplantation and growth characteristics of human fetal lymph node in immunodeficient mice. Exp. Hematol. 2000, 28, 1046–1053. [Google Scholar] [CrossRef]
- Billerbeck, E.; Mommersteeg, M.C.; Shlomai, A.; Xiao, J.W.; Andrus, L.; Bhatta, A.; Vercauteren, K.; Michailidis, E.; Dorner, M.; Krishnan, A.; et al. Humanized mice efficiently engrafted with fetal hepatoblasts and syngeneic immune cells develop human monocytes and NK cells. J. Hepatol. 2016, 65, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Dusseaux, M.; Masse-Ranson, G.; Darche, S.; Ahodantin, J.; Li, Y.; Fiquet, O.; Beaumont, E.; Moreau, P.; Riviere, L.; Neuveut, C.; et al. Viral Load Affects the Immune Response to HBV in Mice With Humanized Immune System and Liver. Gastroenterology 2017, 153, 1647–1661. [Google Scholar] [CrossRef] [Green Version]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.J.; Pulendran, B. The potential of the microbiota to influence vaccine responses. J. Leukoc. Biol. 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagan, T.; Cortese, M.; Rouphael, N.; Boudreau, C.; Linde, C.; Maddur, M.S.; Das, J.; Wang, H.; Guthmiller, J.; Zheng, N.Y.; et al. Antibiotics-Driven Gut Microbiome Perturbation Alters Immunity to Vaccines in Humans. Cell 2019, 178, 1313. [Google Scholar] [CrossRef]
