1. Introduction
Enterovirus 71 (EV71), a human virus that belongs to family
Picornaviridae [
1], has been extensively studied, because it is a causative agent of hand, foot, and mouth disease (HFMD) and it is associated with severe neurological complications [
2]. It was first isolated as the etiological agent of HFMD from a young child in California, United States in 1969 [
3]. Enteroviruses, such as Enterovirus 71 (EV−A71), Coxsackievirus type A16 (CV-A16), and other HFMD-causing entroviruses have caused more than seven-million infections, including 2457 fatalities in China from 2008 to 2012. Most HFMD infections that led to fatality were due to the EV-A71 virus [
1]. Since 1997, EV71 has mainly circulated in Asia Pacific region and most of the countries in this region are developing countries, such as Malaysia, China, Taiwan, Brunei, and Vietnam. The most promising vaccine candidate is currently being tested while using inactivated whole EV71 virus [
4]. Other candidates of inactivated whole virus vaccines are also being studied [
5,
6]. Besides using the whole virus, virus-like particles [
7] or viral protein subunit vaccines [
8] are also being tested. EV71 viral proteins that were expressed in either bacterial or plant system [
9,
10,
11] and synthetic peptides harboring antigenic epitopes of EV71 viral proteins [
12,
13] serve as alternatives for EV71 vaccine production. However, there are several drawbacks of using inactivated whole-virus or live-attenuated virus as a vaccine, such as its biosafety issues, cost of production, tedious procedure of viral preparation, as well as the difficulty of storage and delivery. This has led to the development of other types of vaccines, including epitope peptide vaccine, consisting of the specific immunogenic epitope, which is capable of stimulating an immune response.
The recombinant protein technology enables polypeptide-based vaccine to be produced in bulk, irreversible of their virulent form, easier, and cheaper. Administering the immunogenic protein from an infectious pathogen produces a more targeted and specific immune response. In addition, the risk of active infection that is caused by live attenuated vaccines or inactivated virus vaccines can also be reduced. Despite the specificity, the small polypeptide is often relatively less immunogenic and requires the use of delivery systems, such as carriers or adjuvants, in order to boost up the immunogenicity of the epitope peptide. Nucleocapsid protein (NP) of Newcastle disease virus (NDV) is the most abundant protein on the viral particle [
14]. It heterodimerizes in order to form a herringbone structure, a classical morphology of Paramyxoviruses. Recombinant NP are still able to assemble into this herringbone or ring-like structure when expressed in baculovirus [
15] and
E. coli expression systems [
16]. Additionally, this structure can still be formed by the NP, even though there is a small peptide added onto its C-terminal [
16]. It has been shown that such a fusion protein containing a C-terminal foreign peptide was exposed on the surface of the NP particles [
17,
18]. When the HN and F protein fragments were fused to the C-terminal end of the NP, they were able to induce high immune responses in chickens [
19]. These results showed the potential use of NP as a carrier protein for foreign immunogens.
In the previous study, a truncated nucleocapsid protein (NPt), which was derived from the NDV, was used as a carrier protein to deliver the first 100 amino acid of the N-terminal of VPI (VP1
1–100) into animals. The obtained results showed that the recombinant protein exhibited strong immunogenic properties in rabbits and mice [
20,
21,
22]. However, the newborn mice were only partially protected upon the lethal challenge of EV71. The partial protection was likely due to the insufficient antigenic properties of the N-terminal end of VP1 antigen. Neutralizing epitopes on the VP1 protein of EV71 were shown to be located at its C-terminal hydrophilic moiety [
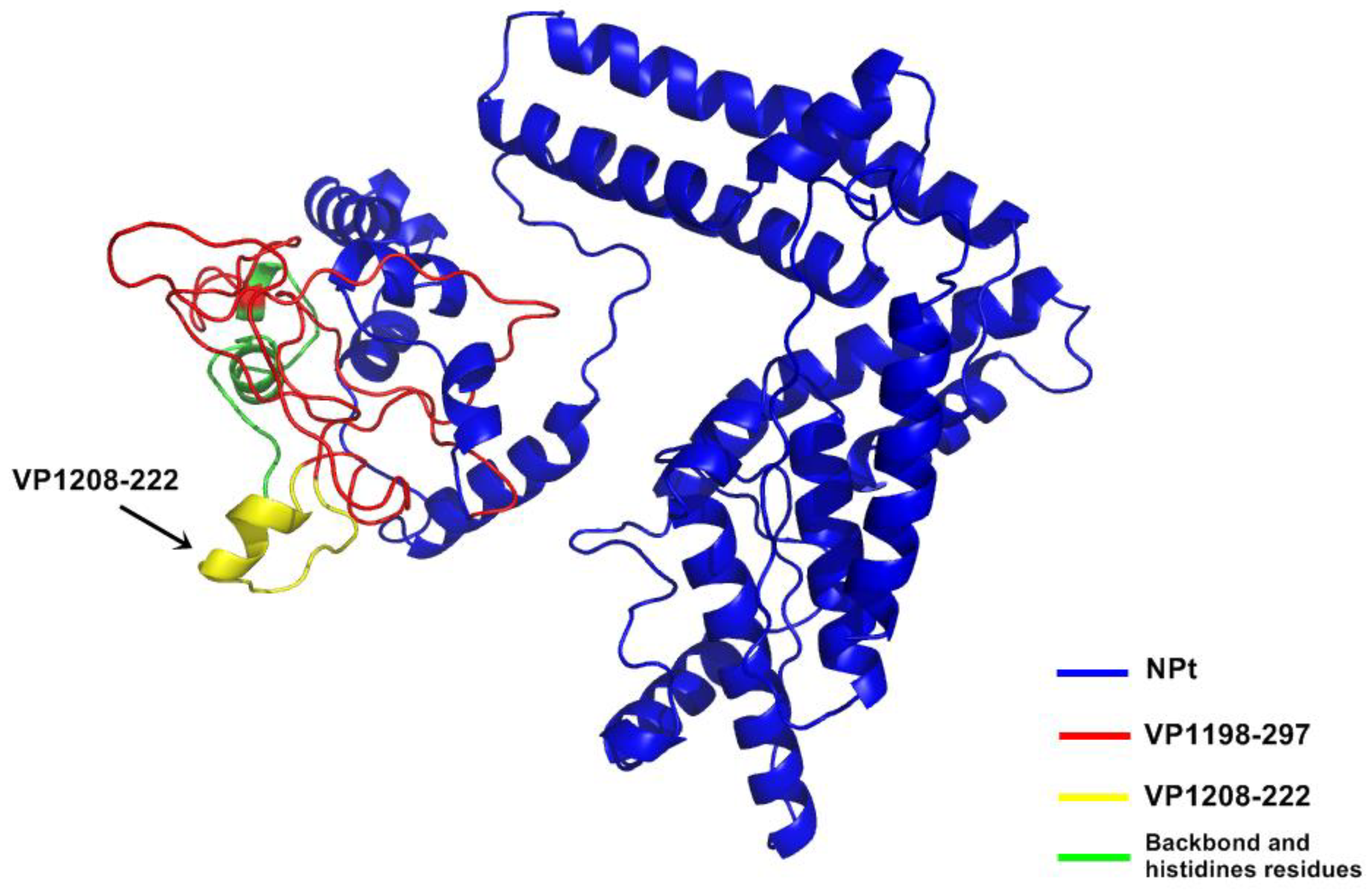
12]. Synthetic peptides, designated as SP70 and SP55, which corresponded to amino acids 163–177 and 208–222 of this C-terminal of VP1, respectively, were capable of inducing strong immune responses against EV71 in mice [
13]. Based on this information, it is hypothesized that hydrophilic fragments of the C-terminal region of EV71 will be able to be expressed as fusions to carrier molecules without affecting its antigenicity. Therefore, in the present study, the antigenicity of the recombinant plasmids carrying the NPt gene as a fusion to VP1 gene regions encoding for VP1
198–297 peptides were evaluated in the mice model.
