Identifying Suspect Bat Reservoirs of Emerging Infections


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Dataset
2.2. Bat Phylogeny
2.3. Phylofactorization of Bat Virus Data
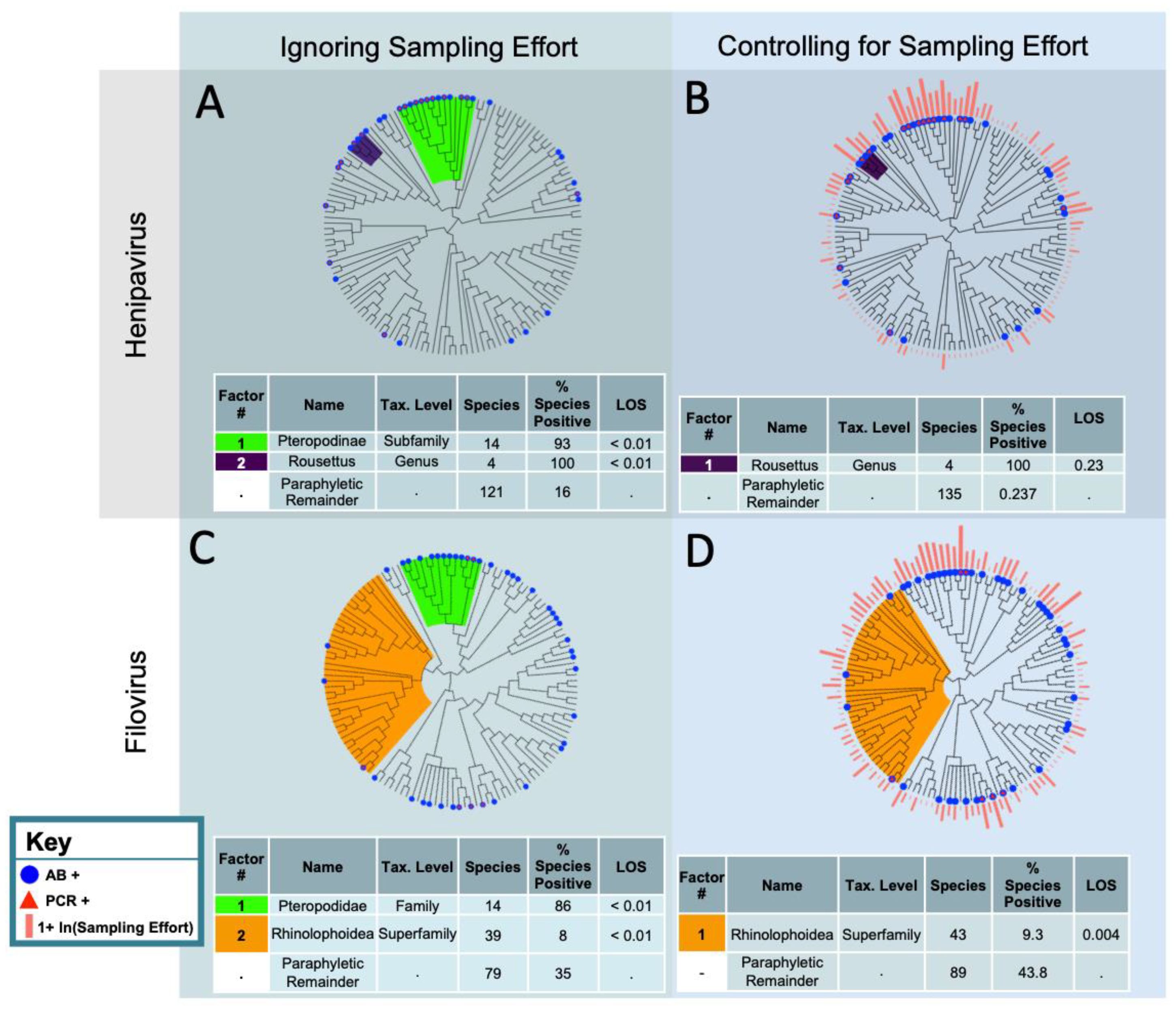
3. Results
3.1. Filovirus Suspect Reservoirs
3.2. Henipavirus Suspect Reservoirs
4. Discussion
4.1. Filoviruses
4.2. Henipaviruses
4.3. Bombali Virus
4.4. Limitations
4.5. Evidence of Infection Metric
4.6. Sampling Effort Metric
4.7. Choice of Phylogenetic Methods
4.8. Physiological Differences between Bat Clades
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Luby, S.P.; Rahman, M.; Hossain, M.J.; Blum, L.S.; Husain, M.M.; Gurley, E.; Khan, R.; Ahmed, B.N.; Rahman, S.; Nahar, N.; et al. Foodborne transmission of Nipah virus, Bangladesh. Emerg. Infect. Dis. 2006, 12, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Leroy, E.M.; Gonzalez, J.P.; Baize, S. Ebola and Marburg haemorrhagic fever viruses: Major scientific advances, but a relatively minor public health threat for Africa. Clin. Microbiol. Infect. 2011, 17, 964–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayman, D.T.; Wang, L.F.; Barr, J.; Baker, K.S.; Suu-Ire, R.; Broder, C.C.; Cunningham, A.A.; Wood, J.L. Antibodies to henipavirus or henipa-like viruses in domestic pigs in Ghana, West Africa. PLoS ONE 2011, 6, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, E.M.; Epelboin, A.; Mondonge, V.; Pourrut, X.; Gonzalez, J.P.; Muyembe-Tamfum, J.J.; Formenty, P. Human Ebola Outbreak Resulting from Direct Exposure to Fruit Bats in Luebo, Democratic Republic of Congo, 2007. Vector-Borne Zoonotic Dis. 2009, 9, 723–728. [Google Scholar] [CrossRef]
- Amman, B.R.; Carroll, S.A.; Reed, Z.D.; Sealy, T.K.; Balinandi, S.; Swanepoel, R.; Kemp, A.; Erickson, B.R.; Comer, J.A.; Campbell, S.; et al. Seasonal Pulses of Marburg Virus Circulation in Juvenile Rousettus aegyptiacus Bats Coincide with Periods of Increased Risk of Human Infection. PLoS Pathog. 2012, 8, e1002877. [Google Scholar] [CrossRef]
- Leendertz, S.A.J.; Gogarten, J.F.; Düx, A.; Calvignac-Spencer, S.; Leendertz, F.H. Assessing the evidence supporting fruit bats as the primary reservoirs for ebola viruses. Ecohealth 2016, 13, 18–25. [Google Scholar] [CrossRef]
