Function and Modulation of Type I Interferons during Respiratory Syncytial Virus Infection



{kind=link}
Abstract
:1. Introduction
2. Type I IFN and RSV
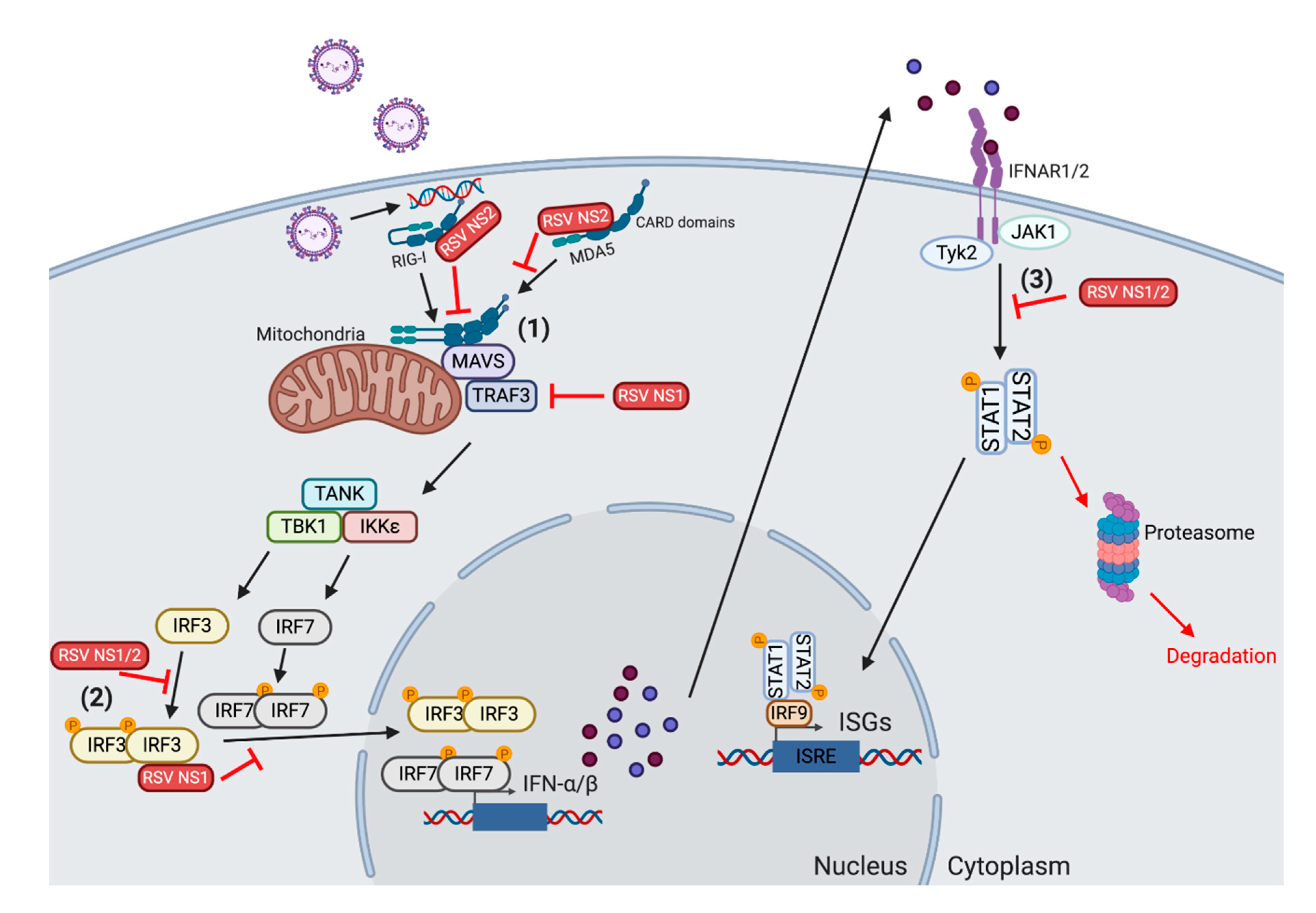
2.1. The Role of Type I IFN and the Innate Immune Response to RSV
2.2. Impairments in Neonatal Type I IFN Responses
2.3. Modulation of Type 1 IFN by RSV
2.4. Alterations in the Adaptive Immune Response by Type 1 IFNs
3. Use of Type I IFNs in Therapeutic and Preventative Strategies for RSV
3.1. Type 1 IFN as a Therapeutic for RSV
3.2. Implications for Vaccine Design
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scheltema, N.M.; Gentile, A.; Lucion, F.; Nokes, D.J.; Munywoki, P.K.; Madhi, S.A.; Groome, M.J.; Cohen, C.; Moyes, J.; Thorburn, K.; et al. Global respiratory syncytial virus-associated mortality in young children (RSV GOLD): A retrospective case series. Lancet Glob. Health 2017, 5, e984–e991. [Google Scholar] [CrossRef] [Green Version]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef] [Green Version]
- Paramore, L.C.; Ciuryla, V.; Ciesla, G.; Liu, L. Economic impact of respiratory syncytial virus-related illness in the US: An analysis of national databases. Pharmacoeconomics 2004, 22, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.B.; Walsh, E.E.; Long, C.E.; Schnabel, K.C. Immunity to and frequency of reinfection with respiratory syncytial virus. J. Infect. Dis. 1991, 163, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Henderson, F.W.; Collier, A.M.; Clyde, W.A., Jr.; Denny, F.W. Respiratory-syncytial-virus infections, reinfections and immunity. A prospective, longitudinal study in young children. N. Engl. J. Med. 1979, 300, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Coenjaerts, F.E.J.; Houspie, L.; Viveen, M.C.; van Bleek, G.M.; Wiertz, E.J.H.J.; Martin, D.P.; Lemey, P. The comparative genomics of human respiratory syncytial virus subgroups A and B: Genetic variability and molecular evolutionary dynamics. J. Virol. 2013, 87, 8213–8226. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.; Baviskar, P.; Duncan-Decocq, R.R.; Patel, D.; Oomens, A.G. The human respiratory syncytial virus matrix protein is required for maturation of viral filaments. J. Virol. 2012, 86, 4432–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cowton, V.M.; McGivern, D.R.; Fearns, R. Unravelling the complexities of respiratory syncytial virus RNA synthesis. J. Gen. Virol. 2006, 87, 1805–1821. [Google Scholar] [CrossRef]
- Batonick, M.; Wertz, G.W. Requirements for Human Respiratory Syncytial Virus Glycoproteins in Assembly and Egress from Infected Cells. Adv. Virol. 2011. [Google Scholar] [CrossRef] [Green Version]
- Gan, S.W.; Tan, E.; Lin, X.; Yu, D.; Wang, J.; Tan, G.M.Y.; Vararattanavech, A.; Yeo, C.Y.; Soon, C.H.; Soong, T.W.; et al. The small hydrophobic protein of the human respiratory syncytial virus forms pentameric ion channels. J. Biol. Chem. 2012, 287, 24671–24689. [Google Scholar] [CrossRef] [Green Version]
