Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing


Abstract
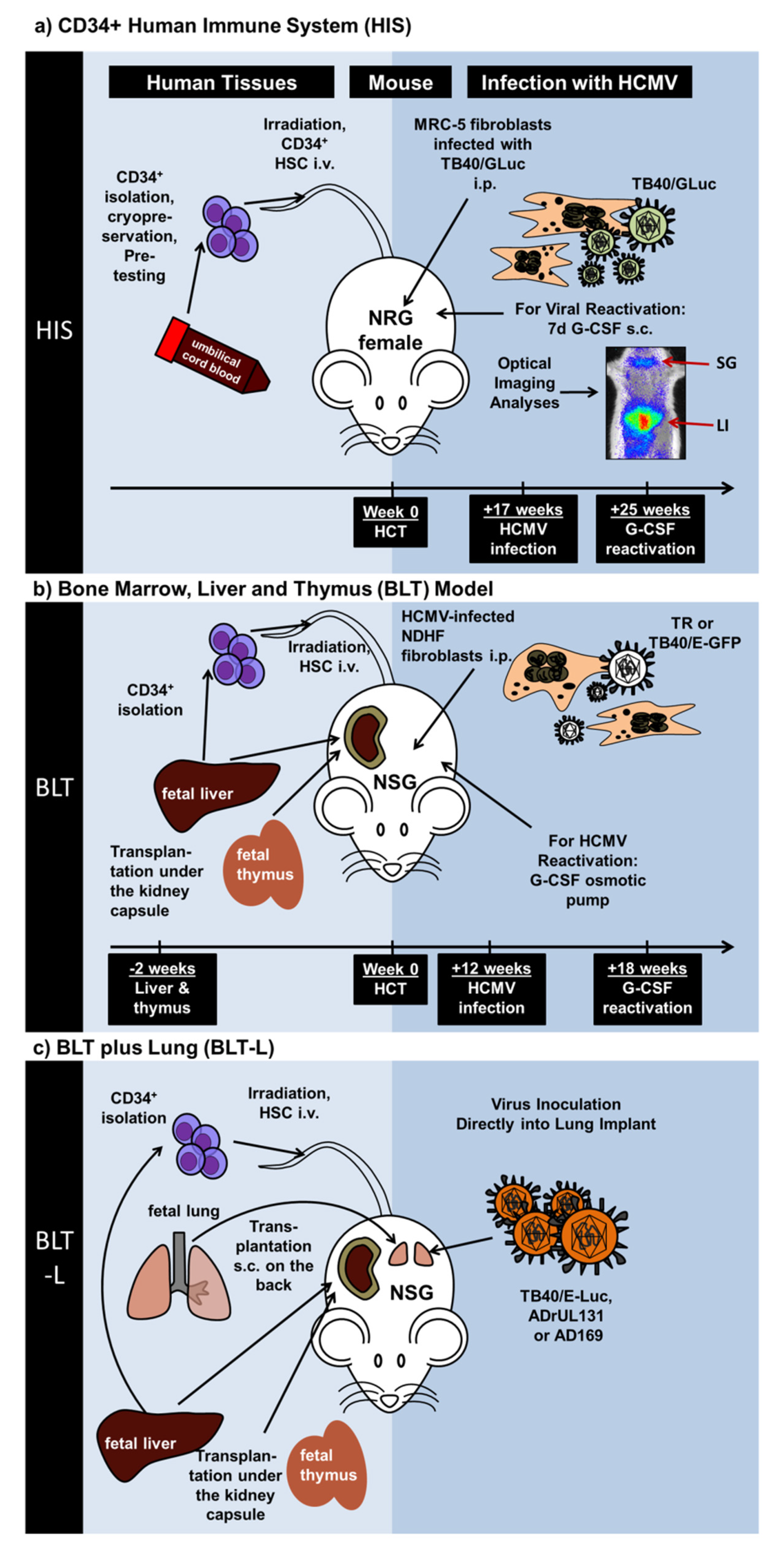
:1. Introduction
2. Mouse Models of HCMV Infections and Human Immune Responses
3. Preclinical Testing of Anti-HCMV Vaccines in HIS Mice
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the worldwide seroprevalence of cytomegalovirus: A systematic review and meta-analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [Green Version]
- Britt, W.J. Congenital human cytomegalovirus infection and the enigma of maternal immunity. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Cobbs, C. Cytomegalovirus is a tumor-associated virus: Armed and dangerous. Curr. Opin. Virol. 2019, 39, 49–59. [Google Scholar] [CrossRef]
- Su, B.Y.; Su, C.Y.; Yu, S.F.; Chen, C.J. Incidental discovery of high systemic lupus erythematosus disease activity associated with cytomegalovirus viral activity. Med. Microbiol. Immunol. 2007, 196, 165–170. [Google Scholar] [CrossRef]
- Streblow, D.N.; Nelson, J.A. Models of hcmv latency and reactivation. Trends Microbiol. 2003, 11, 293–295. [Google Scholar] [CrossRef]
- Sinclair, J. Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J. Clin. Virol. 2008, 41, 180–185. [Google Scholar] [CrossRef]
- Suessmuth, Y.; Mukherjee, R.; Watkins, B.; Koura, D.T.; Finstermeier, K.; Desmarais, C.; Stempora, L.; Horan, J.T.; Langston, A.; Qayed, M.; et al. Cmv reactivation drives posttransplant t-cell reconstitution and results in defects in the underlying tcrbeta repertoire. Blood 2015, 125, 3835–3850. [Google Scholar] [CrossRef]
- Kaminski, H.; Fishman, J.A. The cell biology of cytomegalovirus: Implications for transplantation. Am. J. Transplant 2016, 16, 2254–2269. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef] [Green Version]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [Green Version]
- Ljungman, P.; Brand, R.; Hoek, J.; de la Camara, R.; Cordonnier, C.; Einsele, H.; Styczynski, J.; Ward, K.N.; Cesaro, S.; Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Donor cytomegalovirus status influences the outcome of allogeneic stem cell transplant: A study by the european group for blood and marrow transplantation. Clin. Infect Dis. 2014, 59, 473–481. [Google Scholar] [CrossRef] [Green Version]
- George, B.; Pati, N.; Gilroy, N.; Ratnamohan, M.; Huang, G.; Kerridge, I.; Hertzberg, M.; Gottlieb, D.; Bradstock, K. Pre-transplant cytomegalovirus (cmv) serostatus remains the most important determinant of cmv reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy. Transpl. Infect Dis. 2010, 12, 322–329. [Google Scholar] [CrossRef]
- Ljungman, P.; de la Camara, R.; Cordonnier, C.; Einsele, H.; Engelhard, D.; Reusser, P.; Styczynski, J.; Ward, K.; European Conference on Infections in Leukemia. Management of cmv, hhv-6, hhv-7 and kaposi-sarcoma herpesvirus (hhv-8) infections in patients with hematological malignancies and after sct. Bone Marrow Transplant 2008, 42, 227–240. [Google Scholar] [CrossRef] [Green Version]
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N. Engl. J. Med. 2017, 377, 2433–2444. [Google Scholar] [CrossRef]
