Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Capsid Protein Sequence Analysis
2.2. Linear and Conformational B-Cell Epitope Prediction
2.3. Structure Modelling and Epitope Mapping
2.4. Molecular Dynamics Simulation Analysis
2.5. Molecular Docking
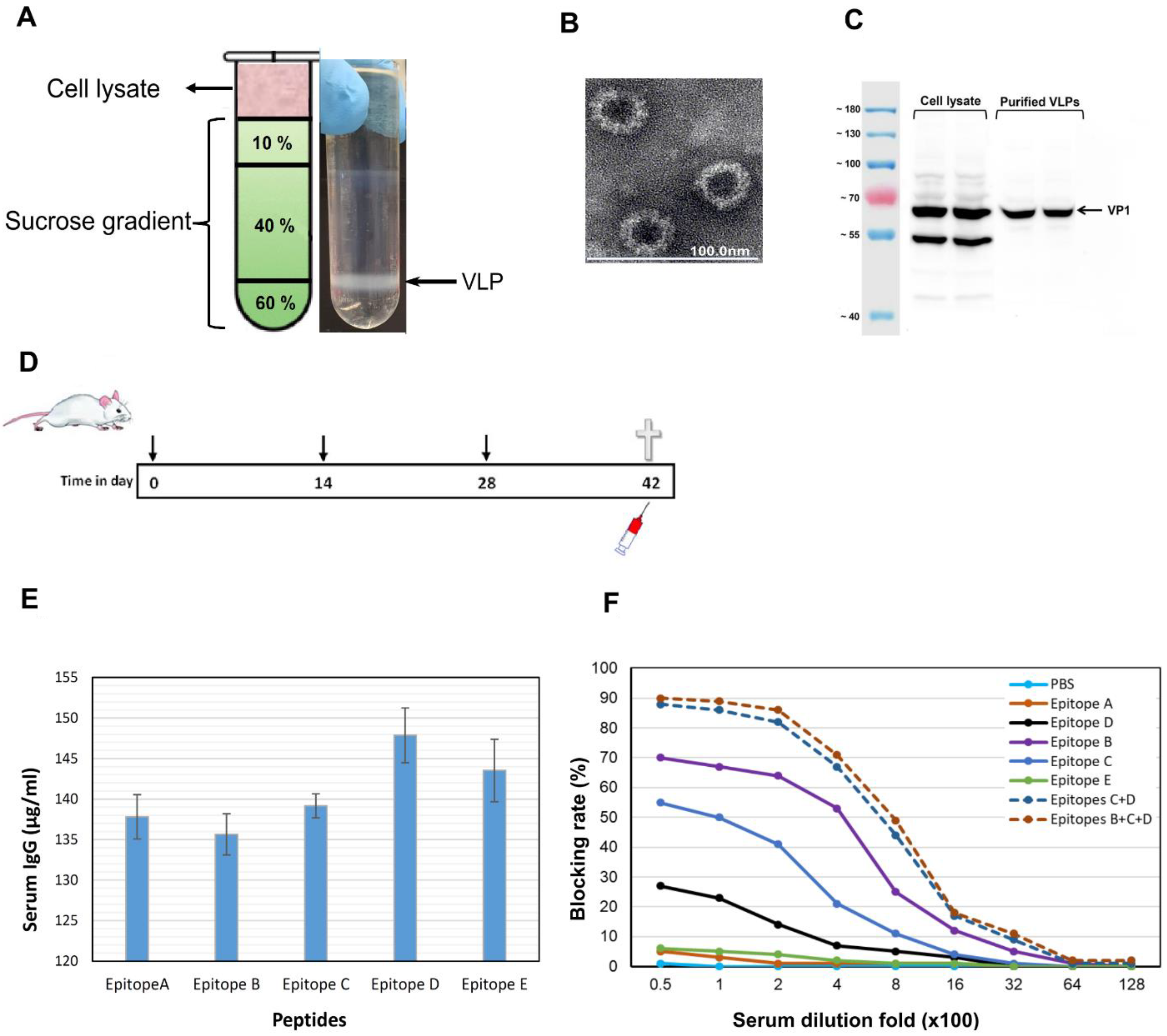
2.6. GII.4 Virus-Like Particle Production and Purification
2.7. Peptide Synthesis, Mouse Immunization, and HBGA Blocking Assay
3. Results
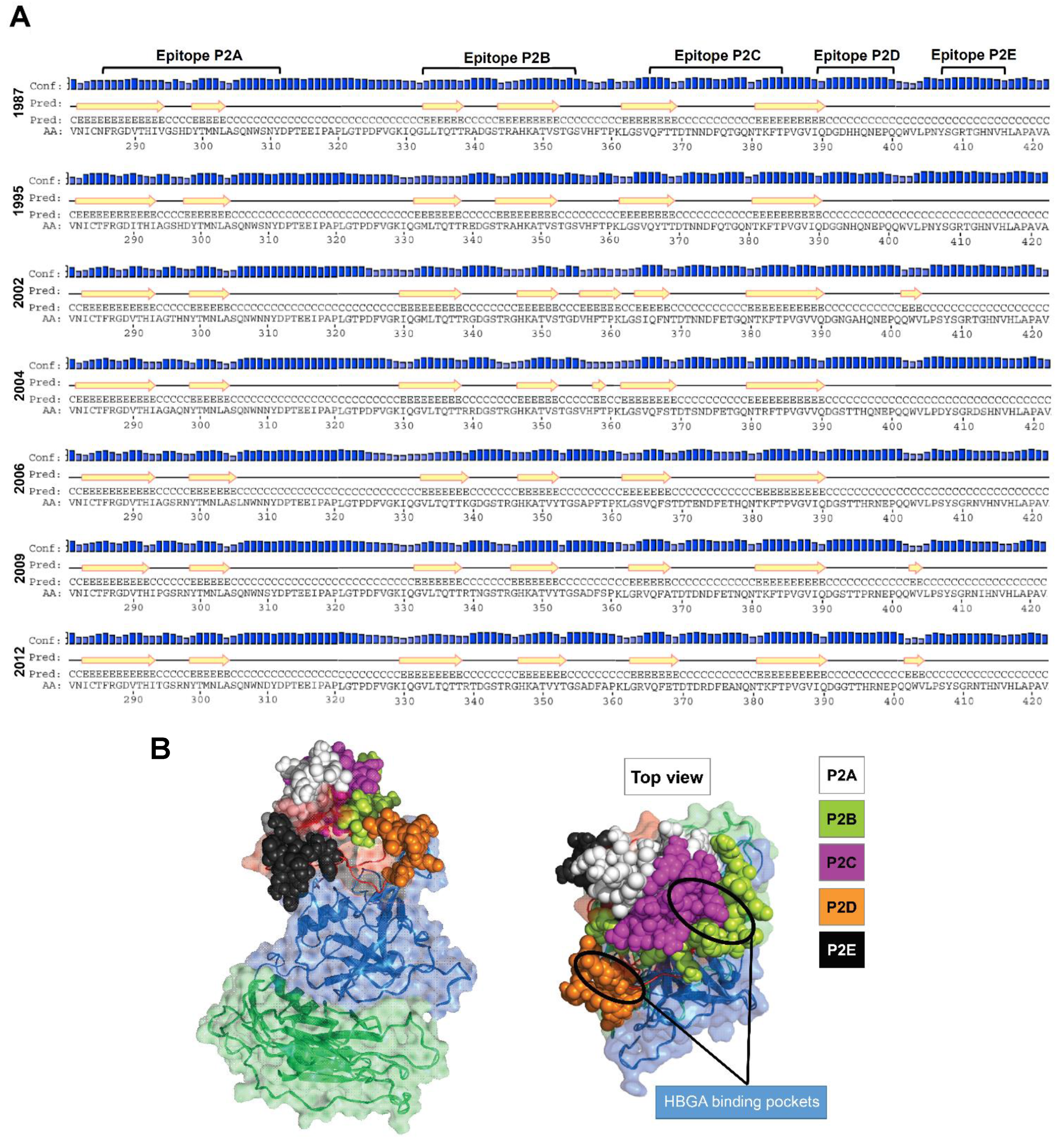
3.1. Putative Liner B-Cell Epitopes Evolve Over Time
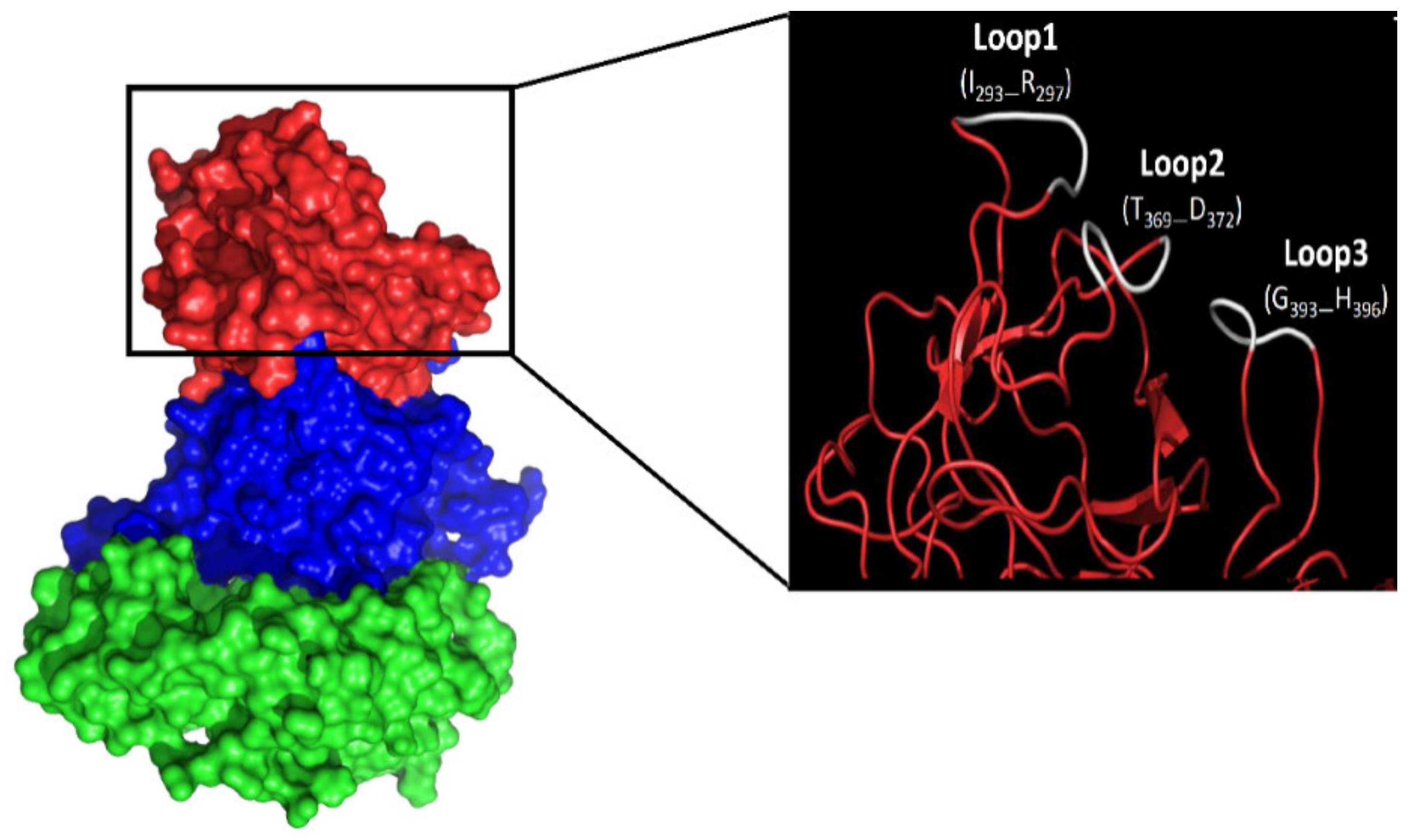
3.2. Amino Acid Substitutions Lead to Conformational Changes in Surface-Exposed Epitopes
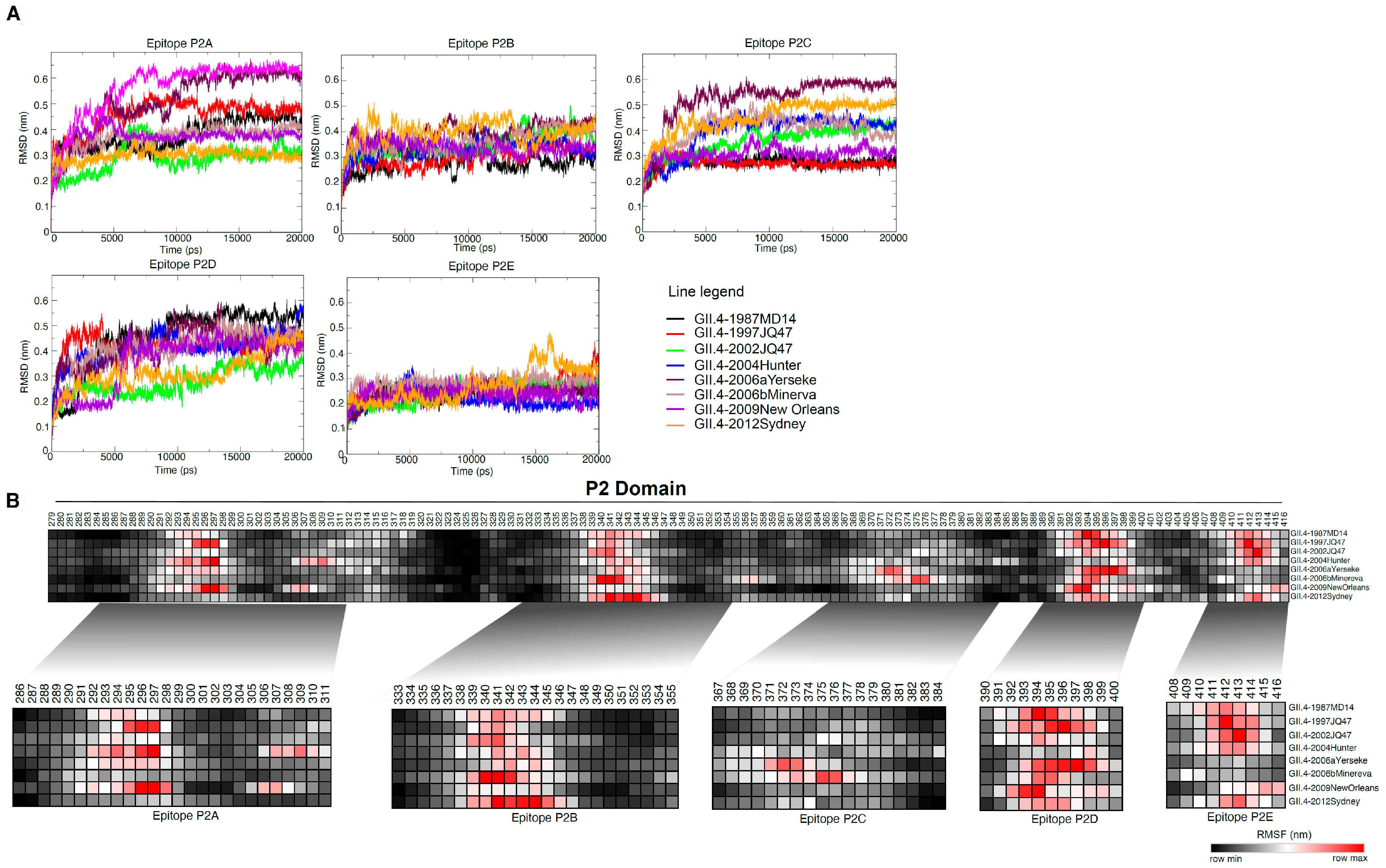
3.3. MD Simulation Revealed Conformational Flexcibilty in the Putative Epitope Sites over Time
3.4. Amino Acid Substitutions in Epitopes P2B, P2C and P2D Affect HBGA Binding
3.5. Epitopes P2B, P2C and P2D Elicited Blocking Antibodies
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hall, A.J.; Glass, R.I.; Parashar, U.D. New insights into the global burden of noroviruses and opportunities for prevention. Expert Rev. Vaccine. 2016, 15, 949–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; De Silva, N.R.; Gargouri, N. World Health Organization Global estimates and regional comparisons of the burden of foodborne disease in 2010. PloS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global economic burden of norovirus gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopman, B.A.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The vast and varied global burden of norovirus: Prospects for prevention and control. PloS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef]