- Spann, K.M.; Tran, K.C.; Chi, B.; Rabin, R.L.; Collins, P.L. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J. Virol. 2004, 78, 4363–4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goritzka, M.; Makris, S.; Kausar, F.; Durant, L.R.; Pereira, C.; Kumagai, Y.; Culley, F.J.; Mack, M.; Akira, S.; Johansson, C. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015, 212, 699–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, J.J.; Rudd, B.D.; Lukacs, N.W. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 2006, 203, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Hijano, D.R.; Vu, L.D.; Kauvar, L.M.; Tripp, R.A.; Polack, F.P.; Cormier, S.A. Role of Type I Interferon (IFN) in the Respiratory Syncytial Virus (RSV) Immune Response and Disease Severity. Front. Immunol. 2019, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demoor, T.; Petersen, B.C.; Morris, S.; Mukherjee, S.; Ptaschinski, C.; De Almeida Nagata, D.E.; Kawai, T.; Ito, T.; Akira, S.; Kunkel, S.L.; et al. IPS-1 signaling has a nonredundant role in mediating antiviral responses and the clearance of respiratory syncytial virus. J. Immunol. 2012, 189, 5942–5953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef] [Green Version]
- Goritzka, M.; Durant, L.R.; Pereira, C.; Salek-Ardakani, S.; Openshaw, P.J.M.; Johansson, C. Alpha/beta interferon receptor signaling amplifies early proinflammatory cytokine production in the lung during respiratory syncytial virus infection. J. Virol. 2014, 88, 6128–6136. [Google Scholar] [CrossRef] [Green Version]
- Foster, G.R. Interferons in host defense. Semin. Liver Dis. 1997, 17, 287–295. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Lee, H.K. Innate immune recognition of respiratory syncytial virus infection. BMB Rep. 2014, 47, 184–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; et al. Respiratory syncytial virus fusion protein-induced toll-like receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists Rhodobacter sphaeroides lipopolysaccharide and eritoran (E5564) and requires direct interaction with MD-2. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Marr, N.; Turvey, S.E. Role of human TLR4 in respiratory syncytial virus-induced NF-kappaB activation, viral entry and replication. Innate Immun. 2012, 18, 856–865. [Google Scholar] [CrossRef]
- Awomoyi, A.A.; Rallabhandi, P.; Pollin, T.I.; Lorenz, E.; Sztein, M.B.; Boukhvalova, M.S.; Hemming, V.G.; Blanco, J.C.G.; Vogel, S.N. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 2007, 179, 3171–3177. [Google Scholar] [CrossRef] [Green Version]
- Caballero, M.T.; Serra, M.E.; Acosta, P.L.; Marzec, J.; Gibbons, L.; Salim, M.; Rodriguez, A.; Reynaldi, A.; Garcia, A.; Bado, D.; et al. TLR4 genotype and environmental LPS mediate RSV bronchiolitis through Th2 polarization. J. Clin. Invest. 2015, 125, 571–582. [Google Scholar] [CrossRef] [Green Version]
- Tulic, M.K.; Hurrelbrink, R.J.; Prele, C.M.; Laing, I.A.; Upham, J.W.; Le Souef, P.; Sly, P.D.; Holt, P.G. TLR4 polymorphisms mediate impaired responses to respiratory syncytial virus and lipopolysaccharide. J. Immunol. 2007, 179, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitko, V.; Musiyenko, A.; Bayfield, M.A.; Maraia, R.J.; Barik, S. Cellular La protein shields nonsegmented negative-strand RNA viral leader RNA from RIG-I and enhances virus growth by diverse mechanisms. J. Virol. 2008, 82, 7977–7987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asgari, S.; Schlapbach, L.J.; Anchisi, S.; Hammer, C.; Bartha, I.; Junier, T.; Mottet-Osman, G.; Posfay-Barbe, K.M.; Longchamp, D.; Stocker, M.; et al. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc. Natl. Acad. Sci. USA 2017, 114, 8342–8347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgnaoui, S.M.; Paz, S.; Hiscott, J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011, 23, 564–572. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar]