- Ljungman, P.; Schmitt, M.; Marty, F.M.; Maertens, J.; Chemaly, R.F.; Kartsonis, N.A.; Butterton, J.R.; Wan, H.; Teal, V.L.; Sarratt, K.; et al. A mortality analysis of letermovir prophylaxis for cytomegalovirus (cmv) in cmv-seropositive recipients of allogeneic hematopoietic-cell transplantation. Clin. Infect Dis. 2019. [Google Scholar] [CrossRef]
- Schleiss, M.R.; Permar, S.R.; Plotkin, S.A. Progress toward development of a vaccine against congenital cytomegalovirus infection. Clin. Vaccine Immunol. 2017, 24. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O'Beirne, J.; et al. Cytomegalovirus glycoprotein-b vaccine with mf59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Kharfan-Dabaja, M.A.; Boeckh, M.; Wilck, M.B.; Langston, A.A.; Chu, A.H.; Wloch, M.K.; Guterwill, D.F.; Smith, L.R.; Rolland, A.P.; Kenney, R.T. A novel therapeutic cytomegalovirus DNA vaccine in allogeneic haemopoietic stem-cell transplantation: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis. 2012, 12, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Lemmermann, N.A.W. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: Predictions for medical translation that survived the “test of time”. Viruses 2018, 10, 693. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhang, F.; Wang, R.; London, L.; London, S.D. Protective mcmv immunity by vaccination of the salivary gland via wharton's duct: Replication-deficient recombinant adenovirus expressing individual mcmv genes elicits protection similar to that of mcmv. FASEB J. 2014, 28, 1698–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Huang, C.; Dong, J.; Yao, Y.; Xie, Z.; Liu, X.; Zhang, W.; Fang, F.; Chen, Z. Complete protection of mice against lethal murine cytomegalovirus challenge by immunization with DNA vaccines encoding envelope glycoprotein complex iii antigens gh, gl and go. PLoS ONE 2015, 10, e0119964. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yao, Y.; Huang, C.; Fu, X.; Chen, Q.; Zhang, H.; Chen, J.; Fang, F.; Xie, Z.; Chen, Z. An adjuvanted inactivated murine cytomegalovirus (mcmv) vaccine induces potent and long-term protective immunity against a lethal challenge with virulent mcmv. BMC Infect Dis. 2014, 14, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, H.; Wu, S.; Chang, H.; Liu, L.; Peng, B.; Fang, F.; Chen, Z. Comparison of multiple DNA vaccines for protection against cytomegalovirus infection in balb/c mice. Virol. J. 2014, 11, 104. [Google Scholar] [CrossRef] [Green Version]
- Itell, H.L.; Kaur, A.; Deere, J.D.; Barry, P.A.; Permar, S.R. Rhesus monkeys for a nonhuman primate model of cytomegalovirus infections. Curr. Opin. Virol. 2017, 25, 126–133. [Google Scholar] [CrossRef] [Green Version]
- Wussow, F.; Yue, Y.; Martinez, J.; Deere, J.D.; Longmate, J.; Herrmann, A.; Barry, P.A.; Diamond, D.J. A vaccine based on the rhesus cytomegalovirus ul128 complex induces broadly neutralizing antibodies in rhesus macaques. J. Virol. 2013, 87, 1322–1332. [Google Scholar] [CrossRef] [Green Version]
- Valencia, S.; Gill, R.B.; Dowdell, K.C.; Wang, Y.; Hornung, R.; Bowman, J.J.; Lacayo, J.C.; Cohen, J.I. Comparison of vaccination with rhesus cmv (rhcmv) soluble gb with a rhcmv replication-defective virus deleted for mhc class i immune evasion genes in a rhcmv challenge model. Vaccine 2019, 37, 333–342. [Google Scholar] [CrossRef]