- Vinjé, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Kroneman, A.; Vega, E.; Vennema, H.; Vinjé, J.; White, P.A.; Hansman, G.; Green, K.; Martella, V.; Katayama, K.; Koopmans, M. Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 2013, 158, 2059–2068. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, P.; de Graaf, M.; Parra, G.I.; Chan, M.C.-W.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H. Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2019, 100, 1393. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Costantini, V.; Swanstrom, J.; Debbink, K.; Donaldson, E.F.; Vinjé, J.; Baric, R.S. Emergence of a norovirus GII. 4 strain correlates with changes in evolving blockade epitopes. J. Virol. 2013, 87, 2803–2813. [Google Scholar] [CrossRef] [Green Version]
- Lindesmith, L.C.; Donaldson, E.F.; LoBue, A.D.; Cannon, J.L.; Zheng, D.-P.; Vinje, J.; Baric, R.S. Mechanisms of GII. 4 norovirus persistence in human populations. PloS Med 2008, 5, e31. [Google Scholar] [CrossRef]
- Bull, R.A.; Eden, J.-S.; Rawlinson, W.D.; White, P.A. Rapid evolution of pandemic noroviruses of the GII. 4 lineage. PloS Pathog. 2010, 6, e1000831. [Google Scholar] [CrossRef]
- Allen, D.J.; Gray, J.J.; Gallimore, C.I.; Xerry, J.; Iturriza-Gómara, M. Analysis of amino acid variation in the P2 domain of the GII-4 norovirus VP1 protein reveals putative variant-specific epitopes. PLoS ONE 2008, 3, e1485. [Google Scholar] [CrossRef] [PubMed]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary dynamics of GII. 4 noroviruses over a 34-year period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus GII. 4 strain antigenic variation. J. Virol. 