
Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy


Abstract
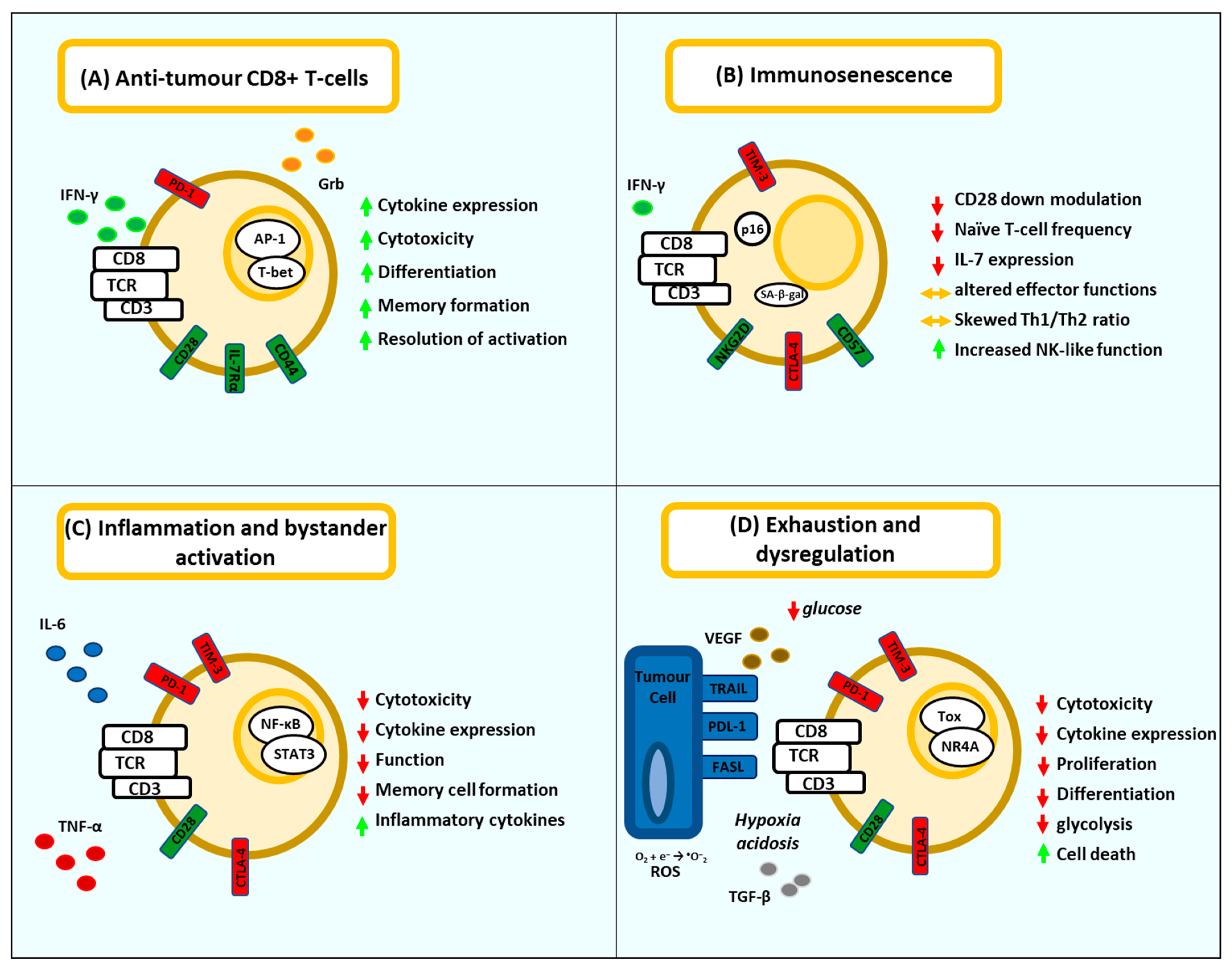
:1. Introduction
2. T-Cell Dysfunction
3. Directing Anti-Tumour T-Cell Responses
4. T-Cell Co-Stimulation

{kind=link}
{kind=link}
{kind=link}
| Co Stimulation | Effect on T-Cells | Questions | Ref |
|---|---|---|---|
| IL-7 | Mitochondrial biogenesis metabolically reprograming T-cells to utilise fatty acid oxidation. improves the vaccine induced anti-tumour response and survival. Increasing the IFN-γ production and cytotoxicity of CD8+ T-cells | IL-7 may not have therapeutic efficacy when used alone and thus requires consideration regarding which immunotherapies may be amplified by IL-7. | [63,64,65] |
| IL-15 | Promising responses in combination with anti-PD-1 therapy in early clinical trial | IL-15 administered as monotherapy proved ineffective with increases in CD8+ T-cells but no objective responses. poor pharmacokinetics need to be addressed. | [69,70,72] |
| IL-21 | Promote the generation of effector T-cells from naïve precursors, enhancing expansion and supporting the development of cytotoxic effector function, may demonstrate efficacy as a single agent | Proliferative responses to IL-21 may be associated with reduced IFN-γ expression. Not yet studied in clinical trials in combination with ICB | [73,79] |
| NKTR | Can induce durable anti-tumour T-cell responses and demonstrates efficacy in poorly immunogenic tumours | Efficacy needs to be demonstrated in a multiarmed trial which is underway | [82,83] |
| Anti-1-44B | Increased cytokine expression and prolonged survival, suppression of tumour growth | Low response rate and potential toxicity need to be addressed | [87,88] |
| Anti-OX40 | Promotes T-cell expansion and survival via enhanced expression of IL-2 and anti-apoptotic proteins such as BCL-2 and promotes the generation of memory T-cells and inhibition of T-regs | Low objective response rate in clinical trials. The need to carefully time administration of OX40 agonist before ICB therapy. | [93,94,95,96,97,98,99] |
| Anti-GITR | Enhances T-lymphocyte activity after suboptimal TCR stimulation by upregulating IL-2 and IFN-γ and enhances T-cell survival by inhibiting TCR activation induced apoptosis. | Low response rate demonstrated in early trials | [101,103,106] |
| Anti-ICOS | Proliferation and cytokine expression in T-cells whilst suppressing T-reg function | A modest patient response in clinical trials thus far | [107,108] |
5. The Gut Microbiome and Dietary Interventions
| T-Cell Signalling | Effect on T-Cells | Questions | Ref |
|---|---|---|---|
| PTPN22 inhibition | small molecule inhibitor of PTPN22, named L-1 has promoted anti-tumour immune responses dependent upon the activation of CD8+ T-cells | Not yet studied in clinical trials | [162] |
| Cholesterol metabolism | Increasing cholesterol availability by blocking ACAT-1 with avasimibe leads to potentiated effector function and enhanced proliferation of CD8+ T-cells | Not yet studied in clinical trials | [164,165] |
| SHP-1 inhibition | inhibition of SHP-1 using the PTP inhibitor sodium stibogluconate results in increased T-cell degranulation and cytotoxicity | Increasing the function T-regs, potential off target effects and autoimmune toxicities | [166,167] |
| SHP-2 | Inhibition of Src Homology Region 2-Containing Protein Tyrosine Phosphatase (SHP)-2 within NSCLC tumours using SHP099 results in increased TIL | Potential to increase intratumoural MDSC via increased production of CXCR2 ligands | [195] |
| Diacylglycerol kinase-RAS | Pharmacological DGKα targeting restores cytotoxic function of chimeric antigen receptor and CD8+ T-cells isolated from solid tumours, suggesting a mechanism to reverse T-cell exhausted phenotypes. | Careful use of DGKα blockade will be required to prevent the inhibition of effector T-cell responses via MAPK pathway inhibition | [194] |
| MEK inhibition | Trametinib upregulate tumour surface expression of MHC and PD-L1 in TNBC cells, resulting in increased TIL in a murine model of breast cancer. Combining MEK inhibition with PD-L1/PD-1 ICB demonstrated enhanced anti-tumour immune responses | MEK inhibition adversely effects of TIL frequency, proliferation and cytokine expression | [175,178,180] |
| P38 inhibition | Pharmacological inhibition of p38 using BIRB796 increased cell expansion and memory while reducing oxidative and genomic stress, improving the efficacy of murine anti-tumour T-cells | Effect on tumour cells is not clear and inhibition may have a protumorigenic effect. | [189,190,191,192,193] |
| Rapalogs | Can increase memory CD8+ T-cells and enhance anti-tumour T-cell based immune responses | May reduce the function of effector T-cells and promote expansion of regulatory CD4+ T-cells. The sequence of mTOR inhibition relative to vaccination or use of ICB is yet to be fully understood. | [197,198,199,200,201,202,203,204,205,206,207,208,209,210] |
