Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
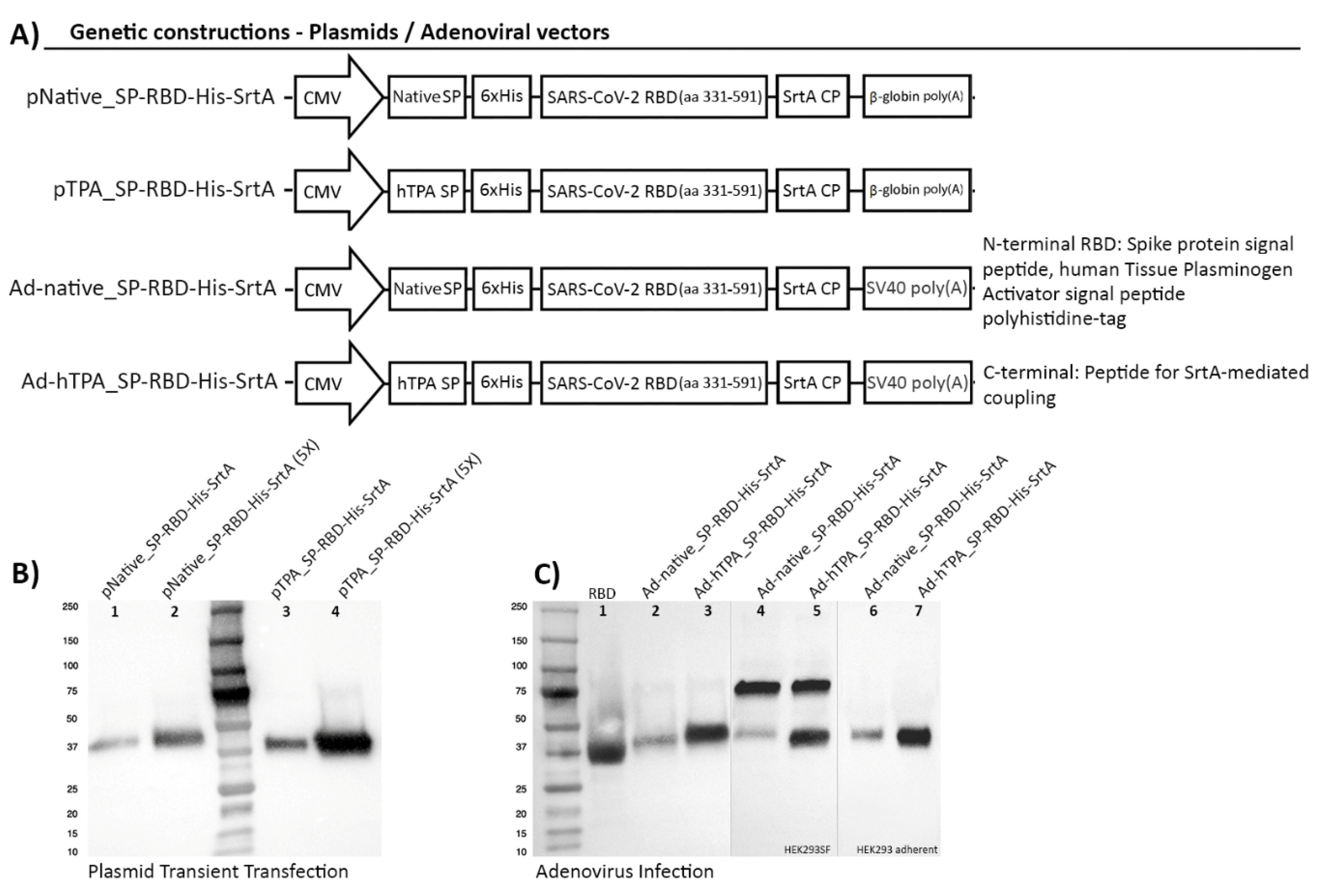
2.1. Plasmids
2.2. Adenovirus Constructs and Seed-Stock Generation
2.3. Cells Lines and Culture Media
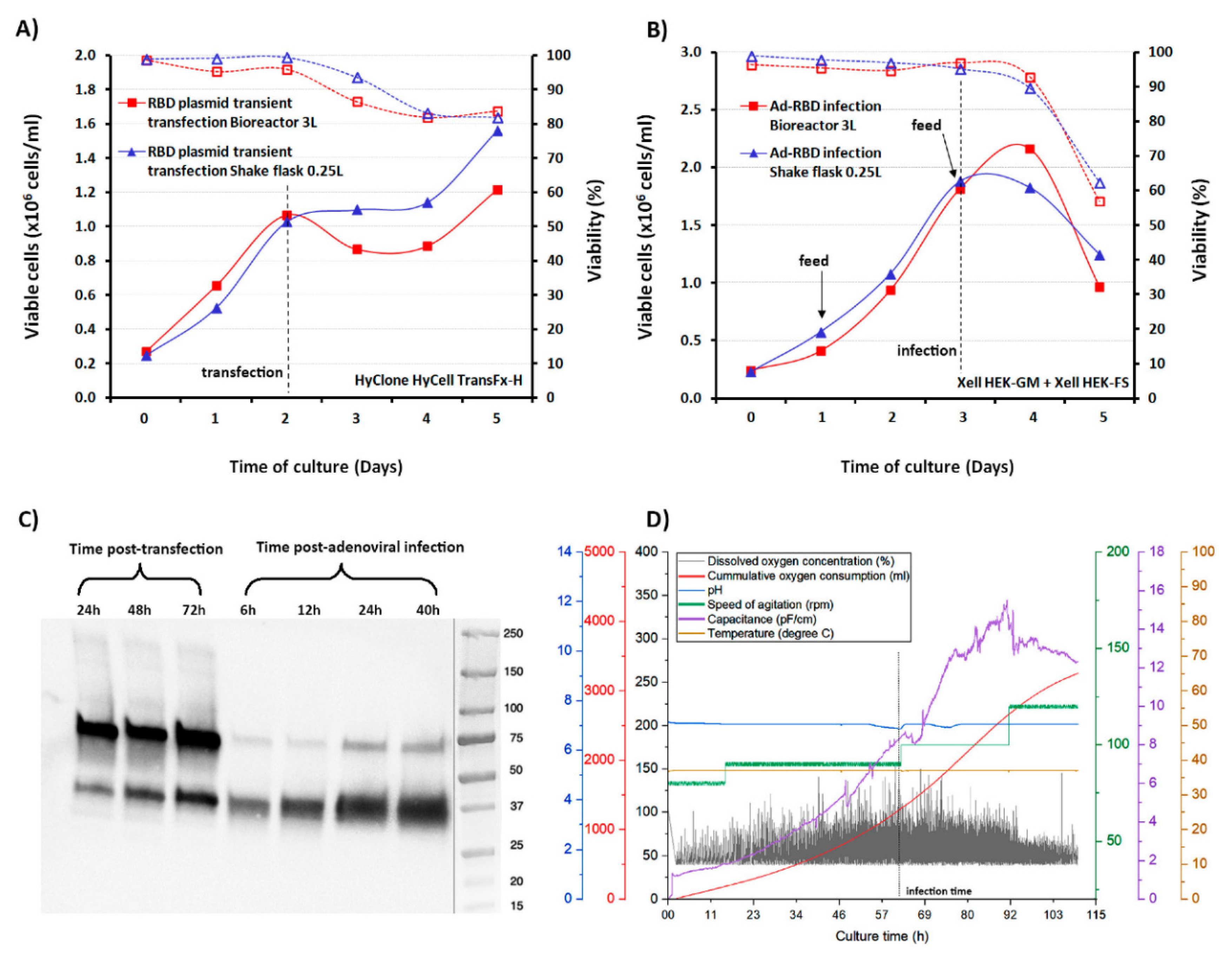
2.4. Production of the Receptor Binding Domain in Shake-Flasks Cultivation Experiments
2.5. Production of the Receptor Binding Domain in 3L-Controlled Bioreactors
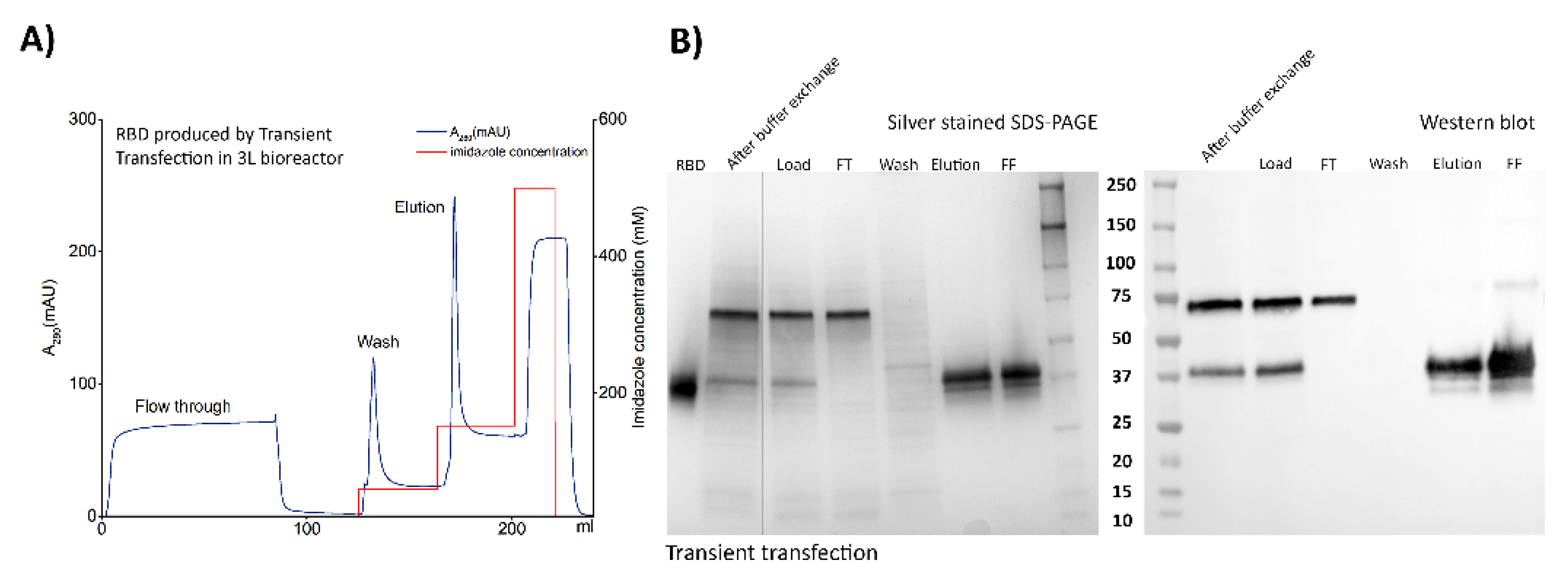
2.6. Downstream Processing
2.6.1. Cells Harvest, Culture Supernatant Clarification and Medium Exchange
2.6.2. Immobilized Metal ion Affinity Chromatography
2.7. Analytical Assays for Characterization of the Recombinant Antigens
2.7.1. Protein Quantification
2.7.2. SDS-PAGE and Western Blot Analyses
2.7.3. Protein Quantification by Enzyme Linked Immunosorbent Assay (ELISA)