2011, 85, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.J.; Noad, R.; Samuel, D.; Gray, J.J.; Roy, P.; Iturriza-Gómara, M. Characterisation of a GII-4 norovirus variant-specific surface-exposed site involved in antibody binding. Virol. J. 2009, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Zakikhany, K.; Allen, D.J.; Brown, D.; Iturriza-Gómara, M. Molecular evolution of GII-4 Norovirus strains. PLoS. ONE. 2012, 7, e41625. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, E.F.; Lindesmith, L.C.; Lobue, A.D.; Baric, R.S. Norovirus pathogenesis: Mechanisms of persistence and immune evasion in human populations. Immunol. Rev. 2008, 225, 190–211. [Google Scholar] [CrossRef]
- Klepeis, J.L.; Lindorff-Larsen, K.; Dror, R.O.; Shaw, D.E. Long-timescale molecular dynamics simulations of protein structure and function. Curr. Opin. Struct. Biol. 2009, 19, 120–127. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting linear B-cell epitopes using string kernels. J. Mol. Recognit. 2008, 21, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M.; Matsushima, Y.; Motoya, T.; Sakon, N.; Shigemoto, N.; Okamoto-Nakagawa, R.; Nishimura, K.; Yamashita, Y.; Kuroda, M.; Saruki, N. Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 2016, 6, 29400. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Wu, D.; Xu, T.; Wang, X.; Xu, X.; Tao, L.; Li, Y.; Cao, Z.-W. SEPPA: A computational server for spatial epitope prediction of protein antigens. Nucleic Acids Res. 2009, 37, W612–W616. [Google Scholar] [CrossRef] [PubMed]
- Ponomarenko, J.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar] [CrossRef] [Green Version]
