IFN-I Independent Antiviral Immune Response to Vesicular Stomatitis Virus Challenge in Mouse Brain


Abstract
:1. Introduction
2. Materials and Methods
2.1. Animal Experimentation
2.2. Animal Ethics Statement
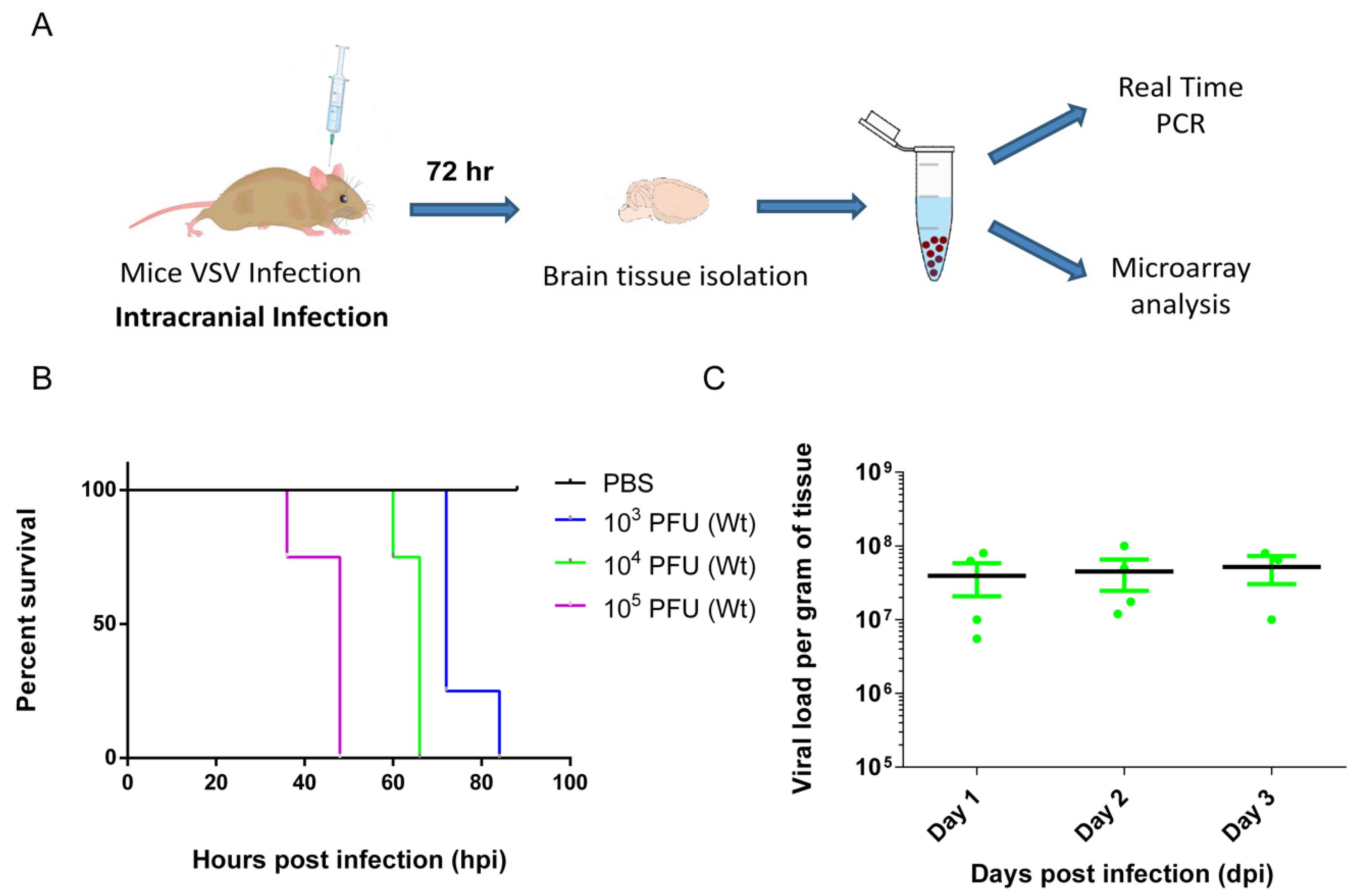
2.3. Virus and Infections
2.4. Real-Time PCR
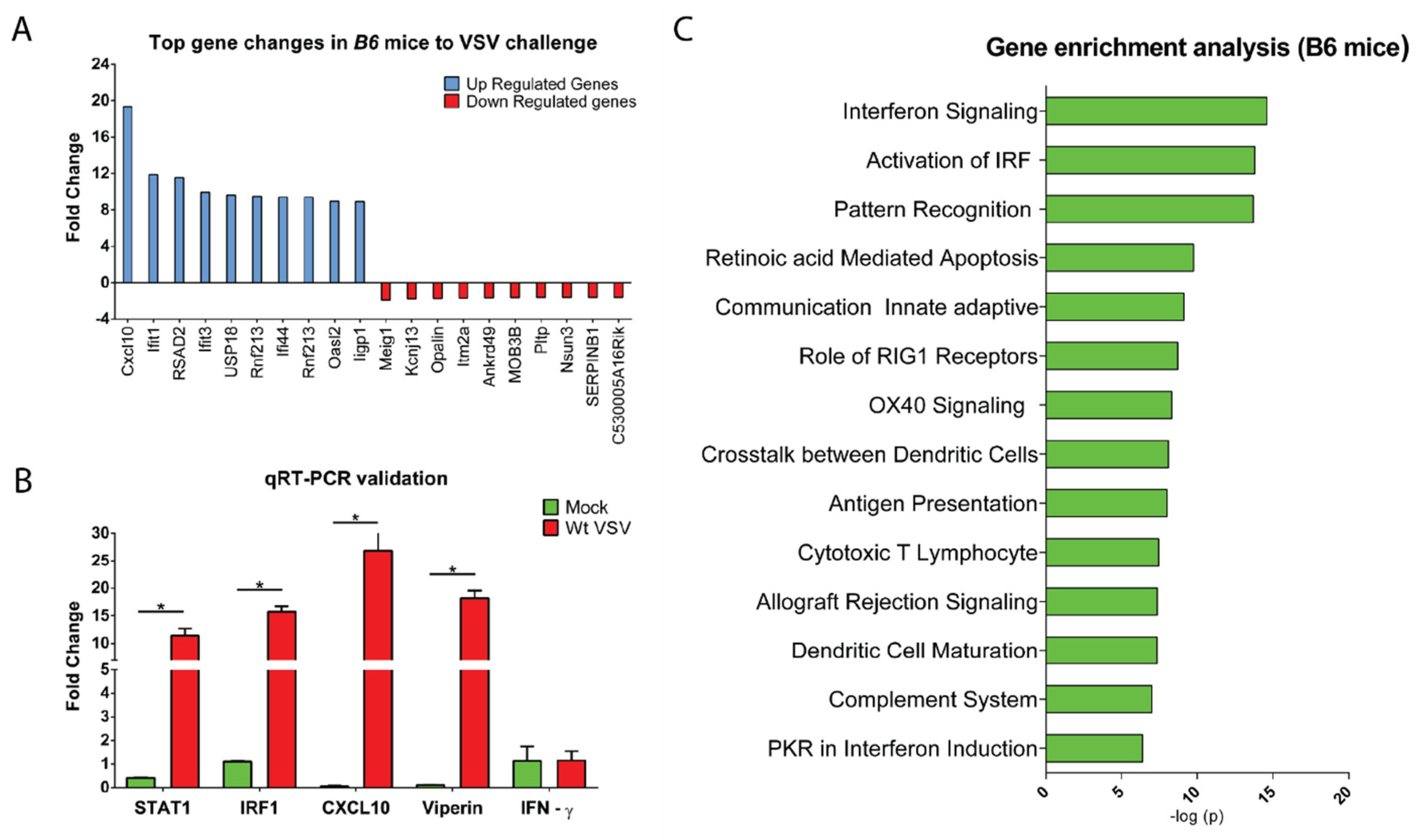
2.5. Microarray Hybridization and Data Analyses
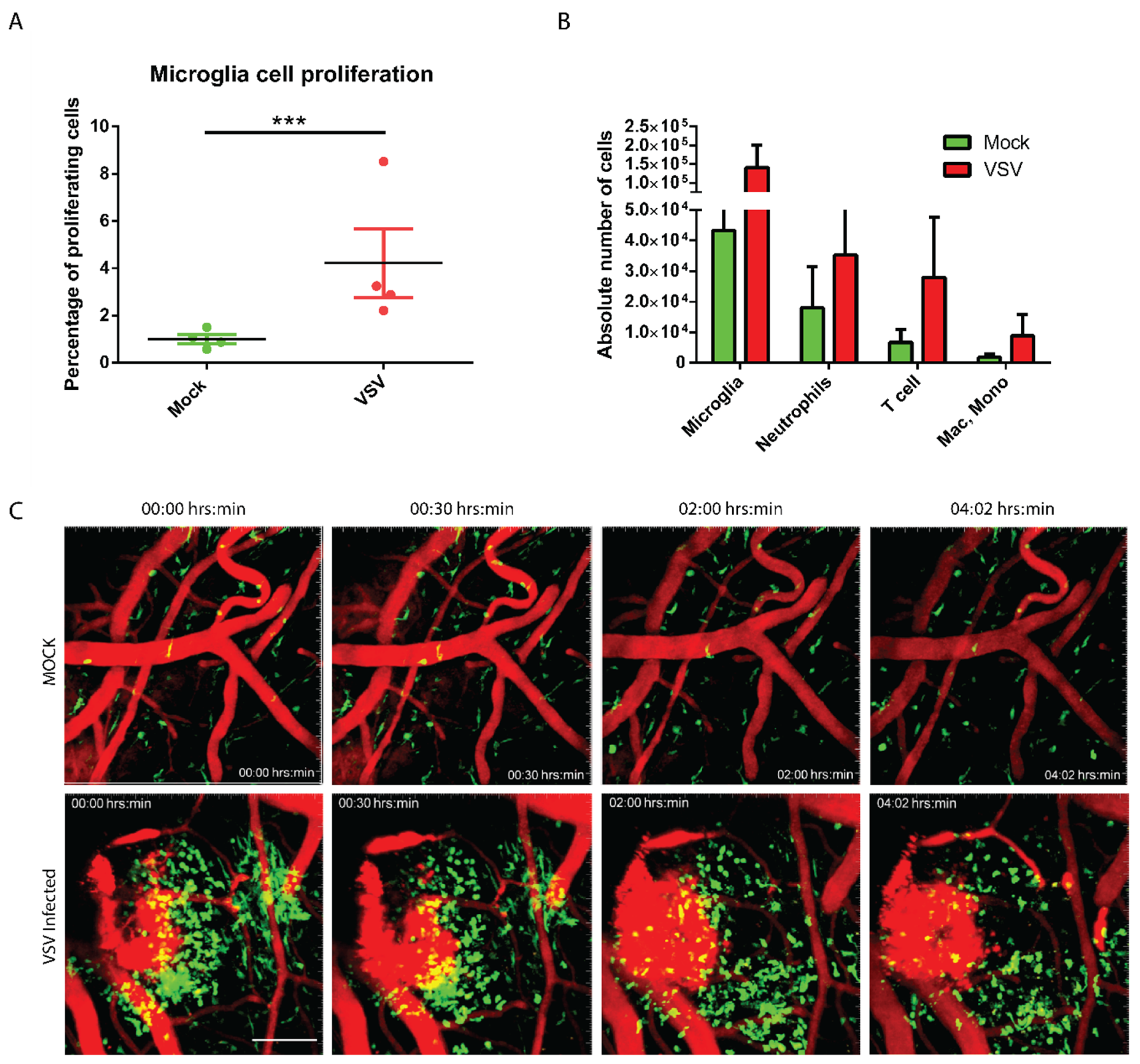
2.6. Intravital Imaging Experiment
2.7. Flow Cytometry
2.8. STRING Database Analysis
2.9. Statistical Analysis
3. Results
3.1. VSV Infection of the CNS Induced Fatality and Brain Viral Load Attained Plateau at Early Stage
3.2. The Virally Infected CNS Showed Microglial Proliferation and Influx of Peripheral Mononuclear Immune Cells
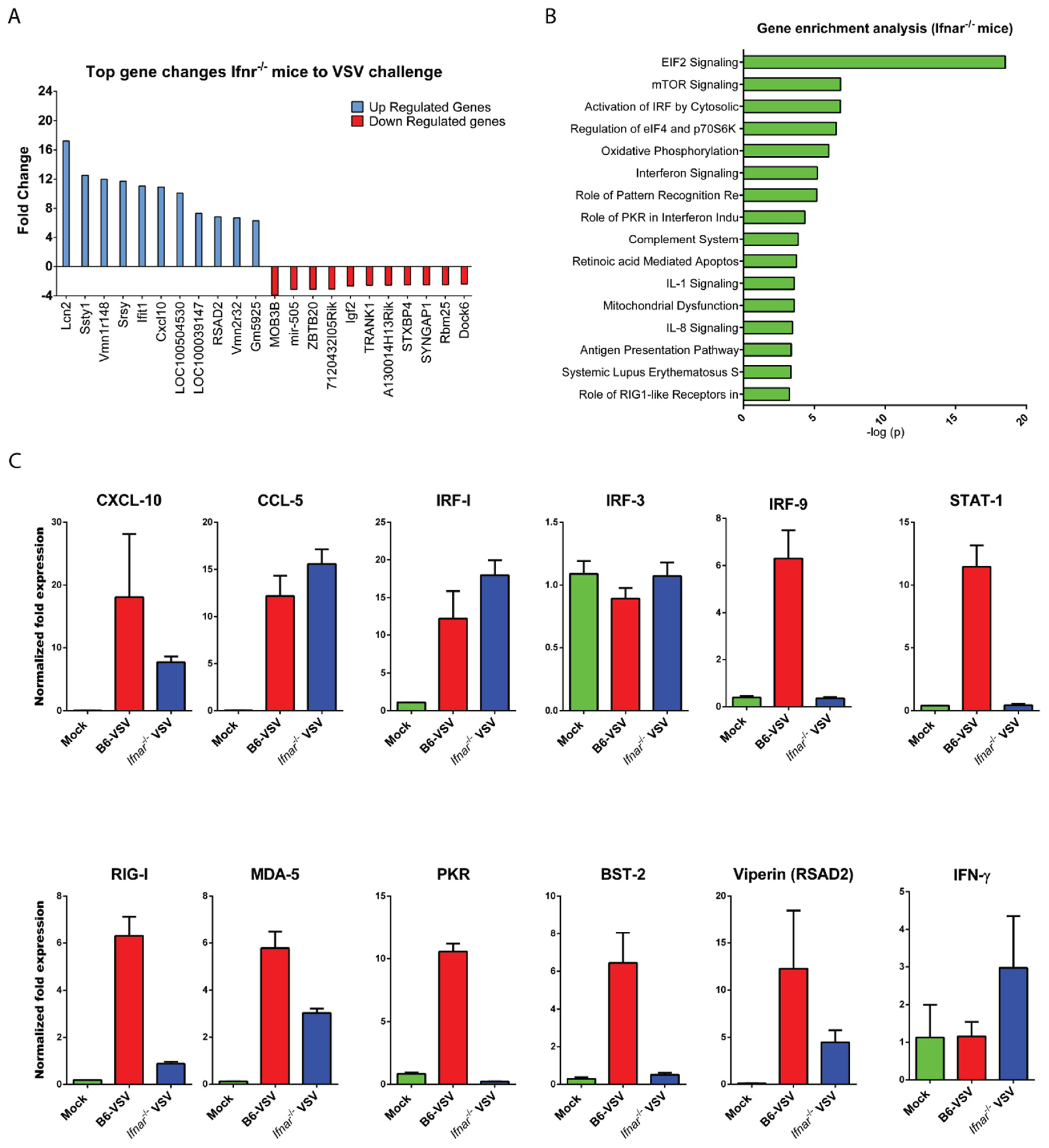
3.3. CNS Gene Expression Profile Reveals the Signature of the IFN-I Signaling Pathway
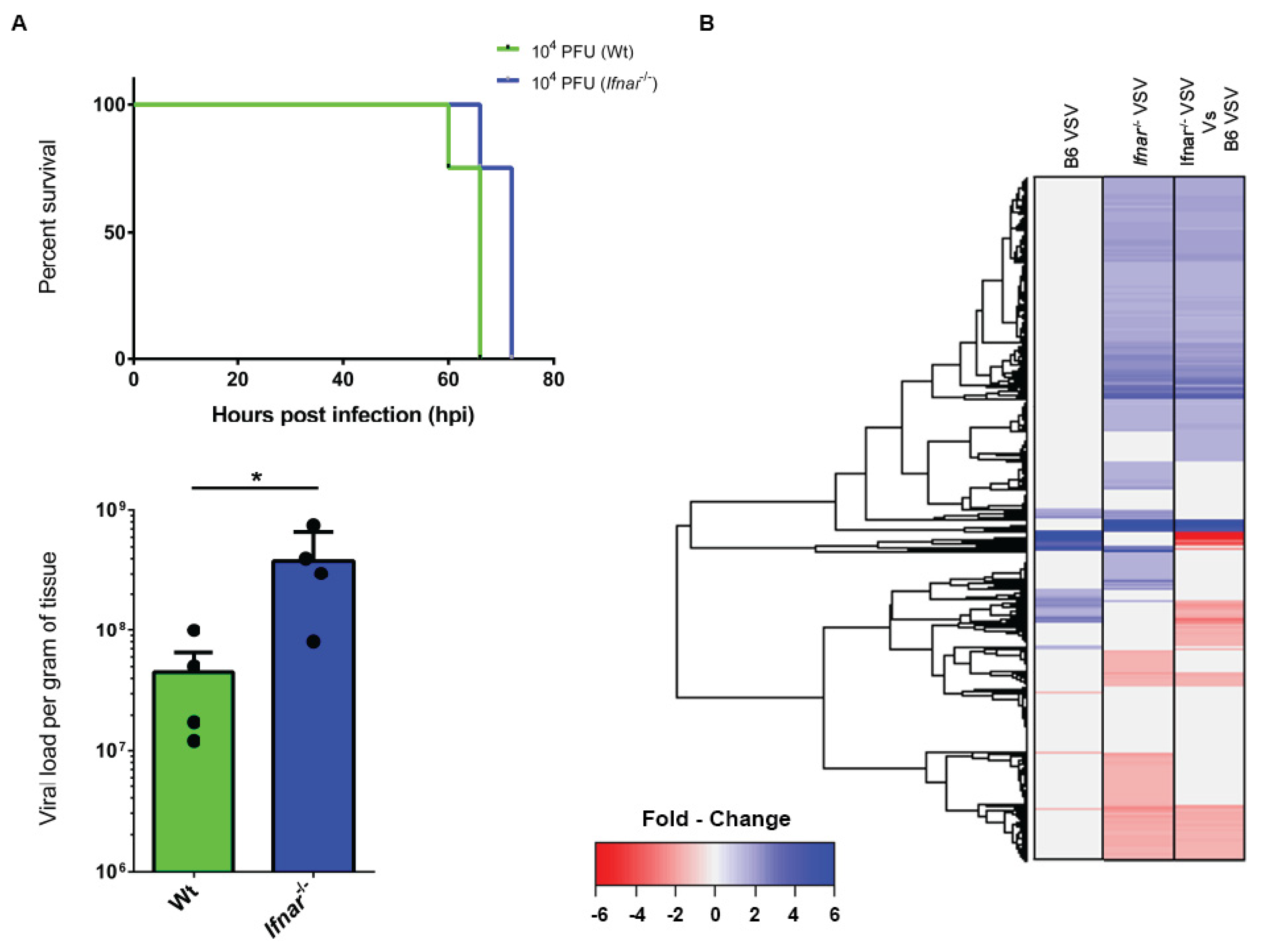
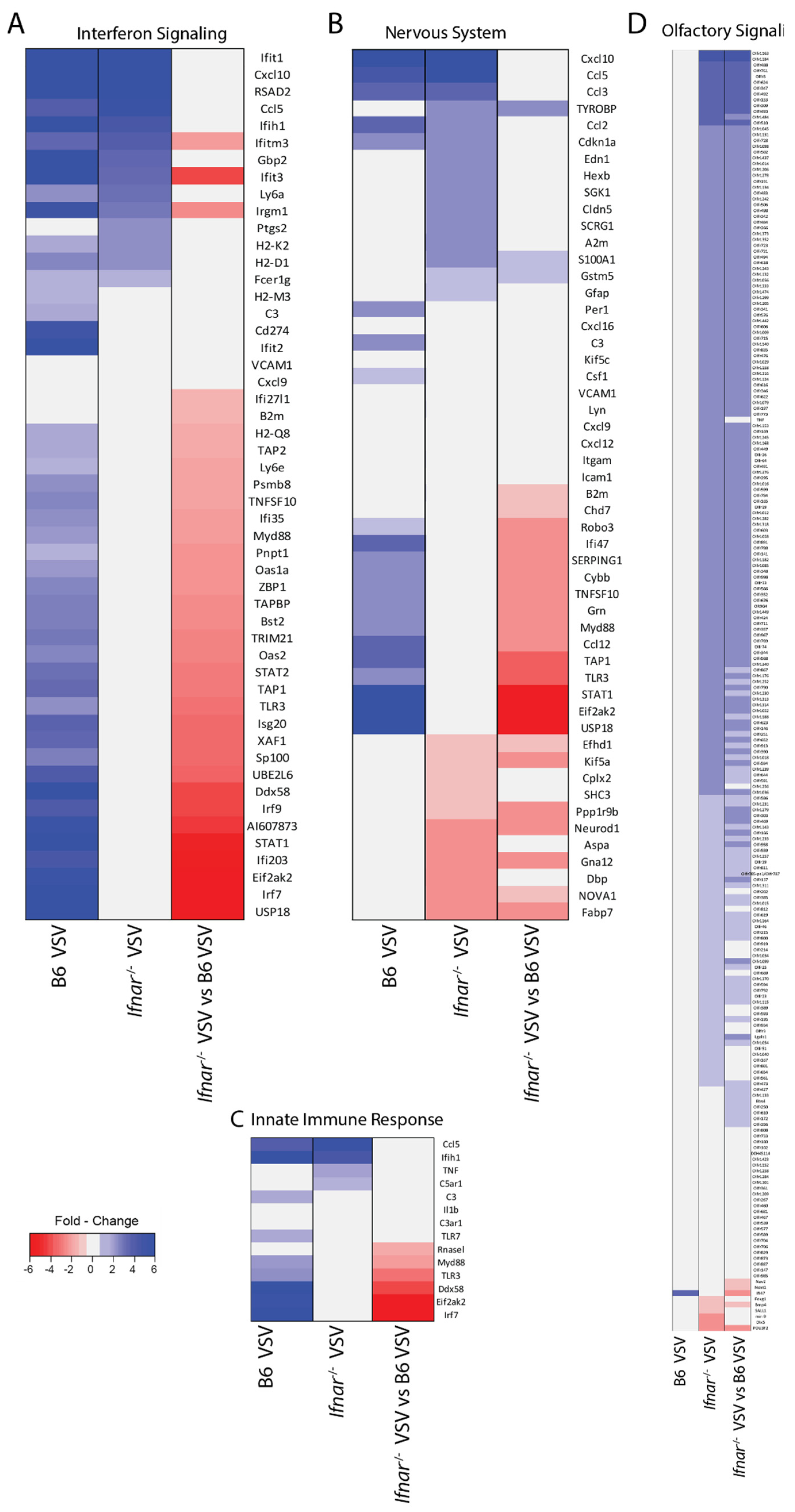
3.4. Loss of IFN-I Signaling Rather Shifted CNS to a Robust Compensatory Mechanism to Mount an Antiviral Response
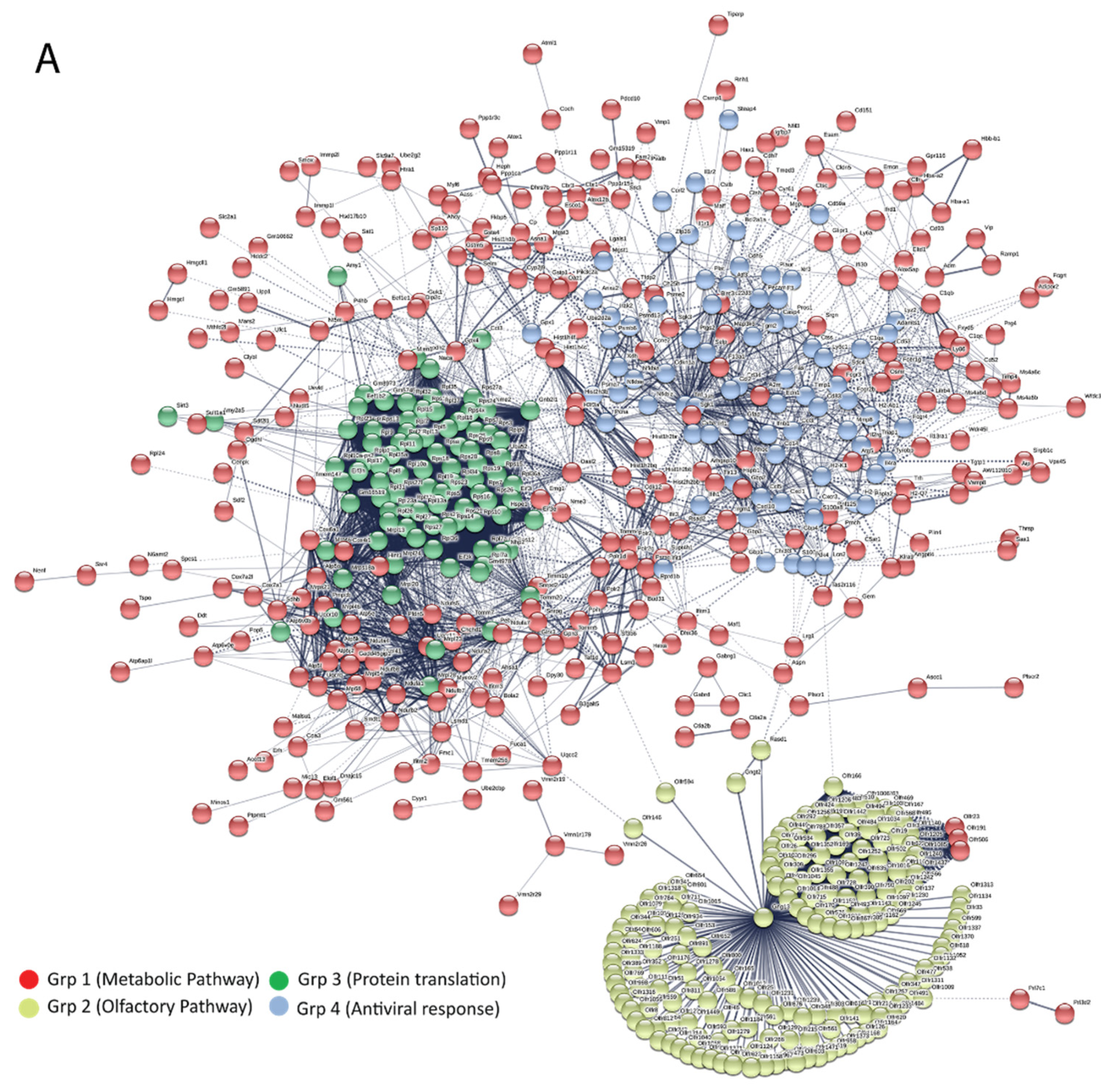
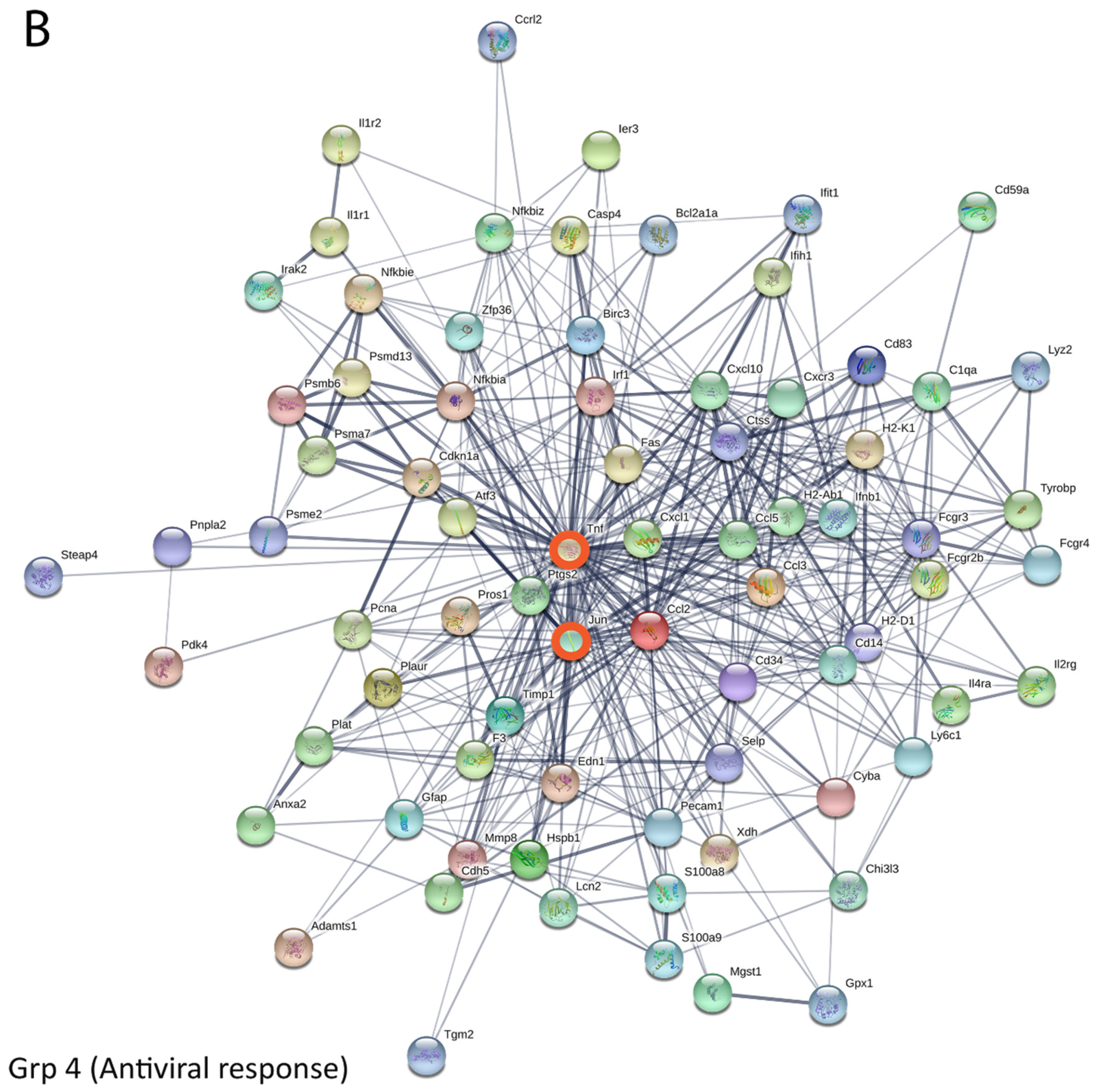
3.5. Bioinformatics and Network Analysis Suggest the Central Role of TNF in the Antiviral Immune Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hašek, M.; Chutna, J.; Sládeček, M.; Lodin, Z. Immunological tolerance and tumour allografts in the brain. Nature 1977, 268, 68–69. [Google Scholar] [CrossRef]
- Medawar, P.B. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar]
- Hickey, W.F.; Kimura, H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 1988, 239, 290–292. [Google Scholar] [CrossRef]