- Makris, S.; Bajorek, M.; Culley, F.J.; Goritzka, M.; Johansson, C. Alveolar Macrophages Can Control Respiratory Syncytial Virus Infection in the Absence of Type I Interferons. J. Innate Immun. 2016, 8, 452–463. [Google Scholar] [CrossRef]
- Jewell, N.A.; Vaghefi, N.; Mertz, S.E.; Akter, P.; Peebles, R.S., Jr.; Bakaletz, L.O.; Durbin, R.K.; Flano, E.; Durbin, J.E. Differential type I interferon induction by respiratory syncytial virus and influenza a virus in vivo. J. Virol. 2007, 81, 9790–9800. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Schlender, J.; Guenthner-Biller, M.; Rothenfusser, S.; Endres, S.; Conzelmann, K.K.; Hartmann, G. Replication-dependent potent IFN-alpha induction in human plasmacytoid dendritic cells by a single-stranded RNA virus. J. Immunol. 2004, 173, 5935–5943. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.R.; Johnson, C.N.; Corbett, K.S.; Edwards, G.C.; Graham, B.S. Primary human mDC1, mDC2, and pDC dendritic cells are differentially infected and activated by respiratory syncytial virus. PLoS ONE 2011, 6, e16458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boogaard, I.; van Oosten, M.; van Rijt, L.S.; Muskens, F.; Kimman, T.G.; Lambrecht, B.N.; Buisman, A.M. Respiratory syncytial virus differentially activates murine myeloid and plasmacytoid dendritic cells. Immunology 2007, 122, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Schijf, M.A.; Lukens, M.V.; Kruijsen, D.; van Uden, N.O.P.; Garssen, J.; Coenjaerts, F.E.J.; van’t Land, B.; van Bleek, G.M. Respiratory syncytial virus induced type I IFN production by pDC is regulated by RSV-infected airway epithelial cells, RSV-exposed monocytes and virus specific antibodies. PLoS ONE 2013, 8, e81695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, Y.; Takeuchi, O.; Kato, H.; Kumar, H.; Matsui, K.; Morii, E.; Aozasa, K.; Kawai, T.; Akira, S. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 2007, 27, 240–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.H.; Oh, D.S.; Jung, H.E.; Chang, J.; Lee, H.K. Plasmacytoid Dendritic Cells Contribute to the Production of IFN-beta via TLR7-MyD88-Dependent Pathway and CTL Priming during Respiratory Syncytial Virus Infection. Viruses 2019, 11, 730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Peters, N.; Schwarze, J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J. Immunol. 2006, 177, 6263–6270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crisler, W.J.; Lenz, L.L. Crosstalk between type I and II interferons in regulation of myeloid cell responses during bacterial infection. Curr. Opin. Immunol. 2018, 54, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Everitt, A.R.; Clare, S.; McDonald, J.U.; Kane, L.; Harcourt, K.; Ahras, M.; Lall, A.; Hale, C.; Rodgers, A.; Young, D.B.; et al. Defining the range of pathogens susceptible to Ifitm3 restriction using a knockout mouse model. PLoS ONE 2013, 8, e80723. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.E.; Busse, D.C.; Binter, S.; Weston, S.; Diaz Soria, C.; Laksono, B.M.; Clare, S.; Van Nieuwkoop, S.; Van den Hoogen, B.G.; Clement, M.; et al. Interferon-Induced Transmembrane Protein 1 Restricts Replication of Viruses That Enter Cells via the Plasma Membrane. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Zhang, L.; Zan, Y.; Du, N.; Yang, Y.; Tien, P. Human respiratory syncytial virus infection is inhibited by IFN-induced transmembrane proteins. J. Gen. Virol. 2015, 96, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.N.