- Kolaskar, A.; Tongaonkar, P.C. A semi-empirical method for prediction of antigenic determinants on protein antigens. Febs Lett. 1990, 276, 172–174. [Google Scholar] [CrossRef] [Green Version]
- Emini, E.A.; Hughes, J.V.; Perlow, D.; Boger, J. Induction of hepatitis A virus-neutralizing antibody by a virus-specific synthetic peptide. J. Virol. 1985, 55, 836–839. [Google Scholar] [CrossRef] [Green Version]
- Karplus, P.; Schulz, G. Prediction of chain flexibility in proteins. Naturwissenschaften 1985, 72, 212–213. [Google Scholar] [CrossRef]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Mian, I.S.; Bradwell, A.R.; Olson, A.J. Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991, 217, 133–151. [Google Scholar] [CrossRef]
- Huang, P.; Farkas, T.; Marionneau, S.; Zhong, W.; Ruvoën-Clouet, N.; Morrow, A.L.; Altaye, M.; Pickering, L.K.; Newburg, D.S.; LePendu, J. Noroviruses bind to human ABO, Lewis, and secretor histo-blood group antigens: Identification of 4 distinct strain-specific patterns. J. Infect. Dis. 2003, 188, 19–31. [Google Scholar] [CrossRef]
- Brinckerhoff, L.H.; Kalashnikov, V.V.; Thompson, L.W.; Yamshchikov, G.V.; Pierce, R.A.; Galavotti, H.S.; Engelhard, V.H.; Slingluff, C.L., Jr. Terminal modifications inhibit proteolytic degradation of an immunogenic mart-127–35 peptide: Implications for peptide vaccines. Int. J. Cancer 1999, 83, 326–334. [Google Scholar] [CrossRef]
- Powell, M.F.; Grey, H.; Gaeta, F.; Sette, A.; Colón, S. Peptide stability in drug development: A comparison of peptide reactivity in different biological media. J. Pharm. Sci. 1992, 81, 731–735. [Google Scholar] [CrossRef]