| Vitamin D | Regulates T-cell activation, proliferation and cytokine expression | How effective is the therapeutic use of vitamin D? | [212,225,226,227] |
| All trans Retinoic acid | Pleiotropic modulation of Cd4+ T-cell differentiation and priming effect on CD8+ T-cells | Delivery of ATRA (systemic or intratumourol?) and potential as a combination with ICB need further study | [228,229,230,231,232,233,234,235,236,237,238] |
6. Mitochondrial Function
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Arruga, F.; Gyau, B.B.; Iannello, A.; Vitale, N.; Vaisitti, T.; Deaglio, S. Immune response dysfunction in chronic lymphocytic leukemia: Dissecting molecular mechanisms and microenvironmental conditions. Int. J. Mol. Sci. 2020, 21, 1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: A key player in cancer development. J. Hematol. Oncol. 2020, 13, 151. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Tan, Y.; Chen, X.; Liu, Y.; Ning, L.; Chen, L.; Li, M.; Liu, Y.; Wang, G.; et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front. Immunol. 2020, 11, 827. [Google Scholar] [CrossRef] [PubMed]
- Kusnadi, A.; Ramírez-Suástegui, C.; Fajardo, V.; Chee, S.J.; Meckiff, B.J.; Simon, H.; Pelosi, E.; Seumois, G.; Ay, F.; Vijayanand, P.; et al. Severely ill COVID-19 patients display impaired exhaustion features in SARS-CoV-2-reactive CD8+ T cells. Sci. Immunol. 2021, 6, eabe4782. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Seo, W.; Jerin, C.; Nishikawa, H. Transcriptional regulatory network for the establishment of CD8+ T cell exhaustion. Exp. Mol. Med. 2021, 53, 202–209. [Google Scholar] [CrossRef]
- Schietinger, A.; Philip, M.; Krisnawan, V.E.; Chiu, E.Y.; Delrow, J.J.; Basom, R.S.; Lauer, P.; Brockstedt, D.G.; Knoblaugh, S.E.; Hämmerling, G.J.; et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 2016, 45, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and novel combinations in the treatment of melanoma-An update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Shrimali, R.K.; Ahmad, S. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauken, K.E.; Sammons, M.A.; Odorizzi, P.M.; Manne, S.; Godec, J.; Khan, O.; Drake, A.M.; Chen, Z.; Sen, D.R.; Kurachi, M.; et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 2016, 354, 1160–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415, Erratum in 2019, 116, 19761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freen-van Heeren, J.J. Using CRISPR to enhance T cell effector function for therapeutic applications. Cytokine 2020, 3, 100049. [Google Scholar] [CrossRef]
- Flores, C.; Fouquet, G.; Moura, I.C.; Maciel, T.T.; Hermine, O. Lessons to learn from low-dose cyclosporin-a: A new approach for unexpected clinical applications. Front. Immunol. 2019, 10, 588. [Google Scholar] [CrossRef] [Green Version]
- Borgoni, S.; Kudryashova, K.S.; Burka, K.; de Magalhães, J.P. Targeting immune dysfunction in aging. Ageing Res. Rev. 2021, 70, 101410. [Google Scholar] [CrossRef]
- Thompson, H.L.; Smithey, M.J.; Surh, C.D.; Nikolich-Žugich, J. Functional and homeostatic impact of age-Related changes in lymph node stroma. Front. Immunol. 2017, 8, 706. [Google Scholar] [CrossRef] [Green Version]
- Lazuardi, L.; Jenewein, B.; Wolf, A.M.; Pfister, G.; Tzankov, A.; Grubeck-Loebenstein, B. Age-related loss of naive T cells and dysregulation of T-cell/B-cell interactions in human lymph nodes. Immunology 2005, 114, 37–43. [Google Scholar] [CrossRef]
- Cusi, M.G.; Martorelli, B.; Di Genova, G.; Terrosi, C.; Campoccia, G.; Correale, P. Age related changes in T cell mediated immune response and effector memory to respiratory syncytial virus (RSV) in healthy subjects. Immun. Ageing 2010, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Saurwein-Teissl, M.; Lung, T.L.; Marx, F.; Gschösser, C.; Asch, E.; Blasko, I.; Parson, W.; Böck, G.; Schönitzer, D.; Trannoy, E.; et al. Lack of antibody production following immunization in old age: Association with CD8(+)CD28(−) T cell clonal expansions and an imbalance in the production of Th1 and Th2 cytokines. J. Immunol. 2002, 168, 5893–5899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro-Garcia, M.A.; Alonso-Arias, R.; Lopez-Larrea, C. When aging reaches CD4+ T-cells: Phenotypic and functional changes. Front. Immunol. 2013, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, K.M.; Fox, A.; Harland, K.L.; Russ, B.E.; Li, J.; Nguyen, T.H.O.; Loh, L.; Olshanksy, M.; Naeem, H.; Tsyganov, K.; et al. Age-related decline in primary CD8(+) T cell responses is associated with the development of senescence in virtual memory CD8(+) T cells. Cell Rep. 2018, 23, 3512–3524. [Google Scholar] [CrossRef]
- Decman, V.; Laidlaw, B.J.; DiMenna, L.; Abdulla, S.; Mozdzanowska, K.; Erikson, J.; Ertl, H.C.J.; Wherry, E.J. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol. 2010, 184, 5151–5159. [Google Scholar] [CrossRef]
- Sceneay, J.; Goreczny, G.J.; Wilson, K.; Morrow, S.; DeCristo, M.J.; Ubellacker, J.M.; Qin, Y.; Laszewski, T.; Stover, D.G.; Barrera, V.; et al. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative breast cancer. Cancer Discov. 2019, 9, 1208–1227. [Google Scholar] [CrossRef] [PubMed]