- Nayak, D.; Zinselmeyer, B.H.; Corps, K.N.; McGavern, D.B. In vivo dynamics of innate immune sentinels in the CNS. Intravital 2012, 1, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Bechmann, I.; Galea, I.; Perry, V.H. What is the blood-brain barrier (not)? Trends Immunol. 2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, M.V.; McGavern, D.B. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Chiocca, E.A.; Rabkin, S.D. Oncolytic Viruses and Their Application to Cancer Immunotherapy. Cancer Immunol. Res. 2014, 2, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Swanson, P.A.; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotbart, H.A. Viral Meningitis. Semin. Neurol. 2000, 20, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.T.; Shuey, H.E.; Buescher, E.L. Epidemic central nervous system disease of mixed enterovirus etiology. Am. J. Epidemiol. 1960, 71, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Canfield, M.A.; Reed, C.M.; Flannery, V.L.; Sullivan, K.H.; Reeve, G.R.; Bailey, R.E.; Poland, J.D.; Tsai, T.F. Epidemiological aspects of a st. Louis encephalitis outbreak in harris county, texas, 1986. J. Infect. Dis. 1988, 157, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.C.; Buescher, E.L.; Artenstein, M.S.; Parkman, P.D. Herpesvirus Infections of the Human Central Nervous System. N. Engl. J. Med. 1967, 277, 1271–1277. [Google Scholar] [CrossRef]
- Hollander, H.; Stringari, S. Human immunodeficiency virus-associated meningitis. Am. J. Med. 1987, 83, 813–816. [Google Scholar] [CrossRef]
- Bang, H.O.; Bang, J. Involvement of the Central Nervous System in Mumps. Acta Med. Scand. 1943, 113, 487–505. [Google Scholar] [CrossRef]
- Balfour, H.H., Jr.; Siem, R.A.; Bauer, H.; Quie, P.G. California Arbovirus (La Crosse) Infections. Pediatrics 1973, 52, 680–691. [Google Scholar] [PubMed]
- Srihongse, S.; Grayson, M.A.; Deibel, R. California serogroup viruses in New York State: The role of subtypes in human infections. Am. J. Trop. Med. Hyg. 1984, 33, 1218–1227. [Google Scholar] [CrossRef]