- Belyakov, I.M.; Derby, M.A.; Ahlers, J.D.; Kelsall, B.L.; Earl, P.; Moss, B.; Strober, W.; Berzofsky, J.A. Mucosal immunization with HIV-1 peptide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl. Acad. Sci. 1998, 95, 1709–1714. [Google Scholar] [CrossRef] [Green Version]
- Ahlers, J.D.; Takeshita, T.; Pendleton, C.D.; Berzofsky, J.A. Enhanced immunogenicity of HIV-1 vaccine construct by modification of the native peptide sequence. Proc. Natl. Acad. Sci. 1997, 94, 10856–10861. [Google Scholar] [CrossRef] [Green Version]
- Afridi, S.Q.; Moeini, H.; Kalali, B.; Wettengel, J.M.; Quitt, O.; Semper, R.; Gerhard, M.; Protzer, U.; Hoffmann, D. Quantitation of norovirus-specific IgG before and after infection in immunocompromised patients. Braz. J. Microbiol. 2019, 1–5. [Google Scholar] [CrossRef]
- Tamminen, K.; Huhti, L.; Koho, T.; Lappalainen, S.; Hytönen, V.P.; Vesikari, T.; Blazevic, V. A comparison of immunogenicity of norovirus GII-4 virus-like particles and P-particles. Immunology 2012, 135, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Lobanov, M.Y.; Bogatyreva, N.; Galzitskaya, O. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Lindesmith, L.C.; LoBue, A.D.; Baric, R.S. Viral shape-shifting: Norovirus evasion of the human immune system. Nat. Reviews. Microbiol. 2010, 8, 231. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Debbink, K.; Swanstrom, J.; Vinjé, J.; Costantini, V.; Baric, R.S.; Donaldson, E.F. Monoclonal antibody-based antigenic mapping of norovirus GII. 4-2002. J. Virol. 2012, 86, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic mapping of a highly variable norovirus GII. 4 blockade epitope: Potential role in escape from human herd immunity. J. Virol. 2012, 86, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Lindesmith, L.; Moe, C.; Marionneau, S.; Ruvoen, N.; Jiang, X.; Lindblad, L.; Stewart, P.; LePendu, J.; Baric, R. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 2003, 9, 548. [Google Scholar] [CrossRef]