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, C.; Resch, B.; Simoes, E.A. Risk factors for severe respiratory syncytial virus lower respiratory tract infection. Open Microbiol. J. 2011, 5, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, D.A.; Rodriguez-Martinez, C.E.; Cardenas, A.C.; Quilaguy, I.E.; Mayorga, L.Y.; Falla, L.M.; Nino, G. Predictors of severity and mortality in children hospitalized with respiratory syncytial virus infection in a tropical region. Pediatr. Pulmonol. 2014, 49, 269–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, T.P.; Sinkin, R.A.; Hall, C.B.; Maniscalco, W.M.; McConnochie, K.M. Respiratory syncytial virus and premature infants born at 32 weeks’ gestation or earlier: Hospitalization and economic implications of prophylaxis. Arch. Pediatr. Adolesc. Med. 2000, 154, 55–61. [Google Scholar] [PubMed]
- Anonymous. Prevention of respiratory syncytial virus infections: Indications for the use of palivizumab and update on the use of RSV-IGIV. American Academy of Pediatrics Committee on Infectious Diseases and Committee of Fetus and Newborn. Pediatrics 1998, 102, 1211–1216. [Google Scholar]
- Beyer, M.; Bartz, H.; Horner, K.; Doths, S.; Koerner-Rettberg, C.; Schwarze, J. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J. Allergy Clin. Immunol. 2004, 113, 127–133. [Google Scholar] [CrossRef]
- Gill, M.A.; Palucka, A.K.; Barton, T.; Ghaffar, F.; Jafri, H.; Banchereau, J.; Ramilo, O. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J. Infect. Dis. 2005, 191, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Cormier, S.A.; Shrestha, B.; Saravia, J.; Lee, G.I.; Shen, L.; DeVincenzo, J.P.; Kim, Y.; You, D. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J. Virol. 2014, 88, 9350–9360. [Google Scholar] [CrossRef] [Green Version]
- Lau-Kilby, A.W.; Turfkruyer, M.; Kehl, M.; Yang, L.; Buchholz, U.J.; Hickey, K.; Malloy, A.M.W. Type I IFN ineffectively activates neonatal dendritic cells limiting respiratory antiviral T-cell responses. Mucosal Immunol. 2019. [Google Scholar] [CrossRef]
- Remot, A.; Descamps, D.; Jouneau, L.; Laubreton, D.; Dubuquoy, C.; Bouet, S.; Lecardonnel, J.; Rebours, E.; Petit-Camurdan, A.; Riffault, S. Flt3 ligand improves the innate response to respiratory syncytial virus and limits lung disease upon RSV reexposure in neonate mice. Eur. J. Immunol. 2016, 46, 874–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marr, N.; Wang, T.I.; Kam, S.H.Y.; Hu, Y.S.; Sharma, A.A.; Lam, A.; Markowski, J.; Solimano, S.; Lavoie, P.M.; Turvey, S.E. Attenuation of respiratory syncytial virus-induced and RIG-I-dependent type I IFN responses in human neonates and very young children. J. Immunol. 2014, 192, 948–957. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, K. Interferon in nasal secretions from infants with viral respiratory tract infections. J. Pediatr. 1978, 93, 33–36. [Google Scholar] [CrossRef]
- Hall, C.B.; Douglas, R.G., Jr.; Simons, R.L.; Geiman, J.M. Interferon production in children with respiratory syncytial, influenza, and parainfluenza virus infections. J. Pediatr. 1978, 93, 28–32. [Google Scholar] [CrossRef]
- Isaacs, D. Production of interferon in respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 1989, 64, 92–95. [Google Scholar] [CrossRef] [Green Version]
- Ruckwardt, T.J.; Malloy, A.M.; Morabito, K.M.; Graham, B.S. Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLoS Pathog. 2014, 10, e1003934. [Google Scholar] [CrossRef] [Green Version]