- Goodpasture, H.C.; Poland, J.D.; Francy, D.B.; Bowen, G.S.; Horn, K.A. Colorado tick fever: Clinical, epidemiologic, and laboratory aspects of 228 cases in Colorado in 1973–1974. Ann. Intern. Med. 1978, 88, 303–310. [Google Scholar] [CrossRef]
- Kim, J.V.; Kang, S.S.; Dustin, M.L.; McGavern, D.B. Myelomonocytic cell recruitment causes fatal CNS vascular injury during acute viral meningitis. Nature 2009, 457, 191–195. [Google Scholar] [CrossRef] [Green Version]
- Nayak, D.; Johnson, K.R.; Heydari, S.; Roth, T.L.; Zinselmeyer, B.H.; McGavern, D.B. Type I Interferon Programs Innate Myeloid Dynamics and Gene Expression in the Virally Infected Nervous System. PLoS Pathog. 2013, 9, e1003395. [Google Scholar] [CrossRef] [Green Version]
- Aurelius, E.; Johansson, B.; Sköldenberg, B.; Forsgren, M. Encephalitis in immunocompetent patients due to herpes simplex virus type 1 or 2 as determined by type-specific polymerase chain reaction and antibody assays of cerebrospinal fluid. J. Med. Virol. 1993. [Google Scholar] [CrossRef]
- Schönberger, K.; Ludwig, M.S.; Wildner, M.; Weissbrich, B. Epidemiology of Subacute Sclerosing Panencephalitis (SSPE) in Germany from 2003 to 2009: A Risk Estimation. PLoS ONE 2013, 8, e68909. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Basu, A. Neuropathogenesis by Chandipura virus: An acute encephalitis syndrome in India. Natl. Med. J. India 2017, 30, 21–25. [Google Scholar] [PubMed]
- Manangeeswaran, M.; Lewkowicz, A.P.; Israely, T.; Ireland, D.D.C.; Verthelyi, D. CpG Oligonucleotides Protect Mice From Alphavirus Encephalitis: Role of NK Cells, Interferons, and TNF. Front. Immunol. 2020, 11, 237. [Google Scholar] [CrossRef] [Green Version]
- Baxter, V.K.; Griffin, D.E. Interferon-gamma modulation of the local T cell response to alphavirus encephalomyelitis. Viruses 2020, 12, 113. [Google Scholar] [CrossRef] [Green Version]