2. Materials and Methods
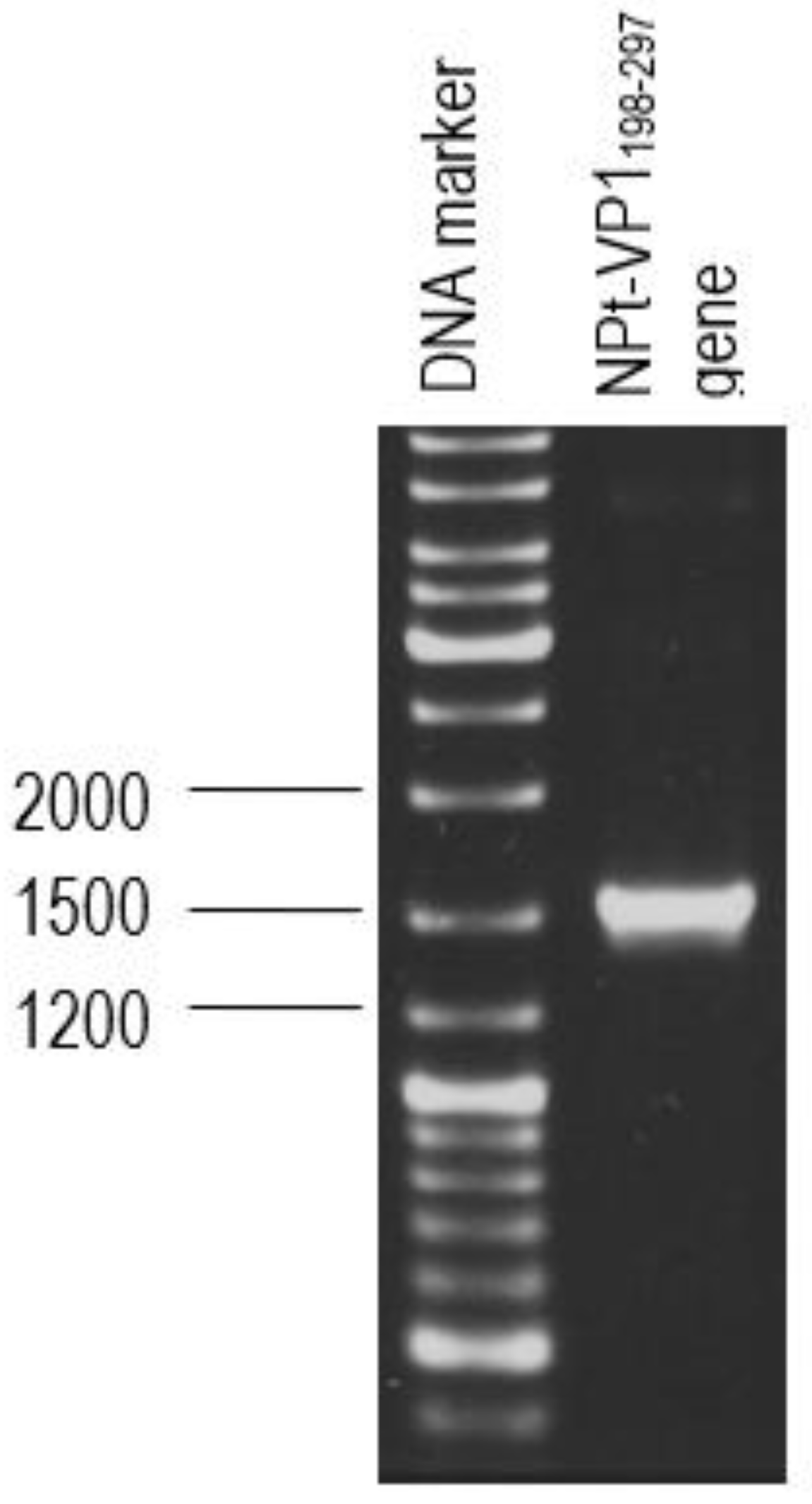
2.1. Synthesis and Amplification of VP1198–297 Gene by Polymerase Chain Reaction (PCR)
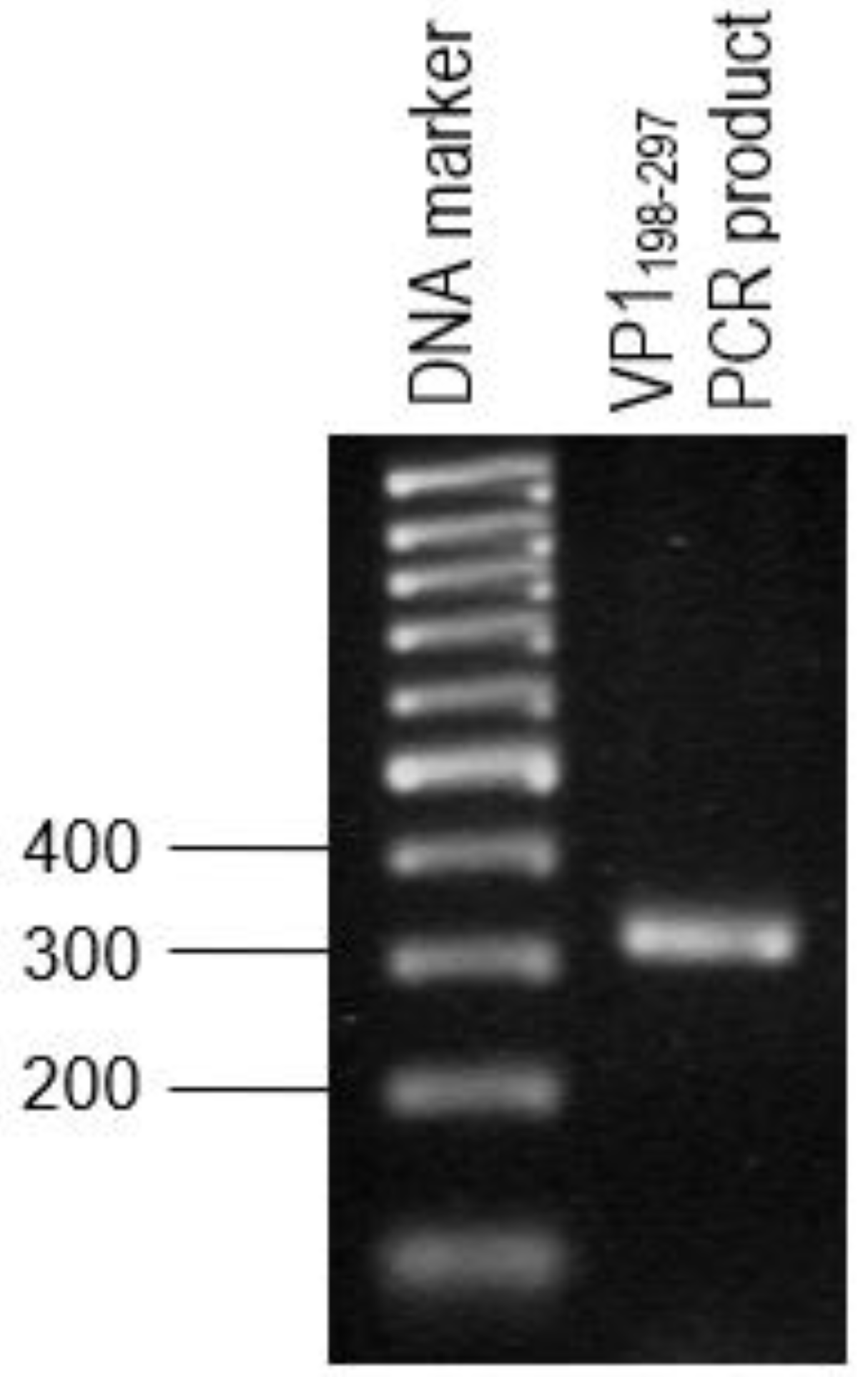
The primer pair was designed for the synthesis of each gene fragment of VP1198–297 based on the nucleotide sequences of the VP1 of EV71 strain MY104/9/SAR/97 (accession no.: AF). DNA sequencing analysis of positive clones was conducted by Macrogen Sequencing Service (Korea). The pTrcHis2-NPt-VP11–100 and pET30a-VP1fl plasmids constructs were extracted using the HiYield plasmid mini kit (Yeastern Biotech, Taipei, Taiwan), according to the manufacturer’s instruction. The synthesis of a gene fragment of VP1 was carried out by PCR. The reaction was performed in 50 µL reaction mixtures containing 1 U DyNAzyme EXT DNA polymerase, 1 X DyNAzyme EXT buffer [50 mM Tris-HCl, 15 mM (NH4)2 SO4, 0.1% Triton X−100; pH 9.0], 1.5 mM MgCl2, 200 µM dNTPs, 0.6 µM forward and reverse primers, and 1 ng of pET30 a-VP1fl plasmid as template. The mixtures were incubated in a Mastercycler® thermal cycler (Eppendorf, Enfield, CT, USA) with a heated lid. The reactions were initiated with pre-denaturation of the DNA template (94 °C, 3 min.), followed by a 35-cycle PCR profile: denaturation (94 °C, 30 s), annealing (60 °C, 1 min.), and extension (72 °C, 1 min.). The reaction was finished with a final extension step (72 °C, 7 min.).