- Plowright, R.K.; Peel, A.J.; Streicker, D.G.; Gilbert, A.T.; McCallum, H.; Wood, J.; Baker, M.L.; Restif, O. Transmission or Within-Host Dynamics Driving Pulses of Zoonotic Viruses in Reservoir–Host Populations. PLoS Negl. Trop. Dis. 2016, 10, e0004796. [Google Scholar] [CrossRef] [Green Version]
- Plowright, R.K.; Parrish, C.R.; McCallum, H.; Hudson, P.J.; Ko, A.I.; Graham, A.L.; Lloyd-Smith, J.O. Pathways to zoonotic spillover. Nat. Rev. Microbiol. 2017, 15, 502–510. [Google Scholar] [CrossRef] [Green Version]
- Washburne, A.D.; Silverman, J.D.; Morton, J.T.; Becker, D.J.; Crowley, D.; Mukherjee, S.; David, L.A.; Plowright, R.K. Phylofactorization: A graph partitioning algorithm to identify phylogenetic scales of ecological data. Ecol. Monogr. 2019, 89, e01353. [Google Scholar] [CrossRef]
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent reservoirs of future zoonotic diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 7039–7044. [Google Scholar] [CrossRef] [Green Version]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Becker, D.J.; Crowley, D.E.; Washburne, A.D.; Plowright, R.K. Temporal and spatial limitations in global surveillance for bat filoviruses and henipaviruses. Biol. Lett. 2019, 15, 20190423. [Google Scholar] [CrossRef] [PubMed]
- Michonneau, F.; Brown, J.W.; Winter, D.J. rotl: An R package to interact with the Open Tree of Life data. Fitzjohn R, editor. Methods Ecol. Evol. 2016, 7, 1476–1481. [Google Scholar] [CrossRef]
- Hinchliff, C.E.; Smith, S.A.; Allman, J.F.; Burleigh, J.G.; Chaudhary, R.; Coghill, L.M.; Crandall, K.A.; Deng, J.; Drew, B.T.; Gazis, R.; et al. Synthesis of phylogeny and taxonomy into a comprehensive tree of life. Proc. Natl. Acad. Sci. USA 2015, 112, 12764–12769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grafen, A. The phylogenetic regression. Philos. Trans. R. Soc. London. B Biol. Sci. 1989, 326, 119. [Google Scholar] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, S.; Szoecs, E.; Foster, Z.; Arendsee, Z.; Boettiger, C.; Ram, K.; Bartomeus, I.; Baumgartner, J.; O’Donnell, J.; Oksanen, J.; et al. Taxize: Taxonomic Information from Around the Web. R Package Version 0.9.95. 2020. Available online: https://github.com/ropensci/taxize (accessed on 15 May 2020).