- Vidaña, B.; Johnson, N.; Fooks, A.R.; Sánchez-Cordón, P.J.; Hicks, D.J.; Nuñez, A. West Nile Virus spread and differential chemokine response in the central nervous system of mice: Role in pathogenic mechanisms of encephalitis. Transbound. Emerg. Dis. 2020, 67, 799–810. [Google Scholar] [CrossRef]
- Choi, J.Y.; Kim, J.H.; Hossain, F.M.A.; Uyangaa, E.; Park, S.O.; Kim, B.; Kim, K.; Eo, S.K. Indispensable role of CX3CR1+ dendritic cells in regulation of virus-induced neuroinflammation through rapid development of antiviral immunity in peripheral lymphoid tissues. Front. Immunol. 2019, 10, 1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashida, E.; Ling, Z.L.; Ashhurst, T.M.; Viengkhou, B.; Jung, S.R.; Songkhunawej, P.; West, P.K.; King, N.J.C.; Hofer, M.J. Zika virus encephalitis in immunocompetent mice is dominated by innate immune cells and does not require T or B cells. J. Neuroinflamm. 2019, 16, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Faust, N.; Varas, F.; Kelly, L.M.; Heck, S.; Graf, T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood 2000, 96, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pan, F.; Parkhurst, C.N.; Grutzendler, J.; Gan, W.-B. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat. Protoc. 2010, 5, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, M.; Wang, J.; Pan, Y.; Wu, F.X. CytoNCA: A cytoscape plugin for centrality analysis and evaluation of protein interaction networks. BioSystems 2015, 127, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, P.; Bao, Y.; Han, Y.; Wang, Y.; Zhang, Q.; Zhan, Z.; Meng, J.; Li, Y.; Li, N.; et al. Zinc finger protein ZBTB20 promotes toll-like receptor-triggered innate immune responses by repressing IκBα gene transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 11097–11102. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.J.; Chen, F.; Liu, Q.G.; Liu, C.C.; Yao, H.; Yu, B.; Zhang, H.-B.; Yan, H.X.; Ye, Y.; Chen, T.; et al. Insulin-like growth factor 2 is a key mitogen driving liver repopulation in mice. Cell Death Dis. 2018. [Google Scholar] [CrossRef] [Green Version]
- Detje, C.N.; Meyer, T.; Schmidt, H.; Kreuz, D.; Rose, J.K.; Bechmann, I.; Prinz, M.; Kalinke, U. Local Type I IFN Receptor Signaling Protects against Virus Spread within the Central Nervous System. J. Immunol. 2009, 182, 2297–2304. [Google Scholar] [CrossRef] [Green Version]
- van den Pol, A.N.; Ding, S.; Robek, M.D. Long-Distance Interferon Signaling within the Brain Blocks Virus Spread. J. Virol. 2014, 88, 3695–3704. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Patro, I.K.; Amit; Shrivastava, M.; Bhumika, S.; Patro, N. Poly I: C induced microglial activation impairs motor activity in adult rats. Indian J. Exp. Biol. 2010, 48, 104–109. [Google Scholar]
- Asensio, V.C.; Maier, J.; Milner, R.; Boztug, K.; Kincaid, C.; Moulard, M.; Phillipson, C.; Lindsley, K.; Krucker, T.; Fox, H.S.; et al. Interferon-Independent, Human Immunodeficiency Virus Type 1 gp120-Mediated Induction of CXCL10/IP-10 Gene Expression by Astrocytes In Vivo and In Vitro. J. Virol. 2001, 75, 7067–7077. [Google Scholar] [CrossRef] [Green Version]
- Brownell, J.; Bruckner, J.; Wagoner, J.; Thomas, E.; Loo, Y.-M.; Gale, M.; Liang, T.J.; Polyak, S.J. Direct, Interferon-Independent Activation of the CXCL10 Promoter by NF- B and Interferon Regulatory Factor 3 during Hepatitis C Virus Infection. J. Virol. 2014, 88, 1582–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C.A. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Baccala, R.; Hoebe, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 2007, 13, 543–551. [Google Scholar] [CrossRef]