2.2. Purification and Transformation of PCR Products
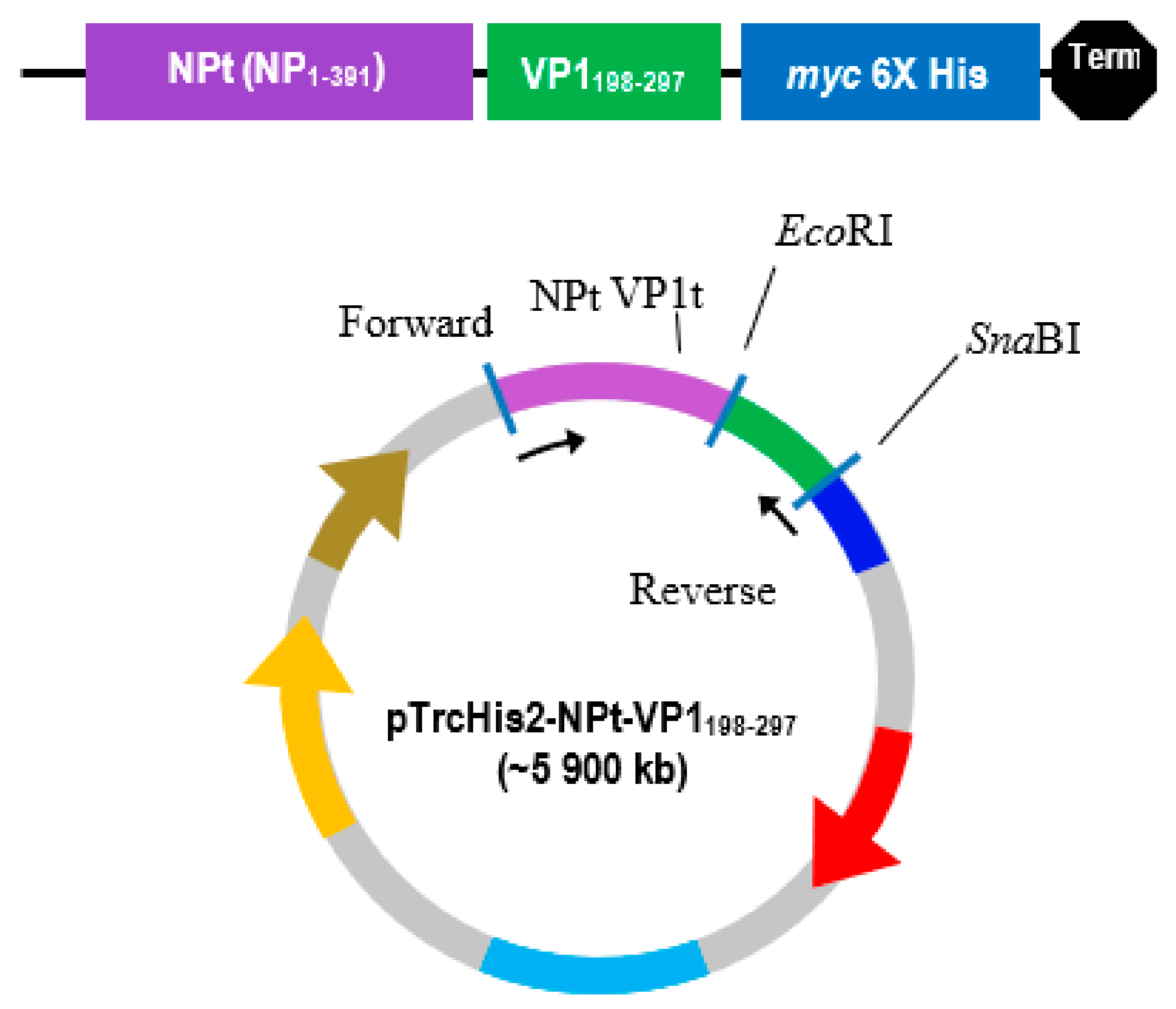
The PCR product was separated by electrophoresis in big wells of a 1% (w/v) TAE agarose gel. The purified VP1 DNA fragments and the pTrcHis2-NPt-VP11–100 plasmid were separately digested in 20 µL of restriction enzyme mixture containing 5–10 U SnaBI, 1X TangoTM buffer. The digested products were separated by agarose gel electrophoresis and purified while using the Gel Extraction Kit (Qiagen, Germantown, MD, USA), according to the manufacturer’s instruction. The VP1 DNA fragments were inserted into SnaBI and EcoRI sites of pTrcHis2-NPt plasmid separately with the ratio 1:3 in a 20 µL of ligation mixtures containing 2.5 U T4 DNA Ligase (Fermentas, Waltham, MA, USA) and 1X T4 DNA Ligase buffer and then incubated at 4 °C for 16 h.
Competent cells of
E. coli TOP10 were prepared while using the calcium chloride (CaCl
2) method. The ligation product was transformed into the competent
E. coli TOP10 cells using the heat-shock method [
23]. This was followed by the addition of 1 mL of Super Optimal broth with catabolite repression (SOC medium) to the mixture and incubated at 37 °C for 1 h with an agitation rate at 250 rpm. Approximately 50–200 µL of each transformation mixture was spread onto LB agar plates containing 50 µg/mL of ampicillin and incubated overnight at 37 °C. Ten pure single colonies of positive transformants were picked for the screening of successful transformations. Each colony was individually inoculated in 10 mL of LB broth containing 50 µg/mL of ampicillin. After overnight incubation, 3 mL of the cultures were used for plasmid extraction using the HiYield plasmid mini kit (Yeastern Biotech, Taipei, Taiwan) according to the manufacturer’s instruction. PCR, restriction enzyme reaction, and DNA sequence analysis confirmed the orientation of inserts.