2.8. RBD Analysis of Oligosaccharides
2.8.1. Deglycosylation with PNGase-F
2.8.2. Chemical and Enzymatic Hydrolysis
2.8.3. High-Performance Anion-Exchange Chromatography with Pulsed Amperometric Detection (HPAEC-PAD)
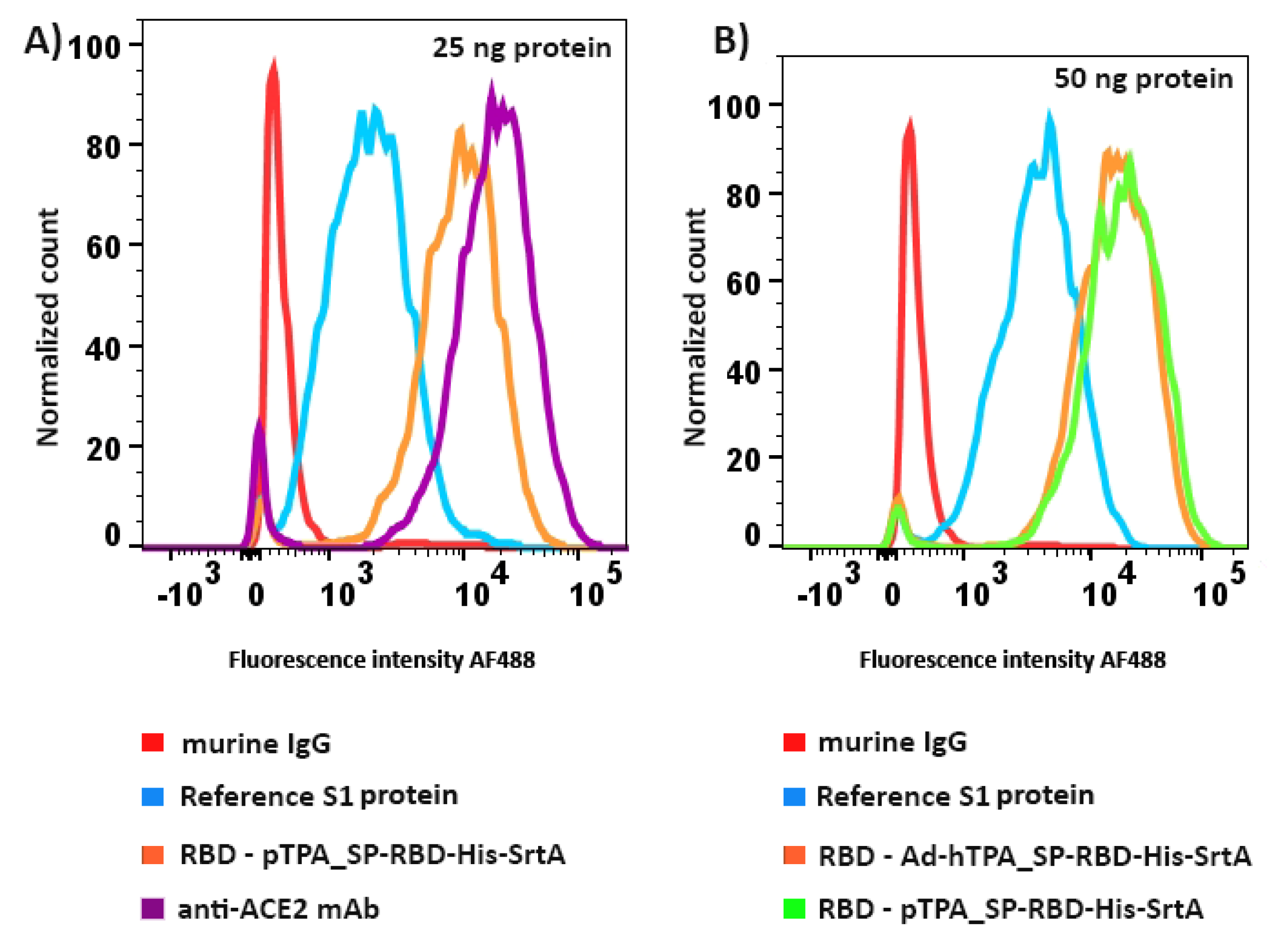
2.9. RBD Binding to Angiotensin-Converting Enzyme 2
3. Results and Discussion
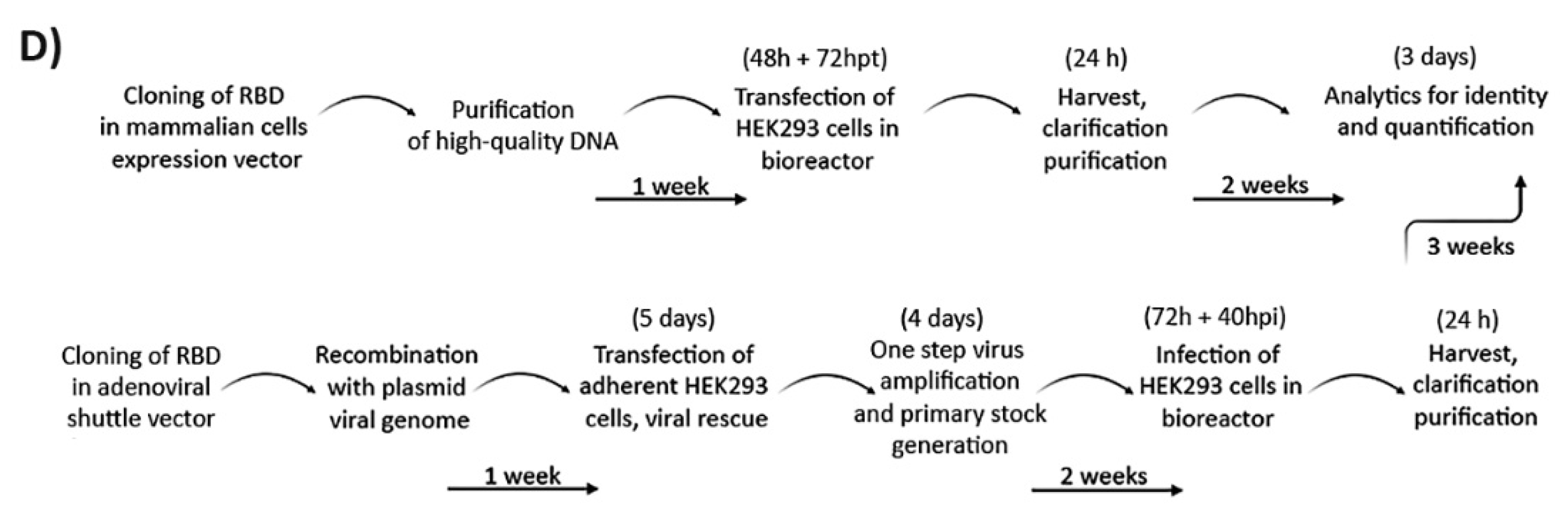
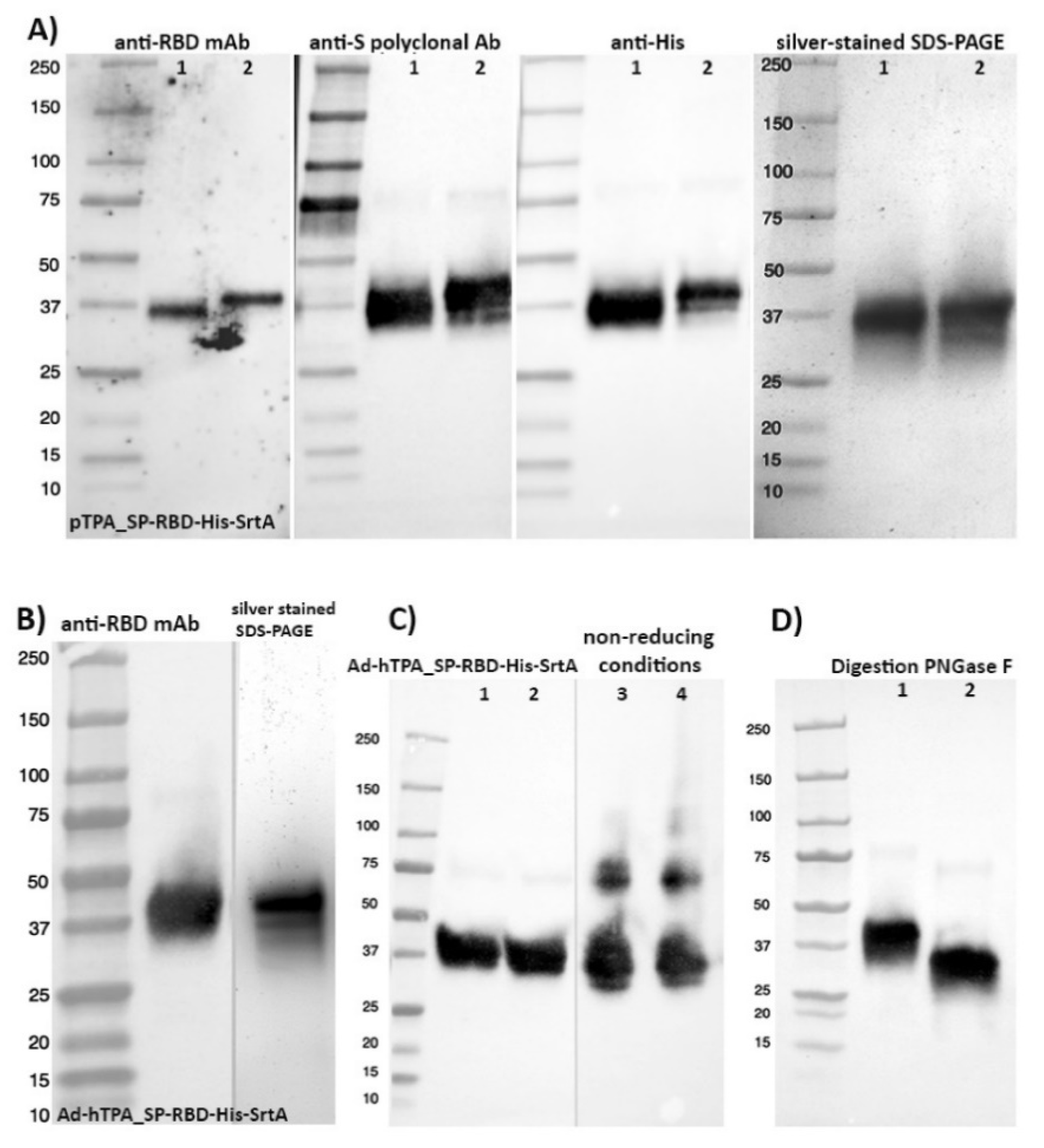
3.1. Design Strategies for Accelerated Expression of SARS-CoV-2 RBD in HEK293SF Suspension Cells
3.2. Scale-Up for Mass Production in Bioreactors
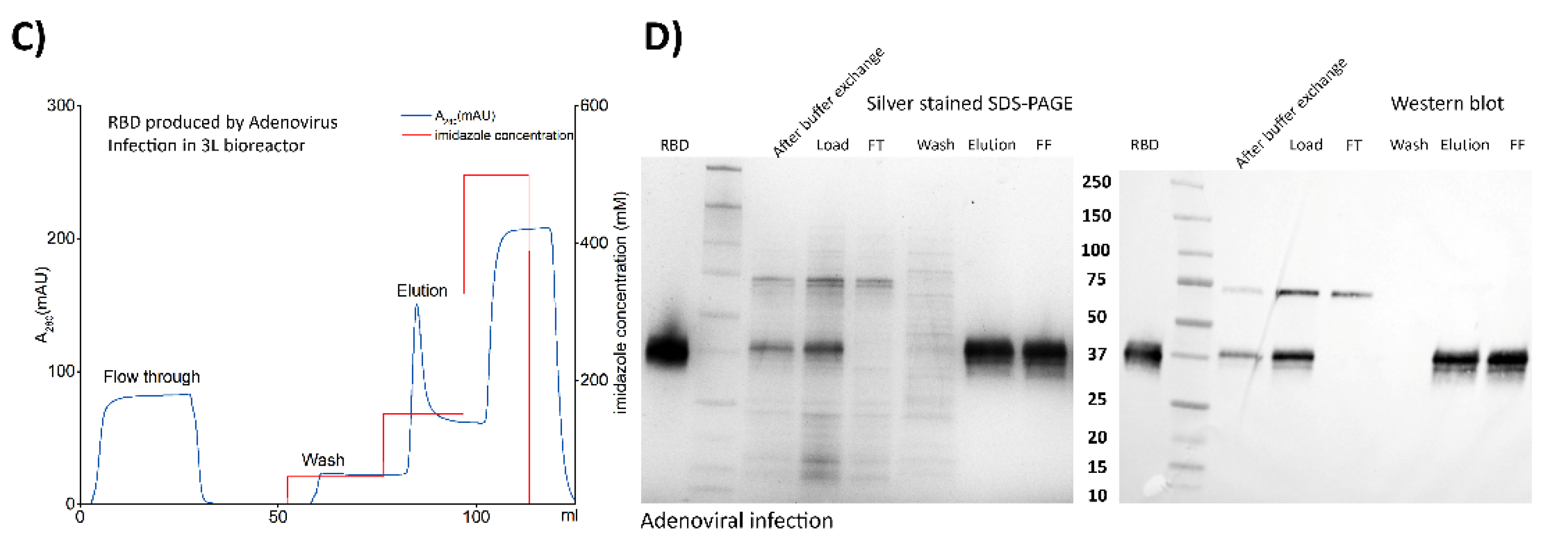
3.3. Downstream Processing and Characterization of the Antigens
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ashour, H.M.; Elkhatib, W.F.; Rahman, M.M.; Elshabrawy, H.A. Insights into the Recent 2019 Novel Coronavirus (SARS-CoV-2) in Light of Past Human Coronavirus Outbreaks. Pathogens 2020, 9, 186. [Google Scholar] [CrossRef] [Green Version]