- Ladomersky, E.; Zhai, L.; Lauing, K.L.; Bell, A.; Xu, J.; Kocherginsky, M.; Zhang, B.; Wu, J.D.; Podojil, J.R.; Platanias, L.C.; et al. Advanced age increases immunosuppression in the brain and decreases immunotherapeutic efficacy in subjects with glioblastoma. Clin. Cancer Res. 2020, 26, 5232–5245. [Google Scholar] [CrossRef] [PubMed]
- Baldini, C.; Romano, P.M.; Voisin, A.-L.; Danlos, F.-X.; Champiat, S.; Laghouati, S.; Kfoury, M.; Vincent, H.; Postel-Vinay, S.; Varga, A.; et al. Impact of aging on immune-related adverse events generated by anti-programmed death (ligand)PD-(L)1 therapies. Eur. J. Cancer 2020, 129, 71–79. [Google Scholar] [CrossRef]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [Green Version]
- Stelekati, E.; Shin, H.; Doering, T.A.; Dolfi, D.V.; Ziegler, C.G.; Beiting, D.P.; Dawson, L.; Liboon, J.; Wolski, D.; Ali, M.A.; et al. Bystander chronic infection negatively impacts development of CD8(+) T cell memory. Immunity 2014, 40, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Barnstorf, I.; Borsa, M.; Baumann, N.; Pallmer, K.; Yermanos, A.; Joller, N.; Spörri, R.; Welten, S.; Kräutler, N.J.; Oxenius, A. Chronic virus infection compromises memory bystander T cell function in an IL-6/STAT1-dependent manner. J. Exp. Med. 2019, 216, 571–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoni, A.; Salvati, L.; Maggi, L.; Capone, M.; Vanni, A.; Spinicci, M.; Mencarini, J.; Caporale, R.; Peruzzi, B.; Antonelli, A.; et al. Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J. Clin. Investig. 2020, 130, 4694–4703. [Google Scholar] [CrossRef]
- Graham, J.B.; Swarts, J.L.; Leist, S.R.; Schäfer, A.; Menachery, V.D.; Gralinski, L.E.; Jeng, S.; Miller, D.R.; Mooney, M.A.; McWeeney, S.K.; et al. Baseline T cell immune phenotypes predict virologic and disease control upon SARS-CoV infection in Collaborative Cross mice. PLoS Pathog. 2021, 17, e1009287. [Google Scholar] [CrossRef]
- Cope, A.P. Studies of T-cell activation in chronic inflammation. Arthritis Res. Ther. 2002, 4 (Suppl. 3), S197–S211. [Google Scholar] [CrossRef] [PubMed]
- Érsek, B.; Molnár, V.; Balogh, A.; Matko, J.; Cope, A.; Buzás, E.; Falus, A.; Nagy, G. CD3ζ-chain expression of human T lymphocytes is regulated by TNF via Src-like adaptor protein-dependent proteasomal degradation. J. Immunol. 2012, 189, 1602–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnstorf, I.; Welten, S.; Borsa, M.; Baumann, N.S.; Pallmer, K.; Joller, N.; Spörri, R.; Oxenius, A. Chronic viral infections impinge on naive bystander CD8 T cells. Immun. Inflamm. Dis. 2020, 8, 249–257. [Google Scholar] [CrossRef]
- Laidlaw, B.J.; Cui, W.; Amezquita, R.A.; Gray, S.M.; Guan, T.; Lu, Y.; Kobayashi, Y.; Flavell, R.A.; Kleinstein, S.H.; Craft, J.; et al. Production of IL-10 by CD4(+) regulatory T cells during the resolution of infection promotes the maturation of memory CD8(+) T cells. Nat. Immunol. 2015, 16, 871–879. [Google Scholar] [CrossRef]
- Ferreira, C.; Barros, L.; Baptista, M.; Blankenhaus, B.; Barros, A.; Figueiredo-Campos, P.; Konjar, Š.; Lainé, A.; Kamenjarin, N.; Stojanovic, A.; et al. Type 1 Treg cells promote the generation of CD8+ tissue-resident memory T cells. Nat. Immunol. 2020, 21, 766–776. [Google Scholar] [CrossRef]
- Naing, A.; Papadopoulos, K.; Infante, J.; Wong, D.J.L.; Autio, K.A.; Ott, P.A.; Falchook, G.S.; Patel, M.R.; Pant, S.; Patniak, A.; et al. Clinical activity and safety of pegylated human IL-10 (AM0010) in combination with anti-PD1. J. Clin. Oncol. 2016, 34, 3018. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M.; Shih, B.B.; Trahair, H.; Subramanian, P.; Devine, O.P.; Glanville, J.; Gilroy, D.; Rustin, M.H.A.; Freeman, T.C.; et al. Recruitment of inflammatory monocytes by senescent fibroblasts inhibits antigen-specific tissue immunity during human aging. Nat Aging 2021, 1, 101–113. [Google Scholar] [CrossRef]
- Pelly, V.S.; Moeini, A.; Roelofsen, L.M.; Bonavita, E.; Bell, C.R.; Hutton, C.; Blanco-Gomez, A.; Banyard, A.; Bromley, C.P.; Flanagan, E.; et al. Anti-inflammatory drugs remodel the tumor immune environment to enhance immune checkpoint blockade efficacy. Cancer Discov. 2021, 24, 2602–2619. [Google Scholar] [CrossRef]
- Wang, D.Y.; McQuade, J.L.; Rai, R.R.; Park, J.; Zhao, S.; Ye, F.; Beckermann, K.E.; Rubinstein, S.M.; Johnpulle, R.; Long, G.; et al. The Impact of nonsteroidal anti-inflammatory drugs, beta blockers, and metformin on the efficacy of anti-PD-1 therapy in advanced melanoma. Oncologist 2020, 25, e602–e605. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, H.; Chen, S.; Li, Z.; Chen, J.; Li, W. The effect of concomitant use of statins, NSAIDs, low-dose aspirin, metformin and beta-blockers on outcomes in patients receiving immune checkpoint inhibitors: A systematic review and meta-analysis. Oncoimmunology 2021, 10, 1957605. [Google Scholar] [CrossRef]