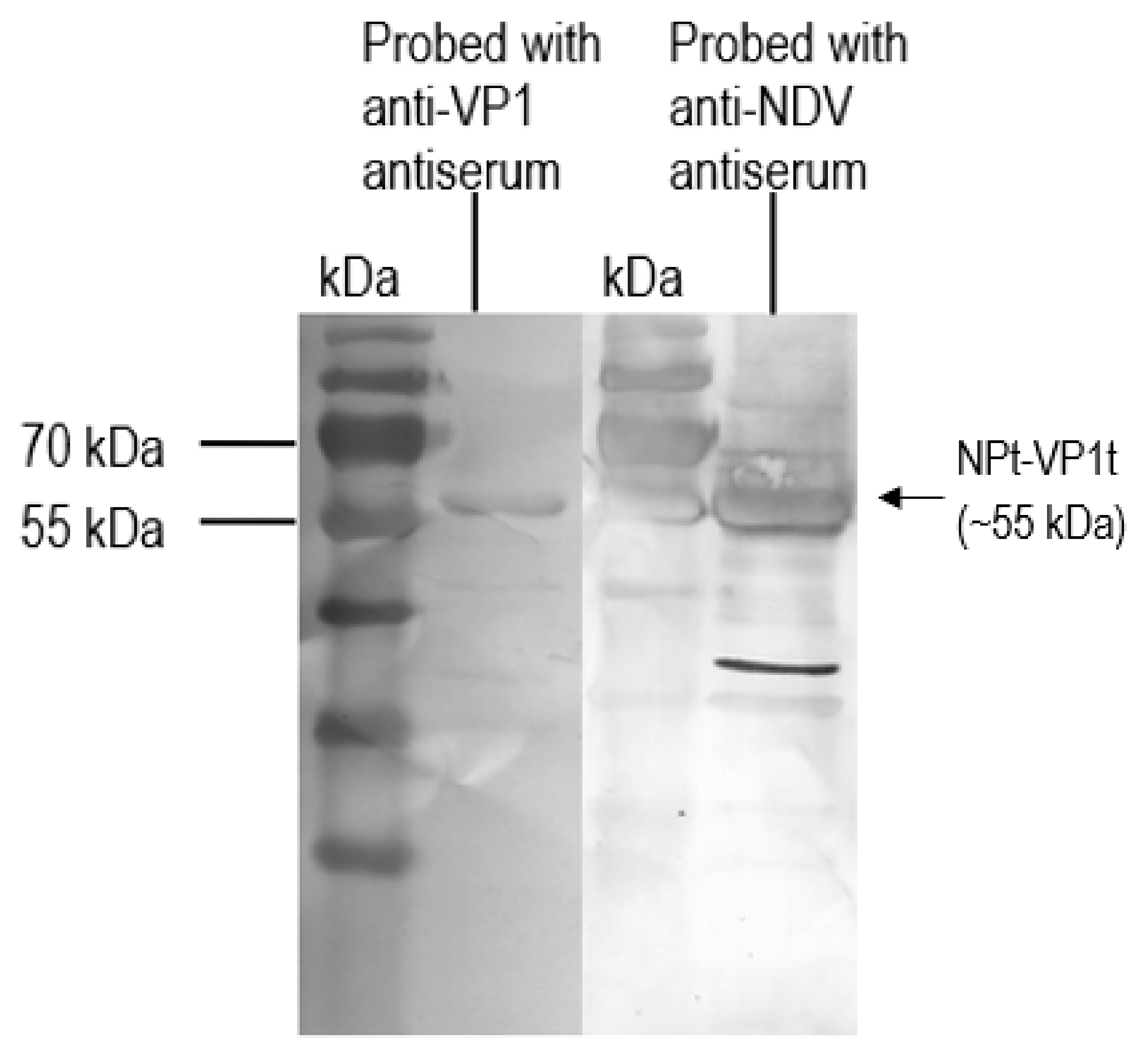
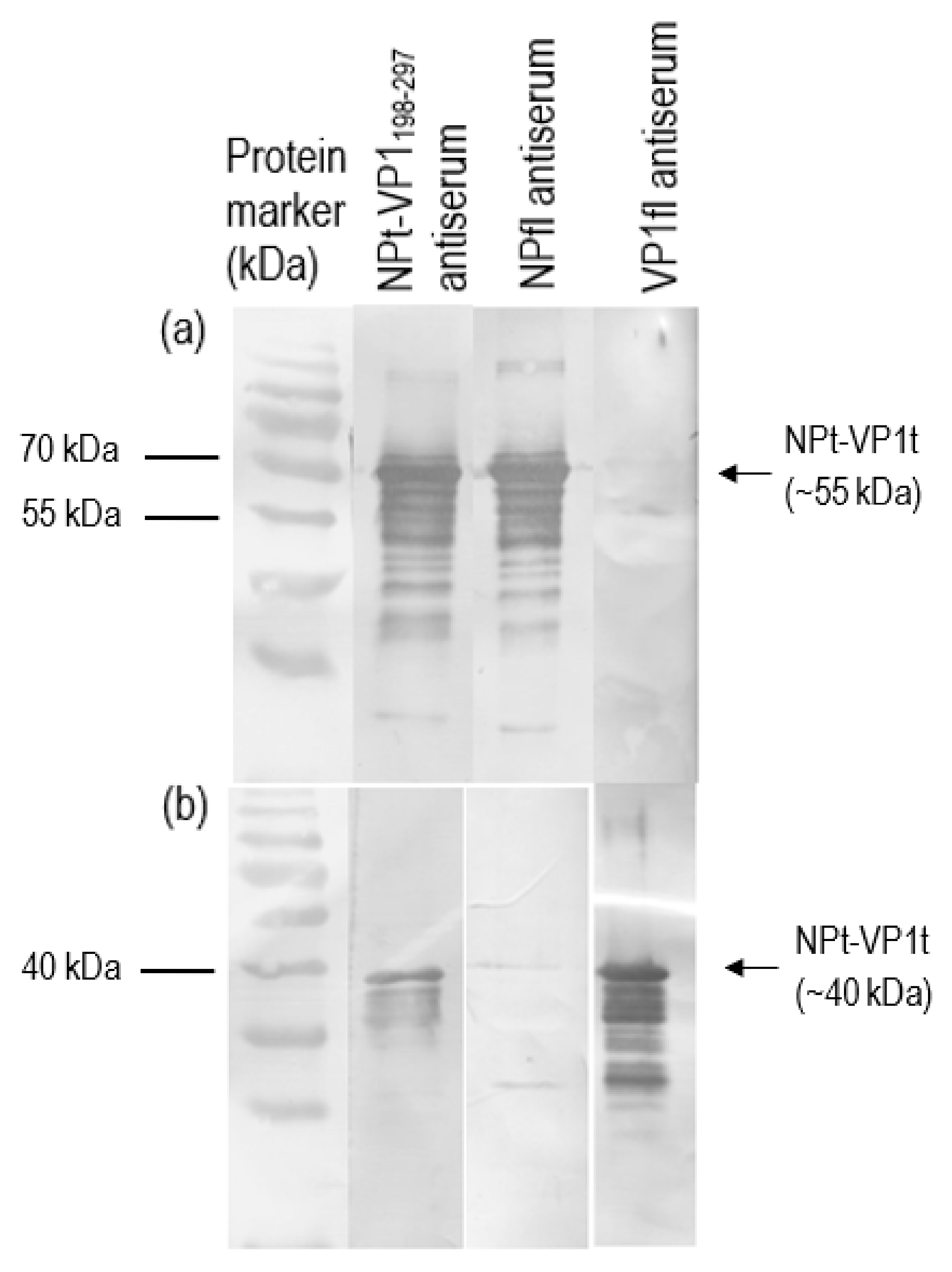
2.3. Expression, SDS-PAGE and Western Blot of Recombinant NPt-VP1198–297 Proteins
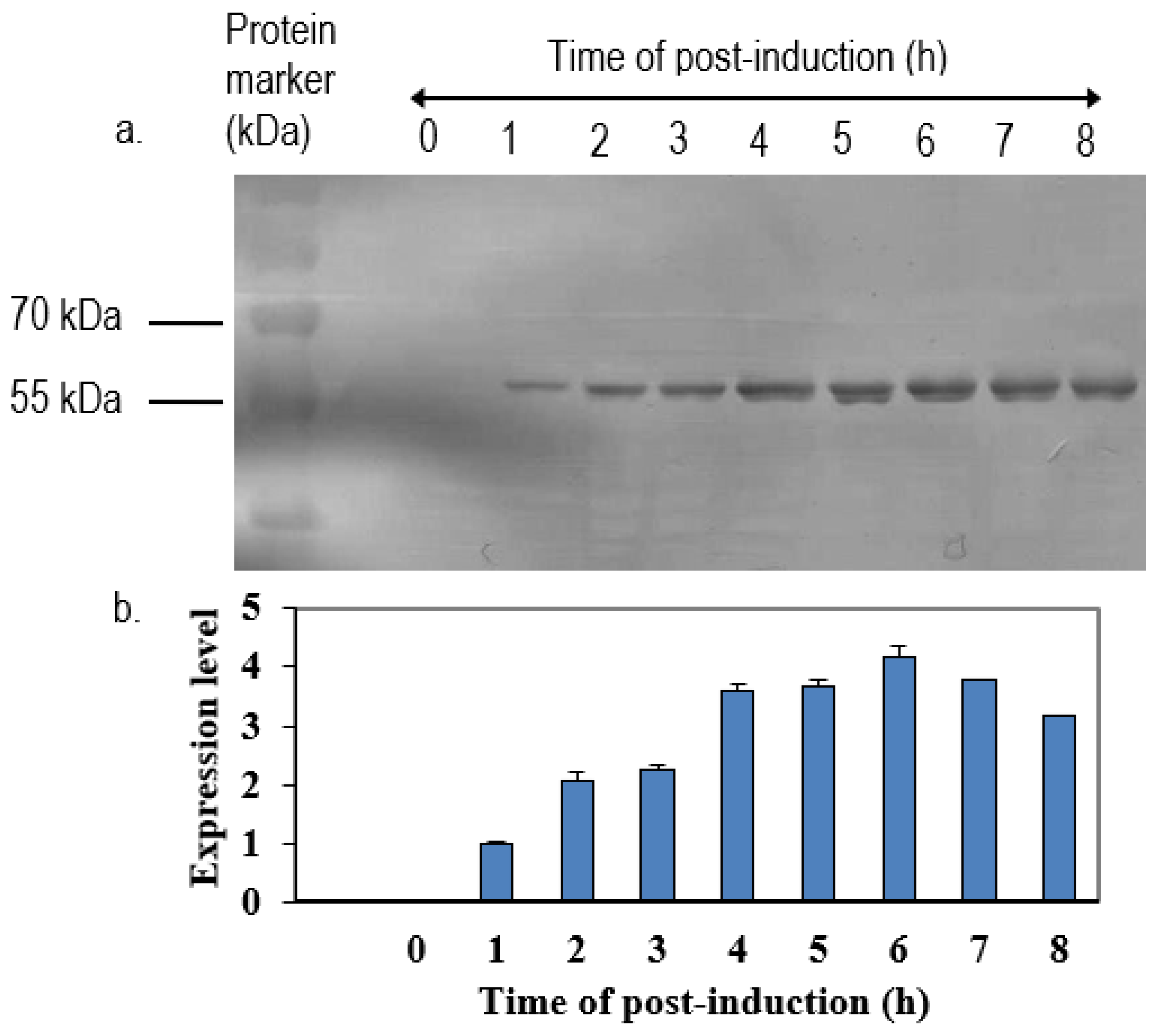
The positive clone was inoculated in 5 mL LB medium containing 50 µg/mL of ampicillin overnight at 37 °C in a shaking incubator. One milliliter of overnight cultures was inoculated in 100 mL of fresh LB medium containing 50 µg/mL of ampicillin. When the OD600 nm of cultures reached 0.6 to 0.8, isopropyl-β-D-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM. Immediately following the addition of IPTG, 1 mL of cultures were collected and left as 0 h induction sample. After that, the remaining cultures were further incubated, and 1 mL of culture was collected at every hour until 8-h incubation. All of the samples were centrifuged at 6000×
g in Sorvall Legend Micro 17 microcentrifuge (Thermo Scientific, Waltham, MA, USA) at 4 °C. The collected cell pellets were then resuspended in 100 µL of sterile phosphate buffer saline (PBS) and analyzed by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and western blotting with anti-His, anti-NDV, and anti-VP1 antibody. The intensities of protein bands were quantified by ImageJ software (National Institutes of Health, Bethesda, MD, USA). The expression level was expressed by the ratio of the readings of band intensities at each time-point divided by the reading of band intensities at 1-h post-induction. For the scale-up expression of recombinant proteins, the volumes of each culture were increased from 0.5 L to 2 L and the samples were harvested after 5 h post-induction with 1.0 M IPTG (
Table 1).
2.4. Purification of Recombinant Protein
Bacterial culture (2 L) expressing recombinant protein was harvested by centrifugation at 6500× g at 4 °C for 10 min. and washed with ice-cold 1X PBS and resuspended in 0.03 volume of lysis buffer, and then incubated at room temperature for 30 min. with gentle mixing. The cell lysate was centrifuged at 12,000× g at 4 °C for 20 min. The pellet was then resuspended in 0.03 volume of lysis buffer containing 1% Triton X−100 and then incubated on ice for 10 min. Subsequently, the suspension was centrifuged at 12,000× g at 4 °C for 20 min. This step