- Washburne, A.D.; Silverman, J.D.; Leff, J.W.; Bennett, D.J.; Darcy, J.L.; Mukherjee, S.; Fierer, N.; David, L.A. Phylogenetic factorization of compositional data yields lineage-level associations in microbiome datasets. PeerJ 2017, 5, e2969. [Google Scholar] [CrossRef]
- Swanepoel, R.; Smit, S.B.; Rollin, P.E.; Formenty, P.; Leman, P.A.; Kemp, A.; Burt, F.J.; Grobbelaar, A.A.; Croft, J.; Bausch, D.G.; et al. Studies of Reservoir Hosts for Marburg Virus. Emerg. Infect. Dis. 2007, 13, 1847–1851. [Google Scholar] [CrossRef]
- Amman, B.R.; Jones, M.E.; Sealy, T.K.; Uebelhoer, L.S.; Schuh, A.J.; Bird, B.H.; Coleman-McCray, J.D.; Martin, B.E.; Nichol, S.T.; Towner, J.S. Oral Shedding of Marburg Viral in Experimentally Infected Egyptian Fruit Bats (Rousettus Aegyptiacus). J. Wildl. Dis. 2015, 51, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.E.; Schuh, A.J.; Amman, B.R.; Sealy, T.K.; Zaki, S.R.; Nichol, S.T.; Towner, J.S. Experimental Inoculation of Egyptian Rousette Bats (Rousettus aegyptiacus) with Viruses of the Ebolavirus and Marburgvirus Genera. Viruses 2015, 7, 3420–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paweska, J.T.; Jansen van Vuren, P.; Fenton, K.A.; Graves, K.; Grobbelaar, A.A.; Moolla, N.; Leman, P.; Weyer, J.; Storm, N.; McCulloch, S.D.; et al. Lack of Marburg Virus Transmission From Experimentally Infected to Susceptible In-Contact Egyptian Fruit Bats. J. Infect. Dis. 2015, 212, S109–S118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amman, B.R.; Nyakarahuka, L.; McElroy, A.K.; Dodd, K.A.; Sealy, T.K.; Schuh, A.J.; Shoemaker, T.R.; Balinandi, S.; Atimnedi, P.; Kaboyo, W.; et al. Marburgvirus resurgence in Kitaka mine bat population after extermination attempts, Uganda. Emerg. Infect. Dis. 2014, 20, 1761–1764. [Google Scholar] [CrossRef] [PubMed]
- Towner, J.S.; Amman, B.R.; Sealy, T.K.; Carroll, S.A.R.; Comer, J.A.; Kemp, A.; Swanepoel, R.; Paddock, C.D.; Balinandi, S.; Khristova, M.L.; et al. Isolation of Genetically Diverse Marburg Viruses from Egyptian Fruit Bats. PLoS Pathog. 2009, 5, e1000536. [Google Scholar] [CrossRef]
- Leroy, E.M.; Kumulungui, B.; Pourrut, X.; Rouquet, P.; Hassanin, A.; Yaba, P.; Délicat, A.; Paweska, J.T.; Gonzalez, J.P.; Swanepoel, R. Fruit bats as reservoirs of Ebola virus. Nature 2005, 438, 575–576. [Google Scholar] [CrossRef]
- Schulz, J.E.; Seifert, S.N.; Thompson, J.T.; Avanzato, V.; Sterling, S.L.; Yan, L.; Letko, M.C.; Matson, M.J.; Fischer, R.J.; Tremeau-Bravard, A.; et al. Serological Evidence for Henipa-like and Filo-like Viruses in Trinidad Bats. J. Infect. Dis. 2020, 221, S375–S382. [Google Scholar] [CrossRef]
- Carroll, S.A.; Towner, J.S.; Sealy, T.K.; McMullan, L.K.; Khristova, M.L.; Burt, F.J.; Swanepoel, R.; Rollin, P.E.; Nichol, S.T. Evolution of Viruses of the Family Filoviridae Based on 97 Whole-Genome Sequences. J. Virol. 2013, 87, 2608–2616. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.J.; Leach, R.W.; Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 2010, 10, 193. [Google Scholar] [CrossRef] [Green Version]
- Han, B.A.; Schmidt, J.P.; Alexander, L.W.; Bowden, S.E.; Hayman, D.T.; Drake, J.M. Undiscovered Bat Hosts of Filoviruses. PLoS Negl. Trop. Dis. 2016, 10, e0004815. [Google Scholar] [CrossRef]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol. 2000, 81, 1927–1932. [Google Scholar] [CrossRef]