- Huemer, H.P. Possible immunosuppressive effects of drug exposure and environmental and nutritional effects on infection and vaccination. Mediat. Inflamm. 2015, 2015, 349176. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Tian, W.; Zhao, F.; Li, M.; Ye, Q.; Wei, Y.; Li, T.; Xie, K. Systemic immune-inflammation index, SII, for prognosis of elderly patients with newly diagnosed tumors. Oncotarget 2018, 9, 35293–35299. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Han, X.; Zha, H.; Tao, H.; Li, X.; Yuan, F.; Chen, G.; Wang, L.; Ma, J.; Hu, Y. Systemic immune-inflammation index and changes of neutrophil-lymphocyte ratio as prognostic biomarkers for patients with pancreatic cancer treated with immune checkpoint blockade. Front. Oncol. 2021, 11, 585271. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Zhang, S.; Tao, H.; Li, X.; Yuan, F.; Chen, G.; Wang, L.; Ma, J.; Hu, Y. Systemic immune-inflammation index, neutrophil-to-lymphocyte ratio, platelet-to-lymphocyte ratio can predict clinical outcomes in patients with metastatic non-small-cell lung cancer treated with nivolumab. J. Clin. Lab. Anal. 2019, 33, e22964. [Google Scholar] [CrossRef] [PubMed]
- Kauffmann-Guerrero, D.; Kahnert, K.; Kiefl, R.; Walter, J.; Behr, J.; Tufman, A. Systemic inflammation and pro-inflammatory cytokine profile predict response to checkpoint inhibitor treatment in NSCLC: A prospective study. Sci. Rep. 2021, 11, 10919. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Tsoi, J.; Onyshchenko, M.; Abril-Rodriguez, G.; Ross-Macdonald, P.; Wind-Rotolo, M.; Champhekar, A.; Medina, E.; Torrejon, D.Y.; Shin, D.S.; et al. Conserved interferon-γ signaling drives clinical response to immune checkpoint blockade therapy in melanoma. Cancer Cell 2020, 38, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, Z.; Chen, L. Memory T cells: Strategies for optimizing tumor immunotherapy. Protein Cell 2020, 11, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, J.; Zeng, H.; Horng, T. Metabolism as a guiding force for immunity. Nat. Cell Biol. 2019, 21, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, M.; Zhang, B.; Sugiura, Y.; Sonomura, K.; Guerrini, M.M.; Tsutsui, Y.; Maruya, M.; Vogelzang, A.; Chamoto, K.; Honda, K.; et al. Metabolic shift induced by systemic activation of T cells in PD-1-deficient mice perturbs brain monoamines and emotional behavior. Nat. Immunol. 2017, 18, 1342–1352. [Google Scholar] [CrossRef]
- Inomata, M.; Kado, T.; Okazawa, S.; Imanishi, S.; Taka, C.; Kambara, K.; Hirai, T.; Tanaka, H.; Tokui, K.; Hayashi, K.; et al. Peripheral PD1-positive CD4 T-lymphocyte count can predict progression-free survival in patients with non-small cell lung cancer receiving immune checkpoint inhibitor. Anticancer Res. 2019, 39, 6887–6893. [Google Scholar] [CrossRef]
- Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Jansen, C.S.; Prokhnevska, N.; Master, V.A.; Sanda, M.G.; Carlisle, J.W.; Bilen, M.A.; Cardenas, M.; et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature 2019, 576, 465–470. [Google Scholar] [CrossRef]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef]
- van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Hinrichs, C.S.; Spolski, R.; Paulos, C.M.; Gattinoni, L.; Kerstann, K.W.; Palmer, D.C.; Klebanoff, C.A.; Rosenberg, S.A.; Leonard, W.J.; Restifo, N.P. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood 2008, 111, 5326–5333. [Google Scholar] [CrossRef] [Green Version]
- Cui, G.; Staron, M.M.; Gray, S.M.; Ho, P.C.; Amezquita, R.A.; Wu, J.; Kaech, S.M. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell 2015, 161, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Lee, K.J.; Lee, S.W. Cancer immunotherapy with T-cell targeting cytokines: IL-2 and IL-7. BMB Rep. 2021, 54, 21–30. [Google Scholar] [CrossRef]
- Pellegrini, M.; Calzascia, T.; Elford, A.; Shahinian, A.; Lin, A.E.; Dissanayake, D.; Dhanji, S.; Nguyen, L.T.; Gronski, M.A.; Morre, M.; et al. Adjuvant IL-7 antagonizes multiple cellular and molecular inhibitory networks to enhance immunotherapies. Nat. Med. 2009, 15, 528–536. [Google Scholar] [CrossRef]
- Zhao, L.; Mei, Y.; Sun, Q.; Guo, L.; Wu, Y.; Yu, X.; Hu, B.; Liu, X.; Liu, H. Autologous tumor vaccine modified with recombinant new castle disease virus expressing IL-7 promotes antitumor immune response. J. Immunol. 2014, 193, 735–745. [Google Scholar] [CrossRef]
- Melchionda, F.; Fry, T.J.; Milliron, M.J.; McKirdy, M.A.; Tagaya, Y.; Mackall, C.L. Adjuvant IL-7 or IL-15 overcomes immunodominance and improves survival of the CD8+ memory cell pool. J. Clin. Investig. 2005, 115, 1177–1187. [Google Scholar] [CrossRef] [Green Version]
- Sportes, C.; Hakim, F.T.; Memon, S.A.; Zhang, H.; Chua, K.S.; Brown, M.R.; Fleisher, T.A.; Krumlauf, M.C.; Babb, R.R.; Chow, C.K.; et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 2008, 205, 1701–1714. [Google Scholar] [CrossRef]
- Tredan, O.; Menetrier-Caux, C.; Ray-Coquard, I.; Garin, G.; Cropet, C.; Verronèse, E.; Bachelot, T.; Rebattu, P.; Heudel, P.E.; Cassier, P.; et al. ELYPSE-7: A randomized placebo-controlled phase IIa trial with CYT107 exploring the restoration of CD4+ lymphocyte count in lymphopenic metastatic breast cancer patients. Ann. Oncol. 2015, 26, 1353–1362. [Google Scholar] [CrossRef]