was repeated with lysis buffer without Triton X−100. The washed pellet was resuspended in phosphate buffer [20 mM sodium phosphate, 500 mM NaCl; pH 7.4] containing 8 M urea, 0.3 mM reduced glutathione (GSH), and 5% glycerol and incubated for 2 h at room temperature with gentle shaking. After centrifugation at 12,000× g for 20 min. at 4 °C, the supernatant was subjected to a HisTrap HP column purification. The column was washed with five volumes of binding buffer containing 8 M urea, 5% glycerol, 3 mM GSH, 0.3 mM glutathione disulfide (GSSG), and 30 mM imidazole. The column was further washed with binding buffer. as mentioned before with a gradual decrease of urea concentration until 2 M and washed with 5 column volumes of binding buffer containing 2 M urea, 5% glycerol, 3 mM GSH, 0.3 mM GSSG, and 30 mM imidazole. After that, the target protein was eluted from the column by elution buffer. The elution product was dialyzed against sodium phosphate buffer containing 1 M of urea overnight at 4 °C and then concentrated by Vivaspin Concentrator.
2.5. Immunization of NPt-VP1198–297 in a Mouse Model
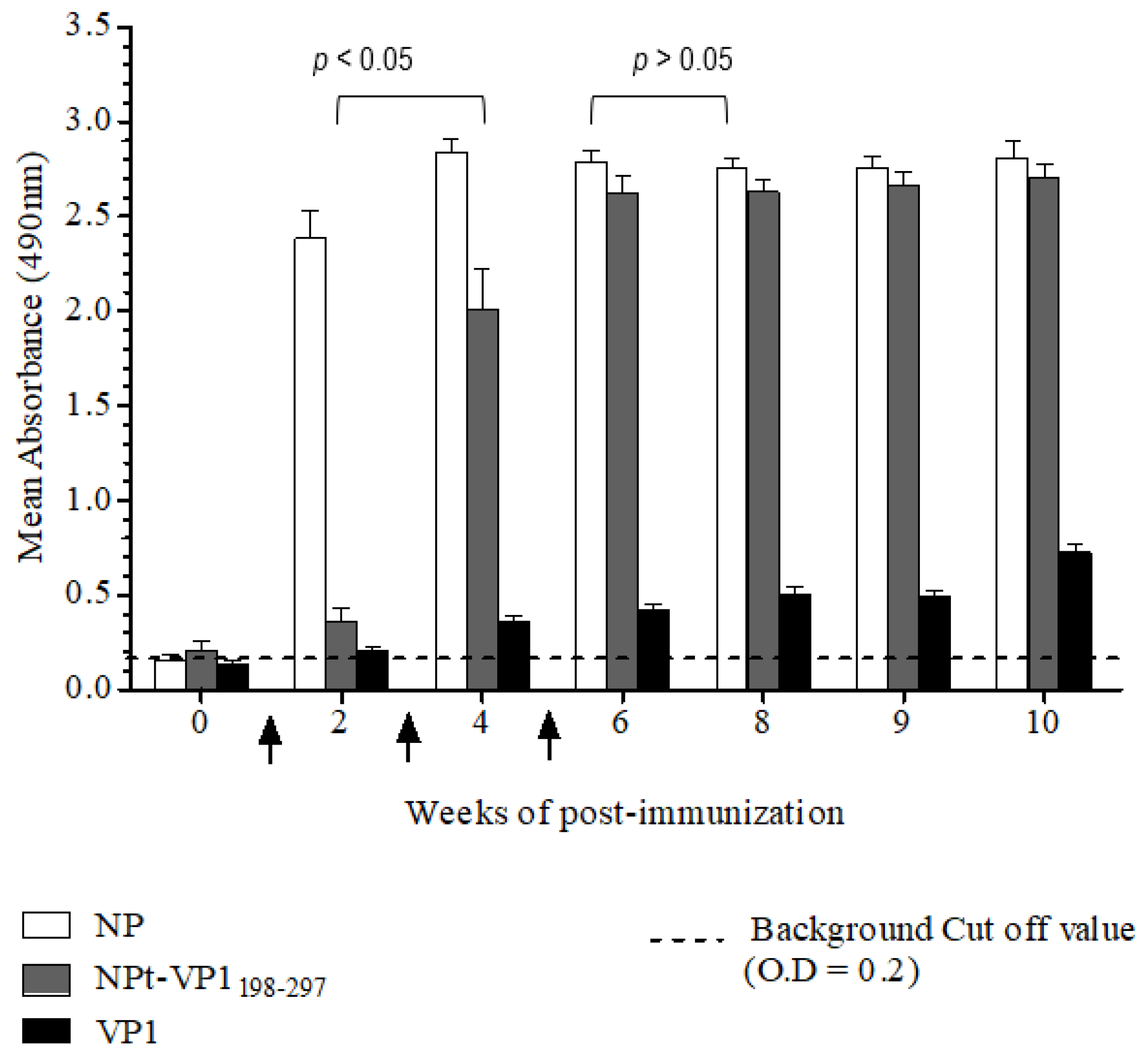
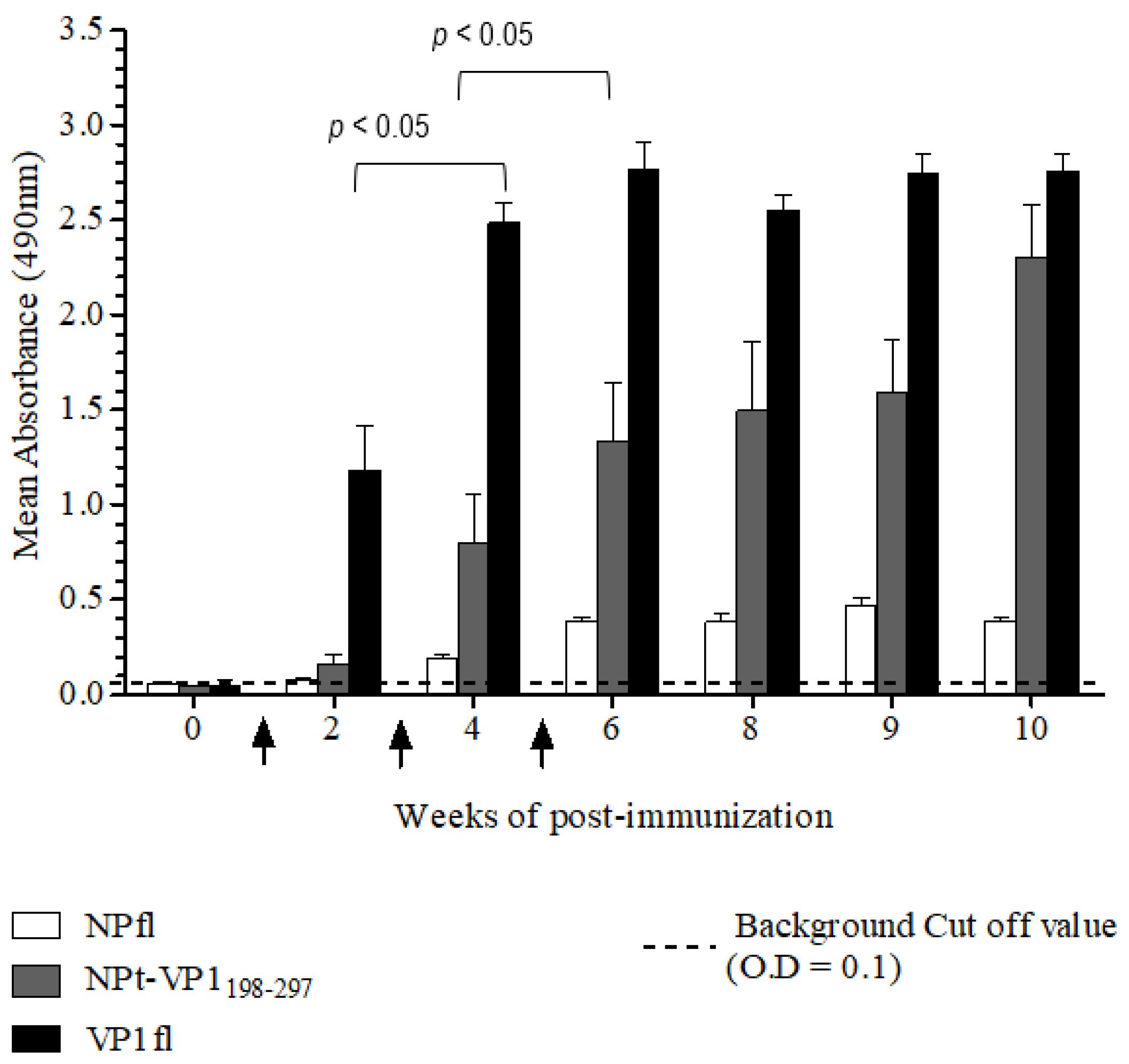
The Animal Care and Use Committee, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Selangor approved all of the animal works in this study (AUP no: 10R84). The animals were raised and cared for according to The Code to Care and Use of Animals in Research. The methods of animal experiments were performed under the guidelines that were prescribed by the Malaysian Association for Accreditation of Laboratory Animal Care. Groups (n = 8) of 6–8-week-old adult female BALB/C mice were immunized with the recombinant NPt-VP1198–297, NPfl (as control), VP1fl (as control) individually. Each group of mice was immunized intraperitoneally with 10 µg of purified protein (in PBS, pH 7.4), which was emulsified with 50% Freund’s complete adjuvant (Sigma, St. Louis, MO, USA). Two boosters (10 µg proteins emulsified with 50% Freund’s incomplete adjuvant) were administered to each mouse every two weeks. The blood samples were collected by bleeding of tail’s vein at weeks 0, 2, 4, 6, 8, 9, and 10. The collected blood samples were stored overnight at 4 °C to allow for the clotting of red blood cells (RBC). On the following day, sera at the upper layer were carefully pipetted out to a new, sterile tube. The remaining blood samples were centrifuged at 2000 rpm for 20 min. and the remaining sera were pipetted out carefully.