- Deere, J.D.; Chang, W.L.W.; Villalobos, A.; Schmidt, K.A.; Deshpande, A.; Castillo, L.D.; Fike, J.; Walter, M.R.; Barry, P.A.; Hartigan-O'Connor, D.J. Neutralization of rhesus cytomegalovirus il-10 reduces horizontal transmission and alters long-term immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 13036–13041. [Google Scholar] [CrossRef] [Green Version]
- Davison, A.J.H.M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative Genomics of Primate Cytomegaloviruses; Caister Academic Press: Norfolk, UK, 2013. [Google Scholar]
- Redwood, A.J.; Shellam, G.; Smith, L.M. Molecular Evolution of Murine Cytomegalovirus Genomes; Caister Academic Press: Wymondham, UK; Norfolk, UK, 2013. [Google Scholar]
- Chiuppesi, F.; Nguyen, J.; Park, S.; Contreras, H.; Kha, M.; Meng, Z.; Kaltcheva, T.; Iniguez, A.; Martinez, J.; La Rosa, C.; et al. Multiantigenic modified vaccinia virus ankara vaccine vectors to elicit potent humoral and cellular immune reponses against human cytomegalovirus in mice. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Kabanova, A.; Perez, L.; Lilleri, D.; Marcandalli, J.; Agatic, G.; Becattini, S.; Preite, S.; Fuschillo, D.; Percivalle, E.; Sallusto, F.; et al. Antibody-driven design of a human cytomegalovirus ghglpul128l subunit vaccine that selectively elicits potent neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2014, 111, 17965–17970. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, C.; Falk, J.J.; Buscher, N.; Penner, I.; Zimmermann, C.; Gogesch, P.; Sinzger, C.; Plachter, B. Dense bodies of a gh/gl/ul128/ul130/ul131 pentamer-repaired towne strain of human cytomegalovirus induce an enhanced neutralizing antibody response. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Loomis, R.J.; Lilja, A.E.; Monroe, J.; Balabanis, K.A.; Brito, L.A.; Palladino, G.; Franti, M.; Mandl, C.W.; Barnett, S.W.; Mason, P.W. Vectored co-delivery of human cytomegalovirus gh and gl proteins elicits potent complement-independent neutralizing antibodies. Vaccine 2013, 31, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Ohrum, K.; Cayatte, C.; Liu, Y.; Wang, Z.; Irrinki, A.; Cataniag, F.; Nguyen, N.; Lambert, S.; Liu, H.; Aslam, S.; et al. Production of cytomegalovirus dense bodies by scalable bioprocess methods maintains immunogenicity and improves neutralizing antibody titers. J. Virol. 2016, 90, 10133–10144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Rosa, C.; Longmate, J.; Martinez, J.; Zhou, Q.; Kaltcheva, T.I.; Tsai, W.; Drake, J.; Carroll, M.; Wussow, F.; Chiuppesi, F.; et al. Mva vaccine encoding cmv antigens safely induces durable expansion of cmv-specific t cells in healthy adults. Blood 2017, 129, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Goldman, D.C.; Bailey, A.S.; Pfaffle, D.L.; Kreklywich, C.N.; Spencer, D.B.; Othieno, F.A.; Streblow, D.N.; Garcia, J.V.; Fleming, W.H.; et al. Granulocyte-colony stimulating factor reactivates human cytomegalovirus in a latently infected humanized mouse model. Cell Host Microbe 2010, 8, 284–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theobald, S.J.; Khailaie, S.; Meyer-Hermann, M.; Volk, V.; Olbrich, H.; Danisch, S.; Gerasch, L.; Schneider, A.; Sinzger, C.; Schaudien, D.; et al. Signatures of t and b cell development, functional responses and pd-1 upregulation after hcmv latent infections and reactivations in nod.Rag.Gamma mice humanized with cord blood cd34 (+) cells. Front. Immunol. 2018, 9, 2734. [Google Scholar] [CrossRef] [Green Version]