- Robinson, T.O.; Schluns, K.S. The potential and promise of IL-15 in immuno-oncogenic therapies. Immunol. Lett. 2017, 190, 159–168. [Google Scholar] [CrossRef]
- Sneller, M.C.; Kopp, W.C.; Yovandich, J.L.; Creekmore, S.P.; Waldmann, T.A.; Lane, H.C. IL-15 administered by continuous infusion to rhesus macaques induces massive expansion of CD8+ T effector memory population in peripheral blood. Blood 2011, 118, 6845–6848. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Morishima, C.; McNeel, D.G.; Patel, M.R.; Kohrt, H.E.K.; Thompson, J.A.; Sondel, P.M.; Wakelee, H.A.; Disis, M.L.; Kaiser, J.C.; et al. A first-in-human phase I study of subcutaneous outpatient recombinant human IL15 (rhIL15) in adults with advanced solid tumors. Clin. Cancer Res. 2018, 24, 1525–1535. [Google Scholar] [CrossRef] [Green Version]
- Waldmann, T.A.; Dubois, S.; Miljkovic, M.D.; Conlon, K.C. IL-15 in the combination immunotherapy of cancer. Front. Immunol. 2020, 11, 868. [Google Scholar] [CrossRef]
- Zhao, M.; Luo, M.; Xie, Y.; Jiang, H.; Cagliero, C.; Li, N.; Ye, H.; Wu, M.; Hao, S.; Sun, T.; et al. Development of a recombinant human IL-15. SIL-15Rα/Fc superagonist with improved half-life and its antitumor activity alone or in combination with PD-1 blockade in mouse model. Biomed. Pharmacother. 2019, 112, 10867. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: A non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef]
- Casey, K.A.; Mescher, M.F. IL-21 promotes differentiation of naive CD8 T cells to a unique effector phenotype. J. Immunol. 2007, 178, 7640–7748. [Google Scholar] [CrossRef]
- Ostiguy, V.; Allard, E.L.; Marquis, M.; Leignadier, J.; Labrecque, N. IL-21 promotes T lymphocyte survival by activating the phosphatidylinositol-3 kinase signaling cascade. J. Leukoc. Biol. 2007, 82, 645–656. [Google Scholar] [CrossRef]
- Zeng, R.; Spolski, R.; Finkelstein, S.E.; Oh, S.; Kovanen, P.E.; Hinrichs, C.S.; Pise-Masison, C.A.; Radonovich, M.F.; Brady, J.N.; Restifo, N.P.; et al. Synergy of IL-21 and IL-15 in Regulating CD8+ T Cell Expansion and Function. J. Exp. Med. 2005, 201, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Zhang, X.; Chibbar, R.; Freyqald, A.; Tikoo, S.; Zheng, C.; Xiang, J. Transgene IL-21-engineered T cell-based vaccine potently converts CTL exhaustion via the activation of the mTORC1 pathway in chronic infection. World, J. Vaccines 2019, 9, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Cong, Y.; Jia, M.; He, Q.; Zhong, H.; Zhao, Y.; Li, H.; Yan, M.; You, J.; Liu, J.; et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 2021, 12, 951. [Google Scholar] [CrossRef]
- Frohlich, A.; Kisielow, J.; Schmitz, I.; Freigang, S.; Shamshiev, A.T.; Weber, J.; Marsland, B.J.; Oxenius, A.; Kopf, M. IL-21R on T Cells Is Critical for Sustained Functionality and Control of Chronic Viral Infection. Science 2009, 324, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Petrella, T.M.; Tozer, R.; Belanger, K.; Savage, K.J.; Wong, R.; Smylie, M.; Kamel-Reid, S.; Tron, V.; Chen, B.E.; Hunder, N.N.; et al. Interleukin-21 has activity in patients with metastatic melanoma: A phase II study. J. Clin. Oncol. 2012, 30, 3396–3401. [Google Scholar] [CrossRef]
- Bolesta, E.; Kowalczyk, A.; Wierzbicki, A.; Eppolito, C.; Kaneko, Y.; Takiguchi, M.; Stamatatos, L.; Shrikant, P.A.; Kozbor, D. Increased level and longevity of protective immune responses induced by DNA vaccine expressing the HIV-1 Env glycoprotein when combined with IL-21 and IL-15 gene delivery. J. Immunol. 2006, 177, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.Z.; Fan, C.W.; Lu, R.; Shao, B.; Sang, Y.X.; Huang, Q.R.; Li, X.; Meng, W.T.; Mo, X.M.; Wei, Y.Q. Forced co-expression of IL-21 and IL-7 in whole-cell cancer vaccines promotes antitumor immunity. Sci. Rep. 2016, 6, 32351. [Google Scholar] [CrossRef]
- Pieper, A.A.; Rakhmilevich, A.L.; Spiegelman, D.V.; Patel, R.B.; Birstler, J.; Jin, W.J.; Carlson, P.M.; Charych, D.H.; Hank, J.A.; Erbe, A.K.; et al. Combination of radiation therapy, bempegaldesleukin, and checkpoint blockade eradicates advanced solid tumors and metastases in mice. J. Immunother. Cancer 2021, 9, e002715. [Google Scholar] [CrossRef]
- Diab, A.; Tannir, N.M.; Bentebibel, S.E.; Hwu, P.; Papadimitrakopoulou, V.; Haymaker, C.; Kluger, H.M.; Gettinger, S.N.; Sznol, M.; Tykodi, S.S.; et al. Bempegaldesleukin (NKTR-214) plus nivolumab in patients with advanced solid tumors: Phase I dose-escalation study of safety, efficacy, and immune activation (PIVOT-02). Cancer Discov. 2020, 10, 1158–1173. [Google Scholar] [CrossRef]
- Thaker, P.H.; Borys, N.; Fewell, J.; Anwer, K. GEN-1 immunotherapy for the treatment of ovarian cancer. Future Oncol. 2019, 15, 421–438. [Google Scholar] [CrossRef]
- Kourko, O.; Seaver, K.; Odoardi, N.; Basta, S.; Gee, K. IL-27, IL-30, and IL-35: A cytokine triumvirate in cancer. Front. Oncol. 2019, 9, 969. [Google Scholar] [CrossRef] [Green Version]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef]