- Daharsh, L.; Zhang, J.S.; Ramer-Tait, A.; Li, Q.S. A Double Humanized BLT-mice Model Featuring a Stable Human-Like Gut Microbiome and Human Immune System. J. Vis. Exp. JoVE 2019. [Google Scholar] [CrossRef] [PubMed]
- Gulden, E.; Vudattu, N.K.; Deng, S.; Preston-Hurlburt, P.; Mamula, M.; Reed, J.C.; Mohandas, S.; Herold, B.C.; Torres, R.; Vieira, S.M.; et al. Microbiota control immune regulation in humanized mice. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]




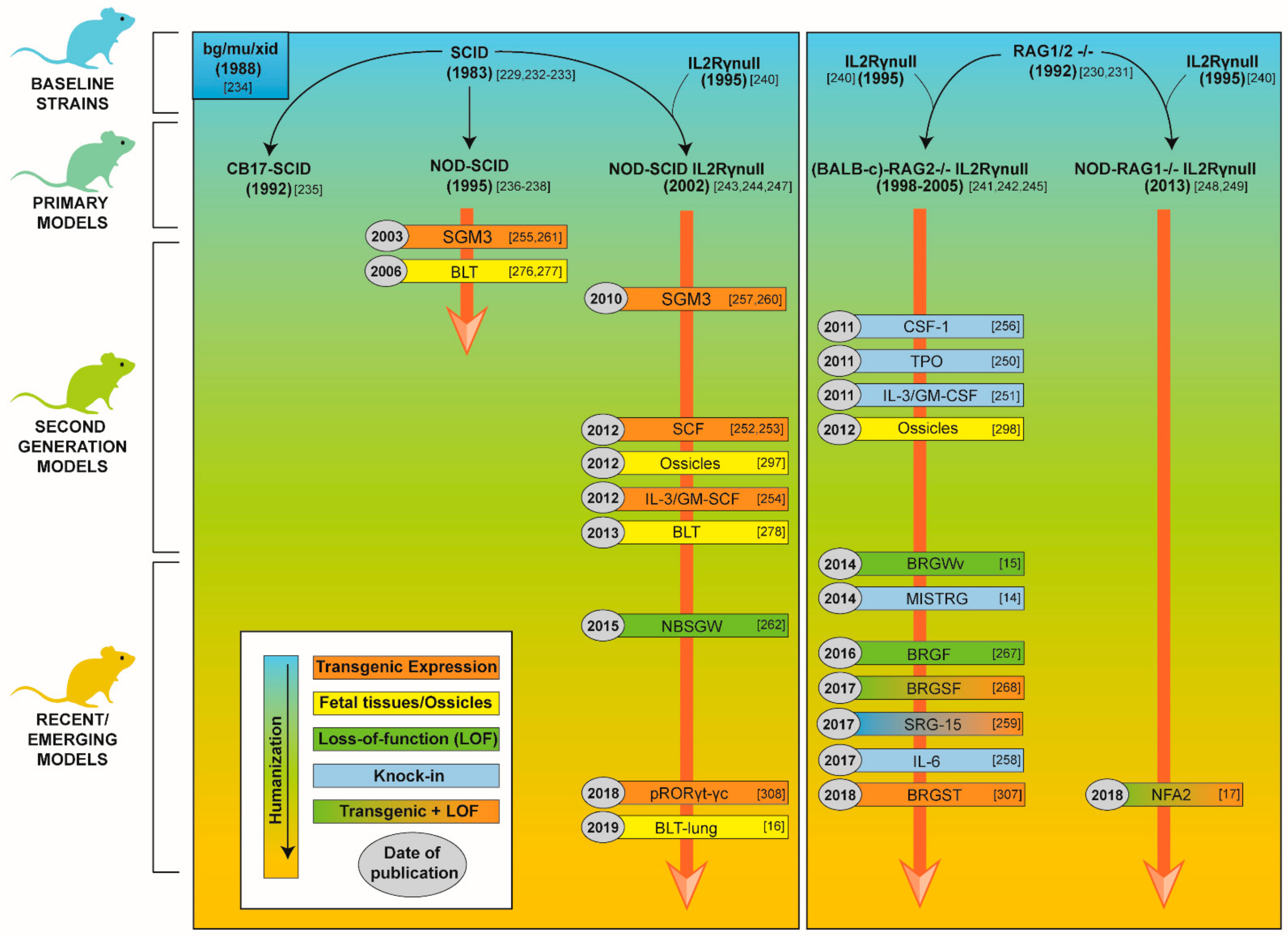{kind=link}
{kind=link}
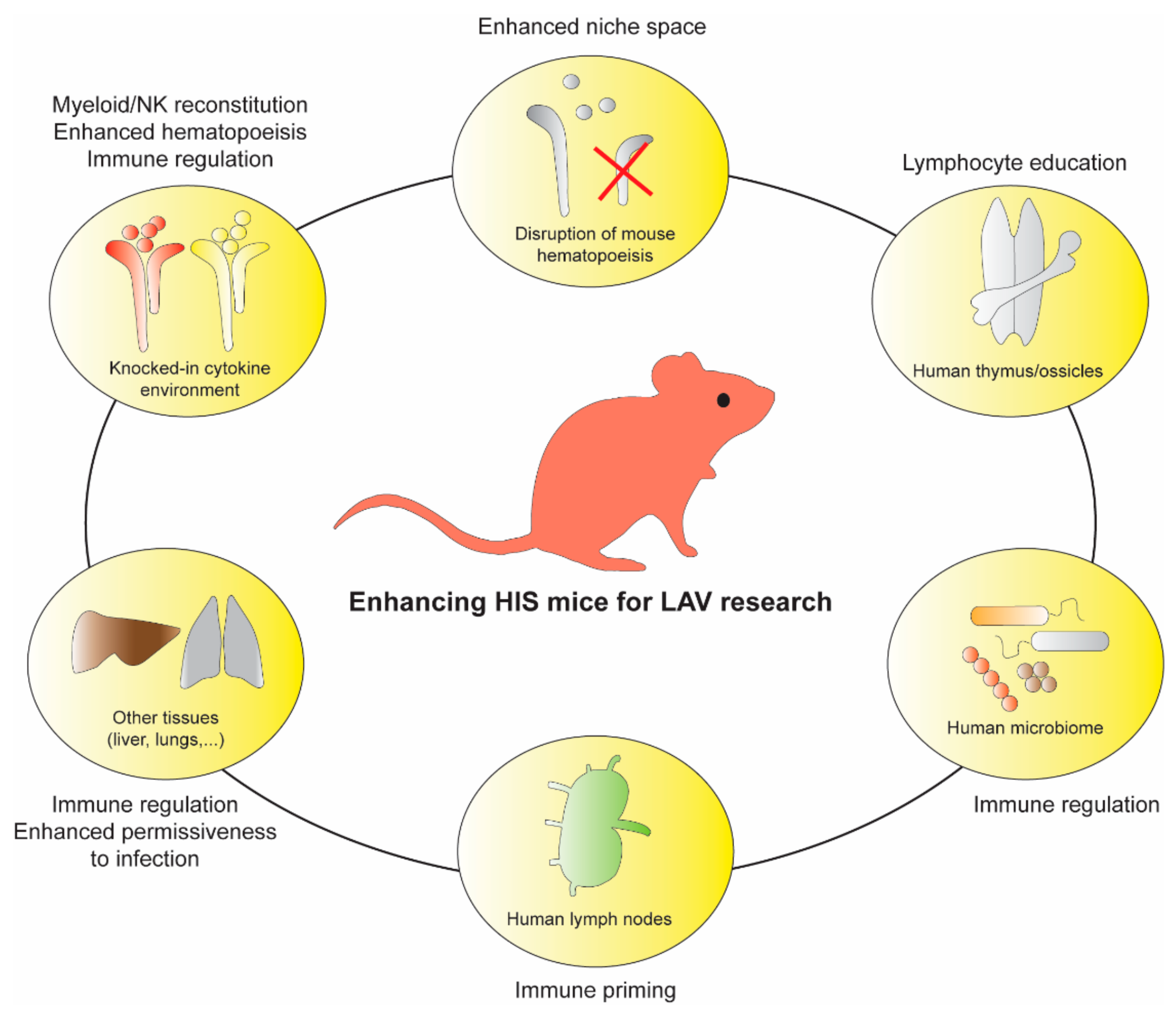{kind=link}
| Vaccine Type | Immunogenicity | Reactogenicity | Safety and Stability |
|---|---|---|---|
| LAV | +++ | +++ | + |
| Inactivated | ++ | ++ | ++ |
| Subunit | + | + | ++ |
| Toxoid | + | + | ++ |
| LAV | History | Attenuation Mechanisms | |||
|---|---|---|---|---|---|
| LAV Type | Method of Generation | Date of Isolation/ Attenuation | Genetic Similarity to Parental/Virulent Strain | In Vitro Findings | Animal Models |
| Smallpox | Isolation of related pathogen | 1798 [25] | n/a (vaccinia vs variola virus) | [154,171,172,173,174] | Mouse [154,155] |
| Bacille–Calmette Guerin (BCG) | Attenuation of related pathogen | 1921 [29] | >99.7% (RD1 deletion from M. bovis) [129,130] | [175,176,177] | Mouse [146,193] |
| Yellow fever virus (YFV) | Attenuation of causative pathogen | 1937 [32] | 99.37% [127] | [133,134,135,136,137,138,139,140,142,143,144,145,178] | Mouse [134,147,148,149]; NHP [151] |