- Coronavirus Disease (COVID-19) Weekly Epidemiological Update and Weekly Operational Update. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 25 September 2020).
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Butler, D. Billion-dollar project aims to prep vaccines before epidemics hit. Nature 2017, 541, 444–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamen, A.A.; Lua, L.H.L.; Mukhopadhyay, T.K. Vaccine Technology VII: Beyond the “decade of vaccines”. Vaccine 2019, 37, 6931–6932. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.; Kawaoka, Y. Predicting the Next Influenza Pandemics. J. Infect. Dis. 2019, 219, S14–S20. [Google Scholar] [CrossRef]
- Jeyanathan, M.; Afkhami, S.; Smaill, F.; Miller, M.S.; Lichty, B.D.; Xing, Z. Immunological considerations for COVID-19 vaccine strategies. Nat. Rev. Immunol. 2020. [Google Scholar] [CrossRef]
- DRAFT Landscape of COVID-19 Candidate Vaccines–15 October 2020. Available online: https://www.who.int/docs/default-source/coronaviruse/novel-coronavirus-landscape-covid-19cf1952c105464714aaaf8c7cd5c5cc8b.pdf?sfvrsn=d6073093_7&download=true (accessed on 31 October 2020).
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, A.M.; Khaleeq, A.; Syeda, H. Elucidation of cellular targets and exploitation of the receptor-binding domain of SARS-CoV-2 for vaccine and monoclonal antibody synthesis. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Baruah, V.; Bose, S. Immunoinformatics-aided identification of T cell and B cell epitopes in the surface glycoprotein of 2019-nCoV. J. Med. Virol. 