- Hakki, M.; Goldman, D.C.; Streblow, D.N.; Hamlin, K.L.; Krekylwich, C.N.; Fleming, W.H.; Nelson, J.A. Hcmv infection of humanized mice after transplantation of g-csf-mobilized peripheral blood stem cells from hcmv-seropositive donors. Biol. Blood Marrow Transplant 2014, 20, 132–135. [Google Scholar] [CrossRef] [Green Version]
- Tomic, A.; Varanasi, P.R.; Golemac, M.; Malic, S.; Riese, P.; Borst, E.M.; Mischak-Weissinger, E.; Guzman, C.A.; Krmpotic, A.; Jonjic, S.; et al. Activation of innate and adaptive immunity by a recombinant human cytomegalovirus strain expressing an nkg2d ligand. PLoS Pathog. 2016, 12, e1006015. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.B.; Vrbanac, V.; Tivey, T.; Tsang, K.; Tager, A.M.; Aliprantis, A.O. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS ONE 2012, 7, e44664. [Google Scholar] [CrossRef]
- Crawford, L.B.; Tempel, R.; Streblow, D.N.; Kreklywich, C.; Smith, P.; Picker, L.J.; Nelson, J.A.; Caposio, P. Human cytomegalovirus induces cellular and humoral virus-specific immune responses in humanized blt mice. Sci. Rep. 2017, 7, 937. [Google Scholar] [CrossRef] [Green Version]
- Mocarski, E.S.; Bonyhadi, M.; Salimi, S.; McCune, J.M.; Kaneshima, H. Human cytomegalovirus in a scid-hu mouse: Thymic epithelial cells are prominent targets of viral replication. Proc. Natl. Acad. Sci. USA 1993, 90, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, A.; De, C.; Abad Fernandez, M.; Lenarcic, E.M.; Xu, Y.; Cockrell, A.S.; Cleary, R.A.; Johnson, C.E.; Schramm, N.J.; Rank, L.M.; et al. Precision mouse models with expanded tropism for human pathogens. Nat. Biotechnol. 2019, 37, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized mouse models of clinical disease. Annu. Rev. Pathol. 2017, 12, 187–215. [Google Scholar] [CrossRef] [Green Version]
- Bosma, G.C.; Custer, R.P.; Bosma, M.J. A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Takenaka, K.; Prasolava, T.K.; Wang, J.C.; Mortin-Toth, S.M.; Khalouei, S.; Gan, O.I.; Dick, J.E.; Danska, J.S. Polymorphism in sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 2007, 8, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Brehm, M.A.; Garcia-Martinez, J.V.; Greiner, D.L. Humanized mice for immune system investigation: Progress, promise and challenges. Nat. Rev. Immunol. 2012, 12, 786–798. [Google Scholar] [CrossRef]
- Hayakawa, J.; Hsieh, M.M.; Anderson, D.E.; Phang, O.; Uchida, N.; Washington, K.; Tisdale, J.F. The assessment of human erythroid output in nod/scid mice reconstituted with human hematopoietic stem cells. Cell Transplant 2010, 19, 1465–1473. [Google Scholar] [CrossRef]