- Chua, K.B.; Koh, C.L.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Halpin, K.; Hyatt, A.D.; Fogarty, R.; Middleton, D.; Bingham, J.; Epstein, J.H.; Rahman, S.A.; Hughes, T.; Smith, C.; Field, H.E.; et al. Pteropid Bats are Confirmed as the Reservoir Hosts of Henipaviruses: A Comprehensive Experimental Study of Virus Transmission. Am. J. Trop. Med. Hyg. 2011, 85, 946–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, H.; Young, P.; Yob, J.M.; Mills, J.; Hall, L.; Mackenzie, J. The natural history of Hendra and Nipah viruses. Microbes Infect. 2001, 3, 307–314. [Google Scholar] [CrossRef]
- Plowright, R.K.; Eby, P.; Hudson, P.J.; Smith, I.L.; Westcott, D.; Bryden, W.L.; Middleton, D.; Reid, P.A.; McFarlane, R.A.; Martin, G.; et al. Ecological dynamics of emerging bat virus spillover. Proc. R. Soc. B Biol. Sci. 2015, 282, 20142124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The discovery of Bombali virus adds further support for bats as hosts of ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.T.; Fooks, A.R.; Hayman, D.T.S.; Horton, D.L.; Müller, T.; Plowright, R.; Peel, A.J.; Bowen, R.; Wood, J.L.N.; Mills, J.; et al. Deciphering Serology to Understand the Ecology of Infectious Diseases in Wildlife. Ecohealth 2013, 10, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Peel, A.J.; McKinley, T.J.; Baker, K.S.; Barr, J.A.; Crameri, G.; Hayman, D.T.; Feng, Y.R.; Broder, C.C.; Wang, L.F.; Cunningham, A.A.; et al. Use of cross-reactive serological assays for detecting novel pathogens in wildlife: Assessing an appropriate cutoff for henipavirus assays in African bats. J. Virol. Methods 2013, 193, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Plowright, R.K.; Becker, D.J.; McCallum, H.; Manlove, K.R. Sampling to elucidate the dynamics of infections in reservoir hosts. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180336. [Google Scholar] [CrossRef] [Green Version]
- Gould, N.E.; Eldredge, N. Puncuated equilibria: An alternative to phyletic gradualism. In Models in Paleobiology; Freeman, Cooper & Company: San Francisco, CA, USA, 1972; pp. 82–115. [Google Scholar]
- Landis, M.J.; Schraiber, J.G.; Liang, M. Phylogenetic analysis using Lévy processes: Finding jumps in the evolution of continuous traits. Syst. Biol. 2013, 62, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Washburne, A.D.; Crowley, D.E.; Becker, D.J.; Olival, K.J.; Taylor, M.; Munster, V.J.; Plowright, R.K. Taxonomic patterns in the zoonotic potential of mammalian viruses. PeerJ 2018, 6, e5979. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crowley, D.; Becker, D.; Washburne, A.; Plowright, R. Identifying Suspect Bat Reservoirs of Emerging Infections. Vaccines 2020, 8, 228. https://doi.org/10.3390/vaccines8020228
Crowley D, Becker D, Washburne A, Plowright R. Identifying Suspect Bat Reservoirs of Emerging Infections. Vaccines. 2020; 8(2):228. https://doi.org/10.3390/vaccines8020228
Chicago/Turabian StyleCrowley, Daniel, Daniel Becker, Alex Washburne, and Raina Plowright. 2020. "Identifying Suspect Bat Reservoirs of Emerging Infections" Vaccines 8, no. 2: 228. https://doi.org/10.3390/vaccines8020228
APA StyleCrowley, D., Becker, D., Washburne, A., & Plowright, R. (2020). Identifying Suspect Bat Reservoirs of Emerging Infections. Vaccines, 8(2), 228. https://doi.org/10.3390/vaccines8020228