- Schlender, J.; Hornung, V.; Finke, S.; Gunthner-Biller, M.; Marozin, S.; Brzozka, K.; Moghim, S.; Endres, S.; Hartmann, G.; Conzelmann, K.K. Inhibition of toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J. Virol. 2005, 79, 5507–5515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Yang, H.; Kong, X.; Mohapatra, S.; San Juan-Vergara, H.; Hellermann, G.; Behera, S.; Singam, R.; Lockey, R.F.; Mohapatra, S.S. Inhibition of respiratory syncytial virus infection with intranasal siRNA nanoparticles targeting the viral NS1 gene. Nat. Med. 2005, 11, 56–62. [Google Scholar] [CrossRef]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef] [Green Version]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef] [Green Version]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-kappaB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef] [PubMed]
- Hastie, M.L.; Headlam, M.J.; Patel, N.B.; Bukreyev, A.B.; Buchholz, U.J.; Dave, K.A.; Norris, E.L.; Wright, C.L.; Spann, K.M.; Collins, P.L.; et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol. Cell. Proteom. 2012, 11, 108–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.S.; Brazas, R.M.; Holtzman, M.J. Respiratory syncytial virus nonstructural proteins NS1 and NS2 mediate inhibition of Stat2 expression and alpha/beta interferon responsiveness. J. Virol. 2005, 79, 9315–9319. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, M.; Shi, L.; Varga, S.M.; Barik, S.; Behlke, M.A.; Look, D.C. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 2006, 344, 328–339. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, M.; Shi, L.; Monick, M.M.; Hunninghake, G.W.; Look, D.C. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2004, 30, 893–900. [Google Scholar] [CrossRef]
- Munir, S.; Nouen, C.L.; Luongo, C.; Buchholz, U.J.; Collins, P.L.; Bukreyev, A. Nonstructural proteins 1 and 2 of respiratory syncytial virus suppress maturation of human dendritic cells. J. Virol. 2008, 82, 8780–8796. [Google Scholar] [CrossRef] [Green Version]
- Reis e Sousa, C. Dendritic cells in a mature age. Nat. Rev. Immunol. 2006, 6, 476–483. [Google Scholar] [CrossRef]
- Montoya, M.; Schiavoni, G.; Mattei, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar] [CrossRef] [Green Version]
- Roman, M.; Calhoun, W.J.; Hinton, K.L.; Avendano, L.F.; Simon, V.; Escobar, A.M.; Gaggero, A.; Diaz, P.V. Respiratory syncytial virus infection in infants is associated with predominant Th-2-like response. Am. J. Respir. Crit. Care Med. 1997, 156, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Bendelja, K.; Gagro, A.; Bace, A.; Lokar-Kolbas, R.; Krsulovic-Hresic, V.; Drazenovic, V.; Mlinaric-Galinovic, G.; Rabatic, S. Predominant type-2 response in infants with respiratory syncytial virus (RSV) infection demonstrated by cytokine flow cytometry. Clin. Exp. Immunol. 2000, 121, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Nenna, R.; Fedele, G.; Frassanito, A.; Petrarca, L.; Di Mattia, G.; Pierangeli, A.; Scagnolari, C.; Papoff, P.; Schiavoni, I.; Leone, P.; et al. Increased T-helper Cell 2 Response in Infants With Respiratory Syncytial Virus Bronchiolitis Hospitalized Outside Epidemic Peak. Pediatr. Infect. Dis. J. 2020, 39, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.E.; Johnson, J.E.; Kuli-Zade, R.K.; Johnson, T.R.; Aung, S.; Parker, R.A.; Graham, B.S. Overexpression of interleukin-4 delays virus clearance in mice infected with respiratory syncytial virus. J. Virol. 1997, 71, 8672–8677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.W.; Graham, B.S. Anti-IL-4 treatment at immunization modulates cytokine expression, reduces illness, and increases cytotoxic T lymphocyte activity in mice challenged with respiratory syncytial virus. J. Clin. Invest. 1994, 94, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.; Caramalho, I.; Demengeot, J. IFN-alpha/beta enhances BCR-dependent B cell responses. Int. Immunol. 2002, 14, 411–419. [Google Scholar] [CrossRef]