- Mombaerts, P.; Iacomini, J.; Johnson, R.S.; Herrup, K.; Tonegawa, S.; Papaioannou, V.E. Rag-1-deficient mice have no mature b and t lymphocytes. Cell 1992, 68, 869–877. [Google Scholar] [CrossRef]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019. [Google Scholar] [CrossRef]
- Falk, J.J.; Laib Sampaio, K.; Stegmann, C.; Lieber, D.; Kropff, B.; Mach, M.; Sinzger, C. Generation of a gaussia luciferase-expressing endotheliotropic cytomegalovirus for screening approaches and mutant analyses. J. Virol. Methods 2016, 235, 182–189. [Google Scholar] [CrossRef]
- Brehm, M.A.; Shultz, L.D.; Luban, J.; Greiner, D.L. Overcoming current limitations in humanized mouse research. J. Infect Dis. 2013, 208 (Suppl. 2), S125–S130. [Google Scholar] [CrossRef] [Green Version]
- Billerbeck, E.; Barry, W.T.; Mu, K.; Dorner, M.; Rice, C.M.; Ploss, A. Development of human cd4+ foxp3+ regulatory t cells in human stem cell factor-, granulocyte-macrophage colony-stimulating factor-, and interleukin-3-expressing nod-scid il2rgamma(null) humanized mice. Blood 2011, 117, 3076–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.; Strowig, T.; Verma, R.; Koduru, S.; Hafemann, A.; Hopf, S.; Kocoglu, M.H.; Borsotti, C.; Zhang, L.; Branagan, A.; et al. Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat. Med. 2016, 22, 1351–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Borsotti, C.; Schickel, J.N.; Zhu, S.; Strowig, T.; Eynon, E.E.; Frleta, D.; Gurer, C.; Murphy, A.J.; Yancopoulos, G.D.; et al. A novel humanized mouse model with significant improvement of class-switched, antigen-specific antibody production. Blood 2017, 129, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.; Ballesteros, A.; Qiu, Q.; Pow Sang, L.; Shashikumar, S.; Casares, S.; Brumeanu, T.D. Generation and testing anti-influenza human monoclonal antibodies in a new humanized mouse model (draga: Hla-a2. Hla-dr4. Rag1 ko. Il-2rgammac ko. Nod). Hum. Vaccin. Immunother. 2018, 14, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Danner, R.; Chaudhari, S.N.; Rosenberger, J.; Surls, J.; Richie, T.L.; Brumeanu, T.D.; Casares, S. Expression of hla class ii molecules in humanized nod.Rag1ko.Il2rgcko mice is critical for development and function of human t and b cells. PLoS ONE 2011, 6, e19826. [Google Scholar] [CrossRef]
- Daenthanasanmak, A.; Salguero, G.; Sundarasetty, B.S.; Waskow, C.; Cosgun, K.N.; Guzman, C.A.; Riese, P.; Gerasch, L.; Schneider, A.; Ingendoh, A.; et al. Engineered dendritic cells from cord blood and adult blood accelerate effector t cell immune reconstitution against hcmv. Mol. Ther. Methods Clin. Dev. 2015, 1, 14060. [Google Scholar] [CrossRef]
- Salguero, G.; Daenthanasanmak, A.; Munz, C.; Raykova, A.; Guzman, C.A.; Riese, P.; Figueiredo, C.; Langer, F.; Schneider, A.; Macke, L.; et al. Dendritic cell-mediated immune humanization of mice: Implications for allogeneic and xenogeneic stem cell transplantation. J. Immunol. 2014, 192, 4636–4647. [Google Scholar] [CrossRef] [Green Version]
- Volk, V.; Reppas, A.I.; Robert, P.A.; Spineli, L.M.; Sundarasetty, B.S.; Theobald, S.J.; Schneider, A.; Gerasch, L.; Deves Roth, C.; Kloss, S.; et al. Multidimensional analysis integrating human t-cell signatures in lymphatic tissues with sex of humanized mice for prediction of responses after dendritic cell immunization. Front. Immunol. 2017, 8, 1709. [Google Scholar] [CrossRef] [Green Version]