- Koelle, K.; Cobey, S.; Grenfell, B.; Pascual, M. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 2006, 314, 1898–1903. [Google Scholar] [CrossRef]
- Koelle, K.; Ratmann, O.; Rasmussen, D.A.; Pasour, V.; Mattingly, J. A dimensionless number for understanding the evolutionary dynamics of antigenically variable RNA viruses. Proc. R. Soc. B: Biol. Sci. 2011, 278, 3723–3730. [Google Scholar] [CrossRef] [Green Version]
- Harrington, P.R.; Lindesmith, L.; Yount, B.; Moe, C.L.; Baric, R.S. Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J. Virol. 2002, 76, 12335–12343. [Google Scholar] [CrossRef] [Green Version]
- Cannon, J.L.; Lindesmith, L.C.; Donaldson, E.F.; Saxe, L.; Baric, R.S.; Vinjé, J. Herd immunity to GII. 4 noroviruses is supported by outbreak patient sera. J. Virol. 2009, 83, 5363–5374. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Fang, P.; Chachiyo, T.; Xia, M.; Huang, P.; Fang, Z.; Jiang, W.; Jiang, X. Noroviral P particle: Structure, function and applications in virus–host interaction. Virology 2008, 382, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LoBue, A.D.; Lindesmith, L.; Yount, B.; Harrington, P.R.; Thompson, J.M.; Johnston, R.E.; Moe, C.L.; Baric, R.S. Multivalent norovirus vaccines induce strong mucosal and systemic blocking antibodies against multiple strains. Vaccine 2006, 24, 5220–5234. [Google Scholar] [CrossRef] [PubMed]