- Gauttier, V.; Judor, J.-P.; Le Guen, V.; Cany, J.; Ferry, N.; Conchon, S. Agonistic anti-CD137 antibody treatment leads to antitumor response in mice with liver cancer. Int. J. Cancer 2014, 135, 2857–2867. [Google Scholar] [CrossRef]
- Segal, N.H.; He, A.R.; Doi, T.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Phase I study of single-agent Utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in patients with advanced cancer. Clin. Cancer Res. 2018, 24, 1816–1823. [Google Scholar] [CrossRef] [Green Version]
- Sznol, M.; Hodi, F.S.; Margolin, K.; McDermott, D.F.; Ernstoff, M.S.; Kirkwood, J.M.; Wojtaszek, C.; Feltquate, D.; Logan, T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (PTS) with advanced cancer (Ca). J. Clin. Oncol. 2008, 26, 3007. [Google Scholar] [CrossRef]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; Levy, R.; Bhatia, S.; Pishvaian, M.J.; Cesari, R.; Chen, Y.; Davis, C.B.; Huang, B.; et al. Results from an integrated safety analysis of Urelumab, an agonist Anti-CD137 monoclonal antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lee, L.-F.; Fisher, T.S.; Jessen, B.; Elliott, M.; Evering, W.; Logronio, K.; Tu, G.H.; Tsaparikos, K.; Li, X.; et al. Combination of 4-1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol. Res. 2015, 3, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Zhao, L.; Hellstrom, I.; Hellstrom, K.E.; Guo, Y. Dual targeting of CD137 co-stimulatory and PD-1 co-inhibitory molecules for ovarian cancer immunotherapy. Oncoimmunology 2014, 3, e28248. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Sznol, M.; Hu-Lieskovan, S.; Papadopoulos, K.P.; Patnaik, A.; Rasco, D.W.; Di Gravio, D.; Huang, B.; Gambhire, D.; Chen, Y.; et al. Phase Ib study of Utomilumab (PF-05082566), a 4-1BB/CD137 agonist, in combination with pembrolizumab (MK-3475) in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 5349–5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol. Rev. 2009, 229, 173–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrison, K.; Hahn, T.; Lee, W.C.; Ling, L.E.; Weinberg, A.D.; Akporiaye, E.T. The small molecule TGF-β signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer Immunol. Immunother. 2012, 61, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruby, C.E.; Yates, M.A.; Hirschhorn-Cymerman, D.; Chlebeck, P.; Wolchok, J.D.; Houghton, A.N.; Offner, H.; Weinberg, A.D. Cutting edge: OX40 agonists can drive regulatory T cell expansion if the cytokine milieu is right. J. Immunol. 2009, 183, 4853–4857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messenheimer, D.J.; Jensen, S.M.; Afentoulis, M.E.; Wegmann, K.W.; Feng, Z.; Friedman, D.J.; Gough, M.J.; Urba, W.J.; Fox, B.A. Timing of PD-1 blockade is critical to effective combination immunotherapy with Anti-OX40. Clin. Cancer Res. 2017, 23, 6165–6177. [Google Scholar] [CrossRef] [Green Version]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. Ox40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [Green Version]
- Glisson, B.S.; Leidner, R.S.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; Schneider, R.; Goel, S.; Ohr, J.P.; Burton, J.; et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 5358–5367. [Google Scholar] [CrossRef]
- Infante, J.R.; Hansen, A.R.; Pishvaian, M.J.; Chow, L.Q.M.; McArthur, G.A.; Bauer, T.M.; Liu, S.V.; Sandhu, S.K.; Tsai, F.Y.-C.; Kim, J.; et al. A phase Ib dose escalation study of the OX40 agonist MOXR0916 and the PD-L1 inhibitor atezolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2016, 34, 101. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Hamid, O.; Thompson, J.A.; Ros, W.; Eskens, F.; Doi, T.; Hu-Leiskovan, S.; Chou, J.; Liao, K.; Ganguly, B.J.; et al. The relationship of pharmacodynamics (PD) and pharmacokinetics (pK) to clinical outcomes in a phase I study of OX40 agonistic monoclonal antibody (mAb) PF-04518600 (PF-8600). J. Clin. Oncol. 2017, 35, 3027. [Google Scholar] [CrossRef]
- Cohen, A.D.; Schaer, D.A.; Liu, C.; Li, Y.; Hirschhorn-Cymmerman, D.; Kim, S.C.; Diab, A.; Rizzuto, G.; Duan, F.; Perales, M.A.; et al. Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation. PLoS ONE 2010, 5, e10436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, B.; Carvajal, R.D.; Marabelle, A.; Patel, S.P.; LoRusso, P.M.; Rasmussen, E.; Juan, G.; Upreti, V.V.; Beers, C.; Ngarmchamnanrith, G.; et al. Dose escalation results from a first-in-human, phase 1 study of glucocorticoid-induced TNF receptor-related protein agonist AMG 228 in patients with advanced solid tumors. J. Immunother. Cancer 2018, 6, 93. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, C.S.; Infante, J.R.; Aljumaily, R.; Naing, A.; Chintakuntlawar, A.V.; Rizvi, N.A.; Ross, H.J.; Gordon, M.; Mallinder, P.R.; Elgeioushi, N.; et al. A phase I study of MEDI1873, a novel GITR agonist, in advanced solid tumors. Ann. Oncol. 2018, 29, viii411. [Google Scholar] [CrossRef]