- Yount, J.S.; Moran, T.M.; López, C.B. Cytokine-Independent Upregulation of MDA5 in Viral Infection. J. Virol. 2007, 81, 7316–7319. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Qifeng, X.; Tohyama, M.; Qureshi, M.H.; Saito, A. Contribution of tumour necrosis factor-alpha (TNF-alpha) in host defence mechanism against Cryptococcus neoformans. Clin. Exp. Immunol. 1996, 106, 468–474. [Google Scholar] [CrossRef]
- Huffnagle, G.B.; Toews, G.B.; Burdick, M.D.; Boyd, M.B.; McAllister, K.S.; McDonald, R.A.; Strieter, R.M.; Kunkel, S.L. Afferent Phase Production of TNF-α Is Required for the Development of Protective T Cell Immunity to Cryptococcus neoformans. J. Immunol. 1996, 157, 4529–4536. [Google Scholar]
- Flynn, J.A.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schrelber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Mestan, J.; Brockhaus, M.; Kirchner, H.; Jacobsen, H. Antiviral activity of tumour necrosis factor. Synergism with interferons and induction of oligo-2′,5′-adenylate synthetase. J. Gen. Virol. 1988, 69, 3113–3120. [Google Scholar] [CrossRef]
- Wada, H.; Saito, K.; Kanda, T.; Kobayashi, I.; Fujii, H.; Fujigaki, S.; Maekawa, N.; Takatsu, H.; Fujiwara, H.; Sekikawa, K.; et al. Tumor Necrosis Factor-α (TNF-α) Plays a Protective Role in Acute Viral Myocarditis in Mice. Circulation 2001, 103, 743–749. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Welander, P.V.; Edwards, C.K.; van Rooijen, N.; Cantin, E. Tumor Necrosis Factor (TNF) Protects Resistant C57BL/6 Mice against Herpes Simplex Virus-Induced Encephalitis Independently of Signaling via TNF Receptor 1 or 2. J. Virol. 2007, 81, 1451–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.H.; Webster, R.G. Tumor necrosis factor alpha exerts powerful anti-influenza virus effects in lung epithelial cells. J. Virol. 