- Herbst-Kralovetz, M.; Mason, H.S.; Chen, Q. Norwalk virus-like particles as vaccines. Expert Rev. Vaccines 2010, 9, 299–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanker, S.; Choi, J.; Sankaran, B.; Atmar, R.; Estes, M.; Prasad, B. Structural Analysis of HBGA Binding Specificity in a Norovirus GII. 4 Epidemic Variant: 632 Implications for Epochal Evolution. J. Virol. 2011, 85, 8635–8645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, S.; Lou, Z.; Tan, M.; Chen, Y.; Liu, Y.; Zhang, Z.; Zhang, X.C.; Jiang, X.; Li, X.; Rao, Z. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 2007, 81, 5949–5957. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.; Huang, P.; Xia, M.; Fang, P.-A.; Zhong, W.; McNeal, M.; Wei, C.; Jiang, W.; Jiang, X. Norovirus P particle, a novel platform for vaccine development and antibody production. J. Virol. 2011, 85, 753–764. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moeini, H.; Afridi, S.Q.; Donakonda, S.; Knolle, P.A.; Protzer, U.; Hoffmann, D. Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies. Vaccines 2021, 9, 52. https://doi.org/10.3390/vaccines9010052
Moeini H, Afridi SQ, Donakonda S, Knolle PA, Protzer U, Hoffmann D. Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies. Vaccines. 2021; 9(1):52. https://doi.org/10.3390/vaccines9010052
Chicago/Turabian StyleMoeini, Hassan, Suliman Qadir Afridi, Sainitin Donakonda, Percy A. Knolle, Ulrike Protzer, and Dieter Hoffmann. 2021. "Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies" Vaccines 9, no. 1: 52. https://doi.org/10.3390/vaccines9010052
APA StyleMoeini, H., Afridi, S. Q., Donakonda, S., Knolle, P. A., Protzer, U., & Hoffmann, D. (2021). Linear B-Cell Epitopes in Human Norovirus GII.4 Capsid Protein Elicit Blockade Antibodies. Vaccines, 9(1), 52. https://doi.org/10.3390/vaccines9010052