- Lu, L.; Xu, X.; Zhang, B.; Zhang, R.; Ji, H.; Wang, X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J. Transl. Med. 2014, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Geva, R.; Voskoboynik, M.; Dobrenkov, K.; Mayawala, K.; Gwo, J.; Wnek, R.; Chartash, E.; Long, G.V. First-in-human phase 1 study of MK-1248, an anti-glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors. Cancer 2020, 126, 4926–4935. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Autio, K.; Golan, T.; Dobrenkov, K.; Chartash, E.; Chen, Q.; Wnek, R.; Long, G.V. Phase I Study of MK-4166, an Anti-human Glucocorticoid-Induced TNF Receptor Antibody, Alone or with Pembrolizumab in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 1904–1911. [Google Scholar] [CrossRef]
- Michaelson, J.S.; Harvey, C.; Elpek, K.; Duong, E.; Shallberg, L.; Wallace, M.; Mabry, R.; Shu, J.; Deshpande, A.; Zi, T.; et al. Abstract SY03-02: Preclinical assessment of JTX-2011, an agonist antibody targeting ICOS, supports evaluation in iconic clinical trial. Cancer Res. 2017, 77, SY03-02. [Google Scholar] [CrossRef]
- Yap, T.A.; Burris, H.A.; Kummar, S.; Falchook, G.S.; Pachynski, R.K.; LoRusso, P.; Tykodi, S.S.; Gibney, G.T.; Gainor, J.F.; Rahma, O.E.; et al. Iconic: Biologic and clinical activity of first in class ICOS agonist antibody JTX-2011 +/− nivolumab (nivo) in patients (PTS) with advanced cancers. J. Clin. Oncol. 2018, 36, 3000. [Google Scholar] [CrossRef]
- Rischin, D.; Groenland, S.L.; Lim, A.M.L.; Balla, M.; Hoos, A.; Angevin, E. Inducible T cell costimulatory (ICOS) receptor agonist, GSK3359609 (GSK609) alone and in combination with pembrolizumab (pembro): Preliminary results from INDUCE-1 expansion cohorts (EC) in head and neck squamous cell carcinoma (HNSCC). Ann. Oncol. 2019, 30, v454–v455. [Google Scholar] [CrossRef]
- Singh, M.; Vianden, C.; Cantwell, M.J.; Dai, Z.; Xiao, Z.; Sharma, M.; Khong, H.; Jaiswal, A.R.; Faak, F.; Hailemichael, Y.; et al. Intratumoral CD40 activation and checkpoint blockade induces T cell-mediated eradication of melanoma in the brain. Nat. Commun. 2017, 8, 1447. [Google Scholar] [CrossRef] [Green Version]
- Piechutta, M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: The role of Cluster of Differentiation 40 (CD40/TNFR5). ESMO Open 2019, 4, e000510. [Google Scholar] [CrossRef] [Green Version]
- French, R.R.; Chan, H.T.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553. [Google Scholar] [CrossRef]
- Bajor, D.L.; Mick, R.; Riese, M.J.; Huang, A.C.; Sullivan, B.; Richman, L.P.; Torigian, D.A.; George, S.M.; Stelekati, E.; Chen, F.; et al. Long-term outcomes of a phase I study of agonist CD40 antibody and CTLA-4 blockade in patients with metastatic melanoma. Oncoimmunology 2018, 7, e1468956. [Google Scholar] [CrossRef] [Green Version]
- Salah, A.; Wang, H.; Li, Y.; Ji, M.; Ou, W.B.; Qi, N.; Wu, Y. Insights into dendritic cells in cancer immunotherapy: From bench to clinical applications. Front. Cell Dev. Biol. 2021, 9, 686544. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; de Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Gopalakrishnan, V.; Daillère, R.; Zitvogel, L.; Wargo, J.A.; Kroemer, G. The gut microbiota influences anticancer immunosurveillance and general health. Nat. Rev. Clin. Oncol. 2018, 15, 382–396. [Google Scholar] [CrossRef]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D.; Enot, D.P.; Pfirschke, C.; Engblom, C.; Pittet, M.J.; et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.L.; et al. Commensal bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, K.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Y.; Yuan, M.; Tao, M.; Kong, C.; Li, H.; Tong, J.; Zhu, H.; Yan, X. Antibiotic administration shortly before or after immunotherapy initiation is correlated with poor prognosis in solid cancer patients: An up-to-date systematic review and meta-analysis. Int. Immunopharmacol. 2020, 88, 106876. [Google Scholar] [CrossRef]
- McKee, A.M.; Kirkup, B.M.; Madgwick, M.; Fowler, W.J.; Price, C.A.; Dreger, S.A.; Ansorge, R.; Makin, K.A.; Caim, S.; Le Gall, G.; et al. Antibiotic-induced disturbances of the gut microbiota result in accelerated breast tumor growth. iScience 2021, 24, 103012. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W. Microbiota-derived short-chain fatty acids promote the memory potential of antigen-activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Spencer, C.N.; Gopalakrishnan, V.; McQuade, J.; Andrews, M.C.; Helmink, B.; Khan, W.; Simans, E.; Haydu, L.; Cogdill, A.; Burton, E.; et al. The gut microbiome (GM) and immunotherapy response are influenced by host lifestyle factors. Cancer Res. 2019, 79, 2838. [Google Scholar]
- Tanoue, T.; Morita, S.; Plichta, D.R.; Skelly, A.N.; Suda, W.; Sugiura, Y.; Narushima, S.; Vlamakis, H.; Motoo, I.; Sugita, K.; et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 2019, 565, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.A.; Kashyap, S.; Ponichtera, H.; Sandy, P.; Parameswaran, P.; Carlson, M.; Sizova, M.; Kravitz, V.; Andrewes, S.; Tory, E.; et al. Monoclonal microbial EDP1503 to induce antitumor responses via gut-mediated activation of both innate and adaptive immunity. J. Clin. Oncol. 2019, 37, e14241. [Google Scholar] [CrossRef]