- Diamond, D.J.; La Rosa, C.; Chiuppesi, F.; Contreras, H.; Dadwal, S.; Wussow, F.; Bautista, S.; Nakamura, R.; Zaia, J.A. A fifty-year odyssey: Prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev. Vaccine 2018, 17, 889–911. [Google Scholar] [CrossRef] [PubMed]
- Gogesch, P.; Penner, I.; Krauter, S.; Buscher, N.; Grode, L.; Aydin, I.; Plachter, B. Production strategies for pentamer-positive subviral dense bodies as a safe human cytomegalovirus vaccine. Vaccines (Basel) 2019, 7, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Model/Species | Virus Strain | Route of Infections | Immune Responses | Vaccine Evaluation |
|---|---|---|---|---|
| Immunocompetent mice | MCMV: e.g., Smith strain | intraplantar or | T, B, and NK cells responses against MV | Recombinant Adenovirus [21]; |
| BALB/c; C57BL/6 | Intraperitoneal (i.p.) injection | DNA vaccines [22,24]; | ||
| (reviewed in [20]) | Attenuated MCMV [23] | |||
| Rhesus macaques (reviewed in [25]) | RhCMV | Subcutaneous (s.c.) or intravenous (i.v.) or vertical transmission | T and B cell responses, neutralizing antibodies against the Rh-Pentamer and gB | RhCMV-gB, |
| -pp65 or -IE1 protein vaccines [36], modified vaccinia Ankara virus (MVA) expressing the RhCMV Pentamer [26]; RhCMV replication-defective virus [27], anti-RhCMV-IL10 vaccination [28] | ||||
| Immunodeficient mice | HCMV TRpM1A [37] or | i.p. injection of in vitro infected fibroblasts | T cell responses against IE1, pp65, gB; | Not reported yet |
| NSG-HIS [37] or | TB40-GLuc [38] | IgGs responses against gB [38] | ||
| NRG-HIS [38] | ||||
| NSG-huPBL CD34 [39] | HCMV from PBL donor | Infected CD34+ cells in PBL | Not reported | Not reported yet |
| NSG-A2-BLT [40] | HCMV Towne strain, | i.p. injection of in vitro infected fibroblasts [41,42] or DCs [40] | T cells against IE1, pp65 [40,42]; | Live attenuated virus strain [40] |
| NSG-BLT [40,42,43] | TR-strain or | Specific IgM [40,42] | ||
| TB40E-GFP [41,42] | Specific IgG [42] | |||
| ULBP2-TB40 [40] | ||||
| NSG-BLT-L [44] | HCMV TB40/E-fLuc; | Lung implants were infected directly with virus injections | T cells against IE1 and pp65; | Not reported yet |
| ADrUL131; | ||||
| AD169 | Specific IgM and IgG [44] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koenig, J.; Theobald, S.J.; Stripecke, R. Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing. Vaccines 2020, 8, 89. https://doi.org/10.3390/vaccines8010089
Koenig J, Theobald SJ, Stripecke R. Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing. Vaccines. 2020; 8(1):89. https://doi.org/10.3390/vaccines8010089
Chicago/Turabian StyleKoenig, Johannes, Sebastian J. Theobald, and Renata Stripecke. 2020. "Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing" Vaccines 8, no. 1: 89. https://doi.org/10.3390/vaccines8010089
APA StyleKoenig, J., Theobald, S. J., & Stripecke, R. (2020). Modeling Human Cytomegalovirus in Humanized Mice for Vaccine Testing. Vaccines, 8(1), 89. https://doi.org/10.3390/vaccines8010089