| Measles | Attenuation of causative pathogen | 1954 [34] | ≥99.7% [128] | [131,132,157,162,179,180,181,182,183,184,185] | Mouse [183,184]; NHP [185,197] |
| Poliovirus | Attenuation of causative pathogen | 1954 [33] | ≥99.9% [168,169,170] | [141,152,153,186,187,188,189,190] | Mouse [153]; NHP [189,190] |
| Influenza | Attenuation of causative pathogen | 1965, 1967 [120,121] | n/a (reassortant vaccine) | [191,192] | Mouse [194]; NHP [195]; Pig [196] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
O’Connell, A.K.; Douam, F. Humanized Mice for Live-Attenuated Vaccine Research: From Unmet Potential to New Promises. Vaccines 2020, 8, 36. https://doi.org/10.3390/vaccines8010036
O’Connell AK, Douam F. Humanized Mice for Live-Attenuated Vaccine Research: From Unmet Potential to New Promises. Vaccines. 2020; 8(1):36. https://doi.org/10.3390/vaccines8010036
Chicago/Turabian StyleO’Connell, Aoife K., and Florian Douam. 2020. "Humanized Mice for Live-Attenuated Vaccine Research: From Unmet Potential to New Promises" Vaccines 8, no. 1: 36. https://doi.org/10.3390/vaccines8010036
APA StyleO’Connell, A. K., & Douam, F. (2020). Humanized Mice for Live-Attenuated Vaccine Research: From Unmet Potential to New Promises. Vaccines, 8(1), 36. https://doi.org/10.3390/vaccines8010036