2002, 76, 1071–1076. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, H.-T.; Tsai, H.-F.; Chyuan, I.-T.; Liao, H.-J.; Chen, C.-J.; Chen, P.-J.; Hsu, P.-N. Tumor Necrosis Factor-Alpha Induced by Hepatitis B Virus Core Mediating the Immune Response for Hepatitis B Viral Clearance in Mice Model. PLoS ONE 2014, 9, e103008. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, N.F.; Sutaria, R.; Jo, J.; Barnes, A.; Blahova, M.; Meredith, L.W.; Cosset, F.L.; Curbishley, S.M.; Adams, D.H.; Bertoletti, A.; et al. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology 2014, 59, 1320–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis C virus infection enhances TNFα-induced cell death via suppression of NF-κB. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway Description | Observed Gene Count | False Discovery Rate | Group |
|---|---|---|---|---|
| 190 | Oxidative phosphorylation | 21 | 6.45 × 10−11 | Group 1 |
| 5012 | Parkinson’s disease | 19 | 1.06 × 10−8 | |
| 5016 | Huntington’s disease | 20 | 5.89 × 10−8 | |
| 5010 | Alzheimer’s disease | 19 | 8.50 × 10−8 | |
| 4932 | Non-alcoholic fatty liver disease (NAFLD) | 17 | 6.37 × 10−7 | |
| 1100 | Metabolic pathways | 42 | 3.44 × 10−2 | |
| 4260 | Cardiac muscle contraction | 7 | 3.58 × 10−2 | |
| 4740 | Olfactory transduction | 175 | 1.55 × 10−229 | Group 2 |
| 3010 | Ribosome | 39 | 4.78 × 10−64 | Group 3 |
| 4668 | TNF signaling pathway | 11 | 2.14 × 10−11 | Group 4 |
| 5168 | Herpes simplex infection | 12 | 3.38 × 10−10 | |
| 4380 | Osteoclast differentiation | 10 | 9.39 × 10−10 | |
| 4060 | Cytokine–cytokine receptor interaction | 12 | 2.32 × 10−9 | |
| 5140 | Leishmaniasis | 8 | 2.36 × 10−9 | |
| 5142 | Chagas disease (American trypanosomiasis) | 9 | 2.77 × 10−9 | |
| 5164 | Influenza A | 10 | 9.71 × 10−9 | |
| 5166 | HTLV-I infection | 11 | 4.71 × 10−8 | |