2020, 92, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Mai, J.; Zhou, W.; Yu, W.; Zhan, Y.; Wang, N.; Epstein, N.D.; Yang, Y. Immunoinformatic Analysis of T- and B-Cell Epitopes for SARS-CoV-2 Vaccine Design. Vaccines 2020, 8, 355. [Google Scholar] [CrossRef]
- Venereo-Sanchez, A.; Gilbert, R.; Simoneau, M.; Caron, A.; Chahal, P.; Chen, W.; Ansorge, S.; Li, X.; Henry, O.; Kamen, A. Hemagglutinin and neuraminidase containing virus-like particles produced in HEK-293 suspension culture: An effective influenza vaccine candidate. Vaccine 2016, 34, 3371–3380. [Google Scholar] [CrossRef] [Green Version]
- Proft, T. Sortase-mediated protein ligation: An emerging biotechnology tool for protein modification and immobilisation. Biotechnol. Lett. 2010, 32, 1–10. [Google Scholar] [CrossRef]
- Farnos, O.; Gelaye, E.; Trabelsi, K.; Bernier, A.; Subramani, K.; Kallel, H.; Yami, M.; Kamen, A.A. Establishing a Robust Manufacturing Platform for Recombinant Veterinary Vaccines: An Adenovirus-Vector Vaccine to Control Newcastle Disease Virus Infections of Poultry in Sub-Saharan Africa. Vaccines 2020, 8, 338. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs. Arch. Exp. Pathol. Pharm. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Cote, J.; Garnier, A.; Massie, B.; Kamen, A. Serum-free production of recombinant proteins and adenoviral vectors by 293SF-3F6 cells. Biotechnol. Bioeng. 1998, 59, 567–575. [Google Scholar]
- Dixon, B.; Department of Biology, University of Waterloo, Ontario, Canada; Rodríguez-Ramos, T.; Department of Biology, University of Waterloo, Ontario, Canada. Personal Communication, 2020.