2.6. Indirect Enzyme-Linked Immunosorbent Assay (ELISA)
Titers of anti-VP1 and anti-NP IgG in mice sera were evaluated by an indirect enzyme-linked immunosorbent assay (ELISA). First, each well of a flat-bottomed, high binding 96 wells ELISA microtiter plate (Greiner Bio-One, Frickenhausen, Germany) was pre-coated with 100 µL of 1.5 µg/mL VP1 or NP in 1X TBS buffer [0.1 M Tris, 0.15 M NaCl; pH 8.0], respectively. Subsequently, the plates were sealed and incubated overnight with shaking at 4 °C in the dark. The wells were coated with pET30, a control protein that served as a negative control. On the following day, the coating solution was removed by gently knocking the plates on a paper towel and washed three times with 1X TBS buffer containing 0.05% (v/v) Tween−20 (TBS-T) at room temperature for 5 min. The remaining buffer in wells was removed by patting the plates. After that, each well was blocked with 250 µL of blocking solution [5% (w/v) bovine serum albumin in TBS] for 2 h at 4 °C. Subsequently, the plates were washed three times with TBS-T. Subsequently, 100 µL of diluted serum samples (1:50) was added to the corresponding wells and the plates were incubated at room temperature for 1 h with gentle shaking. After washing the plates with TBS-T, 100 µL of anti-mouse IgG conjugated with horseradish peroxidase (Cell Signaling, Danvers, MA, USA) (1:1000 in TBS) was added into each well and then incubated at room temperature for 1 h with gentle shaking. Afterwards, the plates were washed with TBS-T for five times and then developed with 100 µL of 0.4 mg/mL o-phenylenediamine dihydrochloride substrate (Acros Organics, Carlsbad, CA, USA) in 0.05 M Citrate Phosphate buffer [Na2 HPO4, citric acid, pH 5.0] containing 20 µL fresh 30% H2 O2. After incubation at room temperature for five min., the reaction was stopped by adding 100 µL of 3 M sulphuric acid (H2 SO4). The absorbance values were then measured at 490 nm on iMark Microplate Absorbance Reader (BioRad, Hercules, CA, USA).