- Hijano, D.R.; Siefker, D.T.; Shrestha, B.; Jaligama, S.; Vu, L.D.; Tillman, H.; Finkelstein, D.; Saravia, J.; You, D.; Cormier, S.A. Type I Interferon Potentiates IgA Immunity to Respiratory Syncytial Virus Infection During Infancy. Sci. Rep. 2018, 8, 11034. [Google Scholar] [CrossRef]
- Anderson, E.J.; Carosone-Link, P.; Yogev, R.; Yi, J.; Simoes, E.A.F. Effectiveness of Palivizumab in High-risk Infants and Children: A Propensity Score Weighted Regression Analysis. Pediatr. Infect. Dis. J. 2017, 36, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Narbona-Lopez, E.; Uberos, J.; Checa-Ros, A.; Rodriguez-Belmonte, R.; Munoz-Hoyos, A. Prevention of syncytial respiratory virus infection with palivizumab: Descriptive and comparative analysis after 12 years of use. Minerva Pediatr. 2018, 70, 513–518. [Google Scholar] [CrossRef]
- Alansari, K.; Toaimah, F.H.; Almatar, D.H.; El Tatawy, L.A.; Davidson, B.L.; Qusad, M.I.M. Monoclonal Antibody Treatment of RSV Bronchiolitis in Young Infants: A Randomized Trial. Pediatrics 2019, 143. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Plata, A.; Baron, S.; Poast, J.S.; Adegboyega, P.A.; Casola, A.; Garofalo, R.P. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 2005, 79, 10190–10199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, R.Y.; Yin, J.; Oppenheimer, S.J.; Tam, J.S.; Lau, J. Treatment of respiratory syncytial virus infection with recombinant interferon alfa-2a. Arch. Dis Child. 1993, 69, 440–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipps, B.E.; Sullivan, W.F.; Portnoy, J.M. Alpha-2A-interferon for treatment of bronchiolitis caused by respiratory syncytial virus. Pediatr. Infect. Dis. J. 1993, 12, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Shi, M.; Deng, Q.; Liu, W.; Li, Q.; Ye, P.; Yu, X.; Zhang, B.; Xu, Y.; Li, X.; et al. A multi-center randomized prospective study on the treatment of infant bronchiolitis with interferon alpha1b nebulization. PLoS ONE 2020, 15, e0228391. [Google Scholar]
- Higgins, P.G.; Barrow, G.I.; Tyrrell, D.A.; Isaacs, D.; Gauci, C.L. The efficacy of intranasal interferon alpha-2a in respiratory syncytial virus infection in volunteers. Antivir. Res. 1990, 14, 3–10. [Google Scholar] [CrossRef]
- Jin, H.; Zhou, H.; Cheng, X.; Tang, R.; Munoz, M.; Nguyen, N. Recombinant respiratory syncytial viruses with deletions in the NS1, NS2, SH, and M2-2 genes are attenuated in vitro and in vivo. Virology 2000, 273, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.N.; Whitehead, S.S.; Bermingham, A.; St. Claire, M.; Elkins, W.R.; Murphy, B.R.; Collins, P.L. Recombinant respiratory syncytial virus that does not express the NS1 or M2-2 protein is highly attenuated and immunogenic in chimpanzees. J. Virol. 2000, 74, 9317–9321. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.N.; Mejias, A.; Ramilo, O.; Peeples, M.E. Live Attenuated Vaccine with a Stabilized Mutation and Gene Deletion for Prevention of Respiratory Syncytial Virus Disease in Young Children. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Munir, S.; Hillyer, P.; Le Nouen, C.; Buchholz, U.J.; Rabin, R.L.; Collins, P.L.; Bukreyev, A. Respiratory syncytial virus interferon antagonist NS1 protein suppresses and skews the human T lymphocyte response. PLoS Pathog. 2011, 7, e1001336. [Google Scholar] [CrossRef] [Green Version]