- Direct Determination of Sialic Acids in Glycoprotein Hydrolyzates by HPAE-PAD. Available online: https://assets.thermofisher.com/TFS-Assets/CMD/Application-Notes/AU-180-IC-Sialic-Acids-Glycoprotein-Hydrolyzates-AU71730-EN.pdf (accessed on 22 September 2020).
- Correa Giron, C.; Laaksonen, A.; Barroso da Silva, F.L. On the interactions of the receptor-binding domain of SARS-CoV-1 and SARS-CoV-2 spike proteins with monoclonal antibodies and the receptor ACE2. Virus Res. 2020, 285, 198021. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell. Mol. Immunol. 2020, 17, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Zhao, G.; Chan, C.C.; Li, L.; He, Y.; Zhou, Y.; Zheng, B.J.; Jiang, S. A 219-mer CHO-expressing receptor-binding domain of SARS-CoV S protein induces potent immune responses and protective immunity. Viral Immunol. 2010, 23, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, E.; Toledo, J.R.; Chiong, M.; Parra, F.; Rodriguez, E.; Montero, C.; Mendez, L.; Capucci, L.; Farnos, O. Single dose adenovirus vectored vaccine induces a potent and long-lasting immune response against rabbit hemorrhagic disease virus after parenteral or mucosal administration. Vet. Immunol. Immunopathol. 2011, 142, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Laliberte-Gagne, M.E.; Bolduc, M.; Therien, A.; Garneau, C.; Casault, P.; Savard, P.; Estaquier, J.; Leclerc, D. Increased Immunogenicity of Full-Length Protein Antigens through Sortase-Mediated Coupling on the PapMV Vaccine Platform. Vaccines 2019, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Bolduc, M.; Baz, M.; Laliberte-Gagne, M.E.; Carignan, D.; Garneau, C.; Russel, A.; Boivin, G.; Savard, P.; Leclerc, D. The quest for a nanoparticle-based vaccine inducing broad protection to influenza viruses. Nanomedicine 2018, 14, 2563–2574. [Google Scholar] [CrossRef]
- Therien, A.; Bedard, M.; Carignan, D.; Rioux, G.; Gauthier-Landry, L.; Laliberte-Gagne, M.E.; Bolduc, M.; Savard, P.; Leclerc, D. A versatile papaya mosaic virus (PapMV) vaccine platform based on sortase-mediated antigen coupling. J. Nanobiotechnol. 2017, 15, 54. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef]
- Iwasaki, A.; Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 2020, 20, 339–341. [Google Scholar] [CrossRef] [Green Version]
- Venereo-Sanchez, A.; Fulton, K.; Koczka, K.; Twine, S.; Chahal, P.; Ansorge, S.; Gilbert, R.; Henry, O.; Kamen, A. Characterization of influenza H1N1 Gag virus-like particles and extracellular vesicles co-produced in HEK-293SF. Vaccine 2019, 37, 7100–7107. [Google Scholar] [CrossRef]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef] [PubMed]
- Schachter, H. The joys of HexNAc. The synthesis and function of N- and O-glycan branches. Glycoconj. J. 2000, 17, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef]
- Watanabe, Y.; Bowden, T.A.; Wilson, I.A.; Crispin, M. Exploitation of glycosylation in enveloped virus pathobiology. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1480–1497. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.S.; Azadi, P. Deducing the N- and O- glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. Glycobiology 2020. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, W.; Mao, Y.; Chen, Y.; Wang, S.; Zhong, Y.; Su, T.; Gong, M.; Du, D.; Lu, X.; et al. Site-specific N-glycosylation Characterization of Recombinant SARS-CoV-2 Spike Proteins. Mol. Cell. Proteom. 2020. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific analysis of the SARS-CoV-2 glycan shield. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Adamo, R.; Sonnino, S. Impact of glycoscience in fighting Covid-19. Glycoconj. J. 2020, 37, 511–512. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive. Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Culture Conditions * | Cell Density at Transfection/Infection | Sampling Time | RBD—ELISA (µg/mL) |