2.7. Immunoblotting Analyses
Recombinant VP1 and NP were separated on 12% SDS-PAGE gel and then transferred onto PVDF membranes as antigen for immunoassay. After blocking with 10% milk diluent, the mice sera containing antibodies to the recombinant VP1 and NP were used as primary antibodies (1:500 in 1X TBS) in order to probe the blotted recombinant proteins. Rabbit anti-VP1 serum (1:2000 in 1X TBS) and rabbit anti-NDV serum (1:9000 in 1X TBS) were used to serve as positive controls. Subsequently, the membranes were incubated with appropriate alkaline phosphatase-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa-Cruz, CA, USA) with a dilution of 1:5000 in 1X TBS at room temperature for 1 h. The mixtures of BCIP/NBT in 10 mL alkaline phosphatase buffer were added for signal development. When the desire bands appeared, the substrate was discarded, and the membrane was washed with distilled water.
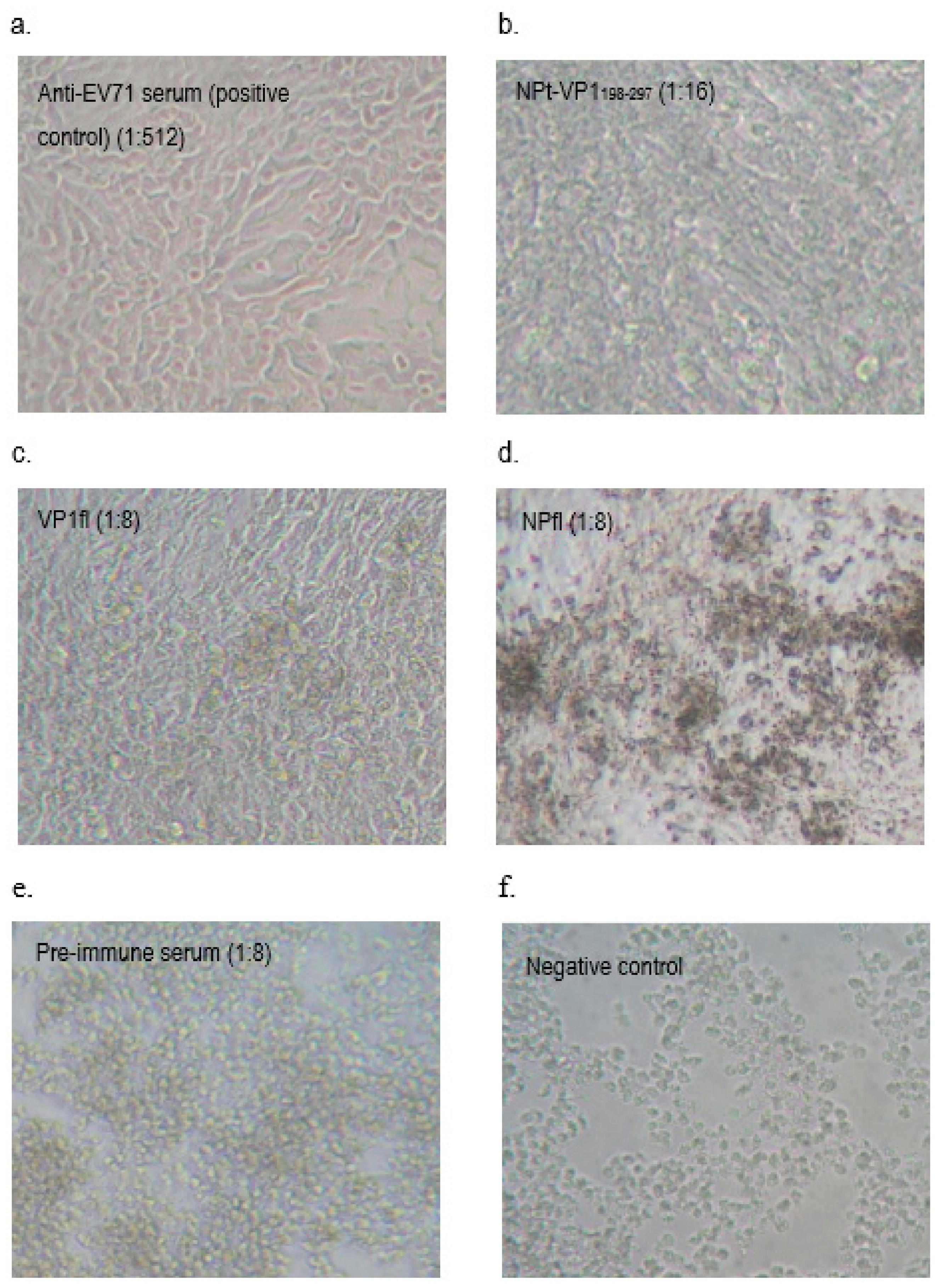
2.8. Neutralization Assay
A neutralization assay was carried out in order to determine the presence of neutralizing antibodies in the immunized mice sera. In brief, 1.5 × 104 of Vero cells were seeded and cultured in a 96-well tissue culture plate (Corning, New York, NY, USA) with DMEM (PAA, Pittsburgh, PA, USA) containing 5% FBS (PAA, USA) at 37 °C with 5% CO2 for 36 h. The pooled mice sera collected from pre-immunized or immunized mice were inactivated at 56 °C for 30 min. Afterwards, each sample was two-fold serially diluted with DMEM media containing 2% FBS (PAA, USA), 1% L-glutamine and 1% antibiotic-antimycotic solution (PAA, USA). The diluted sera were mixed with an equal volume of EV71 strain A104 containing a final 1000 TCID50. After incubation at 37 °C for 2 h, the mixtures of serum and virus were added to the Vero cell culture in the designated wells (well 1:8 to 1:512). The positive control was added with EV71 post-infection serum and a well containing virus only were served as a negative control. After seven days of incubation at 37 °C with 5% CO2, the cytopathic effect (CPE) of cells was observed under an inverted light microscope (Olympus, Tokyo, Japan) and the neutralization titers were determined by calculating the highest dilutions that could result in less than 50% CPE. All of the experimental data in this study were analyzed while using the Student’s t-test and presented as mean ± standard error (SE). Differences with p < 0.05 were considered to be significant.