- McFarland, E.J.; Karron, R.A.; Muresan, P.; Cunningham, C.K.; Valentine, M.E.; Perlowski, C.; Thumar, B.; Gnanashanmugam, D.; Siberry, G.K.; Schappell, E.; et al. Live-Attenuated Respiratory Syncytial Virus Vaccine Candidate With Deletion of RNA Synthesis Regulatory Protein M2-2 is Highly Immunogenic in Children. J. Infect. Dis. 2018, 217, 1347–1355. [Google Scholar] [CrossRef]
- McFarland, E.J.; Karron, R.A.; Muresan, P.; Cunningham, C.K.; Perlowski, C.; Libous, J.; Oliva, J.; Jean-Philippe, P.; Moye, J., Jr.; Schappell, E.; et al. Live-attenuated respiratory syncytial virus vaccine with M2-2 deletion and with SH non-coding region is highly immunogenic in children. J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.C.; Barber, J.; Tripp, R.A. Respiratory syncytial virus (RSV) attachment and nonstructural proteins modify the type I interferon response associated with suppressor of cytokine signaling (SOCS) proteins and IFN-stimulated gene-15 (ISG15). Virol. J. 2008, 5, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starr, R.; Willson, T.A.; Viney, E.M.; Murray, L.J.L.; Rayner, J.R.; Jenkins, B.J.; Gonda, T.J.; Alexander, W.S.; Metcalf, D.; Nicola, N.A.; et al. A family of cytokine-inducible inhibitors of signalling. Nature 1997, 387, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Kiu, H.; Nicholson, S.E. SOCS regulation of the JAK/STAT signalling pathway. Semin. Cell Dev. Biol. 2008, 19, 414–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.F.; Peng, W.M.; Schlee, M.; Barchet, W.; Eis-Hubinger, A.M.; Kolanus, W.; Geyer, M.; Schmitt, S.; Steinhagen, F.; Oldenburg, J.; et al. SOCS1 and SOCS3 Target IRF7 Degradation To Suppress TLR7-Mediated Type I IFN Production of Human Plasmacytoid Dendritic Cells. J. Immunol. 2018, 200, 4024–4035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshansky, C.M.; Krunkosky, T.M.; Barber, J.; Jones, L.P.; Tripp, R.A. Respiratory syncytial virus proteins modulate suppressors of cytokine signaling 1 and 3 and the type I interferon response to infection by a toll-like receptor pathway. Viral Immunol. 2009, 22, 147–161. [Google Scholar] [CrossRef]
- Maher, C.F.; Hussell, T.; Blair, E.; Ring, C.J.; Openshaw, P.J. Recombinant respiratory syncytial virus lacking secreted glycoprotein G is attenuated, non-pathogenic but induces protective immunity. Microbes Infect. 2004, 6, 1049–1055. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stephens, L.M.; Varga, S.M. Function and Modulation of Type I Interferons during Respiratory Syncytial Virus Infection. Vaccines 2020, 8, 177. https://doi.org/10.3390/vaccines8020177
Stephens LM, Varga SM. Function and Modulation of Type I Interferons during Respiratory Syncytial Virus Infection. Vaccines. 2020; 8(2):177. https://doi.org/10.3390/vaccines8020177
Chicago/Turabian StyleStephens, Laura M., and Steven M. Varga. 2020. "Function and Modulation of Type I Interferons during Respiratory Syncytial Virus Infection" Vaccines 8, no. 2: 177. https://doi.org/10.3390/vaccines8020177
APA StyleStephens, L. M., & Varga, S. M. (2020). Function and Modulation of Type I Interferons during Respiratory Syncytial Virus Infection. Vaccines, 8(2), 177. https://doi.org/10.3390/vaccines8020177