|---|---|---|---|---|
| Culture supernatant from adenovirally infected HEK293SF cells | Shake flasks, HEK-GM + HEK-FS | 2 × 106 cells/mL | 40 hpi | 115.3 ± 6.2 |
| 1 × 106 cells/mL | 93.4 ± 17.6 | |||
| Shake flasks, HyCell-TransFx-H + Cell Boost 5 | 2 × 106 cells/mL | 40 hpi | 82.2 ± 3.0 | |
| 1 × 106 cells/mL | 62.6 ± 7.3 | |||
| Culture supernatant from HEK293SF cells adenovirally infected at high cell density | Shake flasks, HEK-GM + HEK-FS | 5 × 106 cells/mL | 40 hpi | 67.5 ± 34.5 |
| Shake flasks, HyCell-TransFx-H + Cell Boost 5 | 5 × 106 cells/mL | 28.4 ± 4.5 | ||
| Culture supernatant from transfected HEK293SF cells | Shake flasks HyCell-TransFx-H | 1 × 106 cells/mL | 24 hpt | 1.5 ± 0.23 |
| 48 hpt | 5.8 ± 0.7 | |||
| 72 hpt | 17.7 ± 5.1 | |||
| Culture supernatant from adenovirally infected HEK293SF cells | Shake flasks Xell HEK-GM + HEK-FS | 2 × 106 cells/mL | 6 hpi | 15.3 ± 1.1 |
| 24 hpi | 62.2 ± 19.3 | |||
| 40 hpi | 131.7 ± 27.2 |
| Sample | Culture Conditions | Cell Density at Transfection/Infection | Sampling Time | RBD—ELISA (µg/mL) |
|---|---|---|---|---|
| Bioreactor harvest broth from transfected HEK293SF cells | 3L-bioreactor operated in batch mode (HyCell-TransFx-H medium) | 1 × 106 cells/mL | 72 hpt | 17.8 |
| Bioreactor harvest broth from adenovirally infected HEK293SF cells | 3L-bioreactor operated in fed-batch mode (Xell HEK-GM medium + HEK-FS supplement) | 2 × 106 cells/mL (MOI = 5) | 40 hpi | 101.7 |
| RBD by Plasmid Transient Transfection 3L Bioreactor | Total Protein Concentration (µg/mL) | RBD—ELISA (µg/mL) | Volume (mL) | Purification Recovery (%) |
|---|---|---|---|---|
| Ni-NTA Load after harvest broth centrifugation, 0.2 µm filtration, medium exchange using 10 kDa cut-off | 345.4 | 34.4 | 118 | 100 |
| Ni-NTA Column flow-through | 231 | below L.O.D *. | 118 | - |
| Wash | 7 | below L.O.D. | 10 | - |
| Elution | 152 | 158.8 | 15 | 58.6 |
| RBD by Adenovirus Infection 3L Bioreactor | Total Protein Concentration (µg/mL) | RBD—ELISA (µg/mL) | Volume (ml) | Purification Recovery (%) |
|---|---|---|---|---|
| Ni-NTA Load after harvest broth centrifugation, ultrafiltration 300 kDa, medium exchange 10 kDa | 392.5 | 112.5 | 26 | 100 |
| Ni-NTA Column flow-through | 212.8 | 8.1 | 26 | 7.2 |
| Wash | 8.4 | below L.O.D *. | 10 | - |
| Elution | 129.8 | 104.4 | 15 | 53.5 |
| Monosaccharide | RBD by Adenoviral Infection | RBD by Transient Transfection |
|---|---|---|
| Fuc | 1.0 | 1.2 |
| GalN | 0.8 | 1.2 |
| GlcN | 4.2 | 4.2 |
| Gal | 1.6 | 1.3 |
| Glc | 13.1 | 5.7 |
| Man | 2.9 | 2.7 |
| Sialic acid | 2.0 | 1.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farnós, O.; Venereo-Sánchez, A.; Xu, X.; Chan, C.; Dash, S.; Chaabane, H.; Sauvageau, J.; Brahimi, F.; Saragovi, U.; Leclerc, D.; et al. Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation. Vaccines 2020, 8, 654. https://doi.org/10.3390/vaccines8040654
Farnós O, Venereo-Sánchez A, Xu X, Chan C, Dash S, Chaabane H, Sauvageau J, Brahimi F, Saragovi U, Leclerc D, et al. Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation. Vaccines. 2020; 8(4):654. https://doi.org/10.3390/vaccines8040654
Chicago/Turabian StyleFarnós, Omar, Alina Venereo-Sánchez, Xingge Xu, Cindy Chan, Shantoshini Dash, Hanan Chaabane, Janelle Sauvageau, Fouad Brahimi, Uri Saragovi, Denis Leclerc, and et al. 2020. "Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation" Vaccines 8, no. 4: 654. https://doi.org/10.3390/vaccines8040654
APA StyleFarnós, O., Venereo-Sánchez, A., Xu, X., Chan, C., Dash, S., Chaabane, H., Sauvageau, J., Brahimi, F., Saragovi, U., Leclerc, D., & Kamen, A. A. (2020). Rapid High-Yield Production of Functional SARS-CoV-2 Receptor Binding Domain by Viral and Non-Viral Transient Expression for Pre-Clinical Evaluation. Vaccines, 8(4), 654. https://doi.org/10.3390/vaccines8040654