4. Discussion
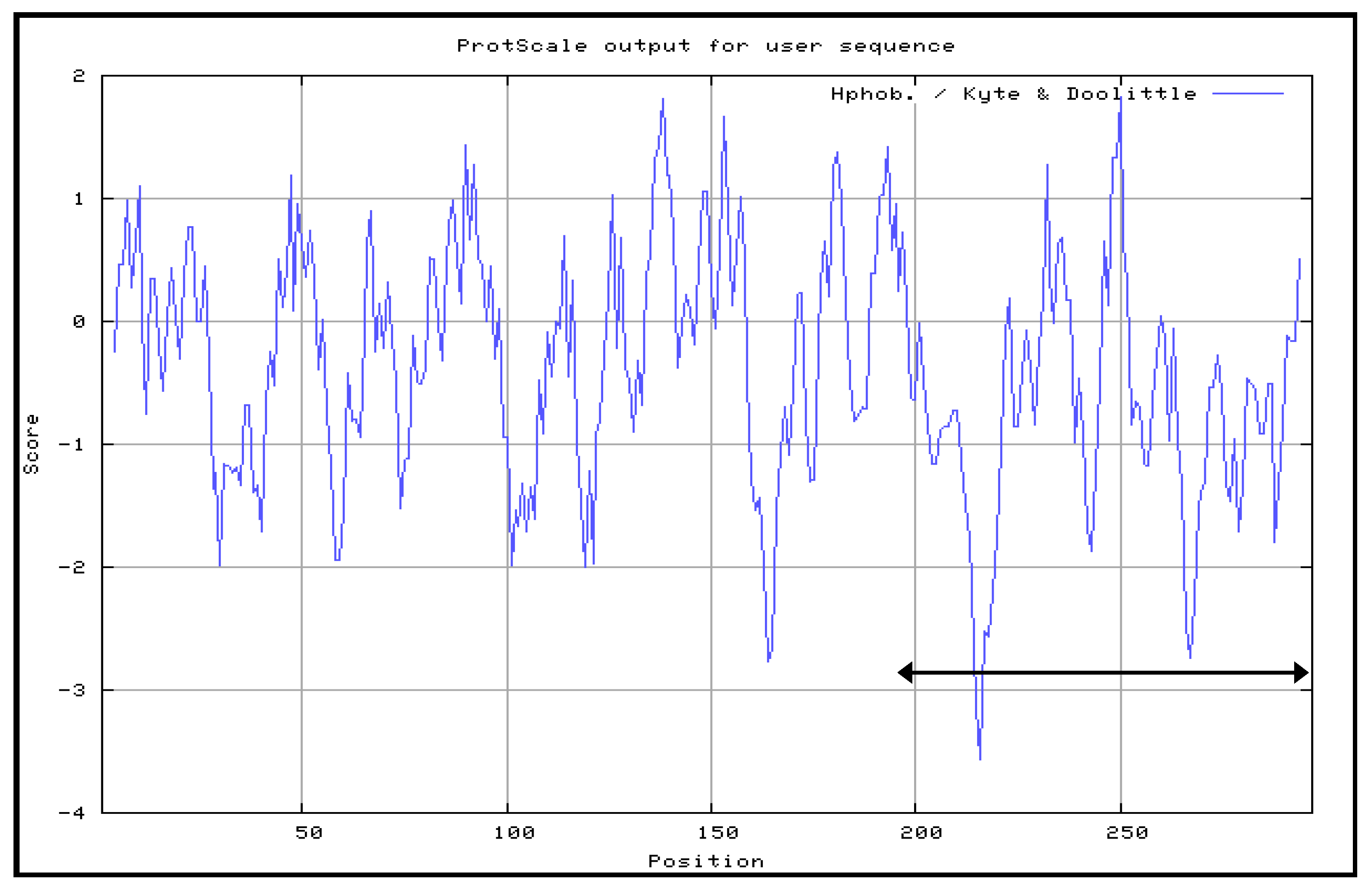
The Kyte and Doolittle hydrophobicity profile was used in order to examine the VP1fl in the present study. Hydrophilic regions of a protein are known to be exposed on the surface of the protein when it is in its tertiary conformation [
28]. It would be correlated between antigenic sites and high hydrophilic regions, as the antigenic sites tend to be exposed on the surfaces of proteins [
29]. B cell epitopes are divided into two types: a conformational epitope, which is recognized by immune cells by its tertiary structure; and, a continuous epitope, which is recognized by its linear sequence of amino acids [
30]. These epitopes are important for the determination of properties of peptides or proteins in vaccine development and diagnostics. Following the confirmation, additional PCR reactions were performed in order to amplify the gene. Each of the primers used was designed to carry specific restriction enzyme sites and it was expected that the PCR-amplified gene fragments would also contain these sites. Each of the gene fragments was designed to contain
EcoRI restriction sites at the 5′ end of DNA fragments, which result in ‘sticky ends’ upon enzyme digestion.
SnaBI restriction sites were introduced at the 3′ termini. This enzyme will leave ‘blunt end’ DNA fragments following digestion [
16,
31]. The ligation mixtures were then transformed into competent
E. coli TOP10 cells. The pTrcHis2-NPt plasmid sequence contained the ampicillin-resistant gene [
16]. This allowed for the selection of transformed cells in media containing ampicillin.
The recombinant plasmid was successfully transformed into
E. coli TOP10 cells. The positive transformants were chosen from ampicillin-resistant colonies of
E. coli TOP10 cells and further analyzed by PCR while using forward primer of pTrcHis2 vector and reverse primers of respective inserts to confirm the correct orientation of the truncated VP1 inserts. The
E. coli system was chosen for this study because post-translational processing, such as glycosylation, is not required for the synthesis of NPfl proteins [
16,
19]. In the present study, we showed that the expression levels of these proteins decreased after their expression peak. This was perhaps due to a degradation of the recombinant proteins inside the cells. Prolonged incubation was previously shown to induce the proteolysis of expressed proteins, leading to yield reduction [
32].
Although the amino acid position 198–297 of VP1 protein mainly contains hydrophilic regions, this truncated VP1 polypeptide that fused to the protein carrier NPt was mainly expressed in inclusion bodies. Similar results were observed with the proteins, such as fragments, f, the Toxoplasma gondii rhoptry protein ROP2 fused with thioredoxin (TRX) or to the maltose-binding protein (MBP), which are well known for improving the solubility of fusion proteins [
33]. It is known that the formation of inclusion bodies is affected by the rate of target protein translation in the bacterial host [
34]. If the rate of protein expression is higher than the rate of their folding into secondary and tertiary structures, then they will likely be improperly folded. These proteins will then accumulate as insoluble aggregates in the bacterial cell [
35] Thus, the recombinant NPt-VP1
198–297 protein was purified under denaturing conditions.
In the present study, the recombinant NPt-VPl
198–297 was purified and immunized into an adult mouse. It was shown that the recombinant vaccine was able to elicit moderately high immune responses. Based on our findings, IgG was the most abundant form of immunoglobulin in serum. IgG plays an important role in complement activation and opsonization [
36]. In most cases, high levels of IgG are formed after secondary immunization. It was also proven that the recombinants vaccine was able to elicit high immune response in adult mice [
21]. This suggests that the NPt-VP1
198–297 protein acted as a strong immunogen for the humoral response. When naive B-cells encounter specific antigen(s), they will rapidly divide and differentiate into immunoglobulin-producing plasma cells [
37]. Consequently, greater magnitudes of antibody response were generated.
It had been shown that the antibodies elicited by VP1 proteins of EV71 were able to neutralize EV71 and inhibited the virus in order to infect the Vero or rhadobdomyosarcoma (RD) cells [
38]. In our study, we showed that the anti-NPt-VP1
198–297 antiserum prevented Vero cells from CPE at titer of 1:16 and the recombinant NPt-VP1
198–297 was able to induce a significant neutralizing immune response against EV71. This result agreed with a previous finding by Xu et al. (2012), which purported that the neutralizing epitopes may locate at the amino acid 208–222 of VP1 proteins. The earlier study reported synthetic peptide, SP70, was able to elicit neutralizing antibodies in order to protect the RD cells from the infection of EV71 at titer of 1:32 [
12]. VP1
1–100 polypeptide, which was carried by NPt carrier (NPt-VP1
1–100) in the prior art, had its efficacy as candidate vaccine tested and showed that it partially protected the neonates from EV71 infection, but has a lack of neutralizing epitope(s) [
21]. The study of Ong and his colleagues (2010) addressed the correlation or neutralizing antibodies with the prevention of viral replication and spreading in skeletal muscle and central nervous system. Thus, this novel observation highlights the potential of NPt-VP1
198–297 proteins as a candidate vaccine of EV71. Because lymphocyte proliferative responses are generally related to cell-mediated immunity [
39] and its association with the viral clearance, we sought to investigate the T-cell responses after immunization with the candidate vaccine constructs. In the present study, based on the titer profiles of neutralizing antibody against EV71, increased levels of splenocyte proliferation in the vaccinated group were observed. According to Wu et al. (2001), the immunization of mice while using the full-length VP1 recombinant protein resulted in a mixed Th1 and Th2 response. Th1 and Th2 subsets of helper T-cells express distinct cytokine patterns, which reflect the different immune response pathways. Th1-cells are known to be involved in cell-mediated immunity, while Th2-cells function as helper T-cells in humoral immunity. A significantly higher titer (1:16) of NPt-VP1
198–297 as compared to VP1fl (1:8) in the assay suggests that NPt-VP1
198–297 promoted a better Th1 immune response when compared to VP1fl. The results suggested that this group was able to generate better immune responses, especially cell-mediated immunity as compared to the control group. The high titer of the serum shows a promising result on the capability of candidate vaccine in providing excellent immune stimulation. It was also suggested that splenocyte proliferation reaction is directly proportional to the cellular immune response that is important for viral killing [
40].
Overall, the results from this study suggested that the NPt-VP1
198–297 is an ideal candidate vaccine against EV71. A study by Wu et al. (2001) using a recombinant VP1 protein expressed in
E. coli BL21 [
8], showed that the VP1 with a complete adjuvant able to elicit a neutralizing antibody response, enhance T helper cell proliferation, and induce high levels of interleukin (IL)−10 and interferon (IFN)- gamma in mice. The findings from the study provide direct evidence that the VP1 contains neutralizing epitopes independent of other viral capsid proteins. This paves the way for the use of VP1 as a backbone antigen for developing subunit vaccines against EV71. IgG level increased in mice immunized with DNA vaccine; in contrast, this level declined after boosting immunization [
41].

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}