| 4620 | Toll-like receptor signaling pathway | 8 | 5.01 × 10−8 | |
| 4145 | Phagosome | 9 | 9.90 × 10−8 | |
| 5152 | Tuberculosis | 9 | 1.45 × 10−7 | |
| 4640 | Hematopoietic cell lineage | 7 | 2.64 × 10−7 | |
| 5150 | Staphylococcus aureus infection | 6 | 3.59 × 10−7 | |
| 4064 | NF-kappa B signaling pathway | 7 | 4.36 × 10−7 | |
| 4621 | NOD-like receptor signaling pathway | 6 | 6.97 × 10−7 | |
| 4650 | Natural killer cell-mediated cytotoxicity | 7 | 1.85 × 10−6 | |
| 4612 | Antigen processinu67t7uygtb hn nng and presentation | 6 | 2.54 × 10−6 | |
| 4610 | Complement and coagulation cascades | 6 | 3.33 × 10−6 | |
| 4210 | Apoptosis | 6 | 4.39 × 10−6 |
| Gene Name | Degree Centrality (Weight) |
|---|---|
| Tnf | 50.06092 |
| Ccl2 | 25.065063 |
| Jun | 24.664934 |
| Ccl5 | 18.67208 |
| Cxcl10 | 17.798105 |
| Ptgs2 | 15.644774 |
| Nfkbia | 14.423865 |
| Cd14 | 13.918694 |
| Irf1 | 13.681601 |
| Fcgr3 | 13.137255 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, A.R.; Byrareddy, S.N.; Nayak, D. IFN-I Independent Antiviral Immune Response to Vesicular Stomatitis Virus Challenge in Mouse Brain. Vaccines 2020, 8, 326. https://doi.org/10.3390/vaccines8020326
Mishra AR, Byrareddy SN, Nayak D. IFN-I Independent Antiviral Immune Response to Vesicular Stomatitis Virus Challenge in Mouse Brain. Vaccines. 2020; 8(2):326. https://doi.org/10.3390/vaccines8020326
Chicago/Turabian StyleMishra, Anurag R., Siddappa N. Byrareddy, and Debasis Nayak. 2020. "IFN-I Independent Antiviral Immune Response to Vesicular Stomatitis Virus Challenge in Mouse Brain" Vaccines 8, no. 2: 326. https://doi.org/10.3390/vaccines8020326
APA StyleMishra, A. R., Byrareddy, S. N., & Nayak, D. (2020). IFN-I Independent Antiviral Immune Response to Vesicular Stomatitis Virus Challenge in Mouse Brain. Vaccines, 8(2), 326. https://doi.org/10.3390/vaccines8020326