- Francisco-Anderson, L.; Shariffudin, S.; Gardner, H.; Sandy, P.; Goldberg, M.; Kashyap, S.; Abdou, M.; Sizova, M.; Kravitz, V.; Ponichtera, H.; et al. A phase I/II clinical trial of EDP1503 with pembrolizumab for triple-negative breast cancer [abstract]. Cancer Res. 2021, 81 (Suppl. 4), PS11-27. [Google Scholar] [CrossRef]
- Araujo, D.V.; Bernal, M.; Tan, T.; Abbas, A.; Heirali, P.; Schneeberger, H.H.; Muniz, T.P.; Chen, B.; Stayner, L.-E.; Xia, K.; et al. First-in-class microbial ecosystem therapeutics 4 (MET4) in metastatic solid cancer patients treated with immunotherapy: MET4-IO. J. Clin. Oncol. 2020, 38, 3098. [Google Scholar] [CrossRef]
- Stevenson, A.; Panzica, A.; Holt, A. Host-microbe interactions mediating antitumorigenic effects of MRX0518, a gut microbiota-derived bacterial strain, in breast, renal and lung carcinoma. J. Clin. Oncol. 2018, 36, e15006. [Google Scholar] [CrossRef]
- Lauté-Caly, D.L.; Raftis, E.J.; Cowie, P.; Hennessy, E.; Holt, A.; Panzica, D.A.; Sparre, C.; Minter, B.; Stroobach, E.; Mulder, I.E. The flagellin of candidate live biotherapeutic Enterococcus gallinarum MRx0518 is a potent immunostimulant. Sci. Rep. 2019, 9, 801. [Google Scholar] [CrossRef]
- Bergerot, P.; Dizman, N.; Ruel, N.; Frankel, P.H.; Hsu, J.; Pal, S.K. A phase I trial to assess the biologic effect of CBM588 (Clostridium butyricum) in combination with nivolumab plus ipilimumab (nivo/ipi) in patients with metastatic renal cell carcinoma (mRCC). J. Clin. Oncol. 2020, 38, TPS764. [Google Scholar] [CrossRef]
- Richard, C.; Benlaifaoui, M.; Ouarzadi, O.E.; Diop, K.; Desilets, A.; Malo, J.; Belkaid, W.; Leblanc, A.; Lamontagne, J.; Messaoudene, M.; et al. High fiber diet modifies gut microbiome, propionate production, intratumor immune response and is associated with outcome in patients with lung cancer treated with immune checkpoint inhibitors. J. ImmunoTherapy Cancer 2020, 8. [Google Scholar] [CrossRef]
- Kim, C.H. Immune regulation by microbiome metabolites. Immunology 2018, 154, 220–229. [Google Scholar] [CrossRef]
- Park, J.; Kim, M.; Kang, S.G.; Jannasch, A.H.; Cooper, B.; Patterson, J.; Kim, C.H. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 2015, 8, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, M.; Nagatomo, R.; Doi, K.; Shimizu, J.; Baba, K.; Saito, T.; Matsumoto, S.; Inoue, K.; Muto, M. Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid Cancer tumors. JAMA Netw. Open 2020, 3, e202895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, N.; Wang, L.; Zhuang, H.; Jiang, G.; Liu, Z. Ultrafine jujube powder enhances the infiltration of immune cells during anti-PD-L1 treatment against murine colon adenocarcinoma. Cancers 2021, 13, 3987. [Google Scholar] [CrossRef]
- Zhang, S.L.; Mao, Y.Q.; Zhang, Z.Y.; Li, Z.M.; Kong, C.Y.; Chen, H.L.; Cai, P.R.; Han, B.; Ye, T.; Wang, L.S. Pectin supplement significantly enhanced the anti-PD-1 efficacy in tumor-bearing mice humanized with gut microbiota from patients with colorectal cancer. Theranostics 2021, 11, 4155–4170. [Google Scholar] [CrossRef]
- Li, Y.; Elmén, L.; Segota, I.; Xian, Y.; Tinoco, R.; Feng, Y.; Fujita, Y.; Segura Muñoz, R.R.; Schmaltz, R.; Bradley, L.M.; et al. Prebiotic-induced anti-tumor immunity attenuates tumor growth. Cell Rep. 2020, 30, 1753–1766.e6. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, M.; Bassaganya-Riera, J.; Mohede, I.C. Immunomodulatory properties of conjugated linoleic acid. Am. J. Clin. Nutr. 2004, 79, 1199S–1206S. [Google Scholar] [CrossRef] [Green Version]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Zhang, Y.; Zheng, K.; Xiang, Q.; Chen, N.; Chen, Z.; Zhang, N.; Zhu, J.; He, Q. Antibiotic-induced disruption of gut microbiota alters local metabolomes and immune responses. Front. Cell Infect. Microbiol. 2019, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, I.G.; Di Filippo, A.; Scirocchi, F.; Di Pietro, F.R.; Rahimi, H.; Ugolini, A.; Scagnoli, S.; Vernocchi, P.; Del Chierico, F.; Putignani, L. Soluble immune checkpoints, gut metabolites and performance status as parameters of response to nivolumab treatment in NSCLC patients. J. Pers. Med. 2020, 10, 208. [Google Scholar] [CrossRef]
- Ferrere, G.; Tidjani Alou, M.; Liu, P.; Goubet, A.G.; Fidelle, M.; Kepp, O.; Durand, S.; Iebba, V.; Fluckiger, A.; Daillère, R.; et al. Ketogenic diet and ketone bodies enhance the anticancer effects of PD-1 blockade. JCI Insight 2021, 6, e145207. [Google Scholar] [CrossRef] [PubMed]
- Wanders, D.; Hobson, K.; Ji, X. Methionine restriction and cancer biology. Nutrients 2020, 12, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, D.G.; Chen, J.; Mamane, V.; Ma, E.H.; Muhire, B.M.; Sheldon, R.D.; Shorstova, T.; Koning, R.; Johnson, R.M.; Esaulova, E.; et al. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming. Cell Metab. 2020, 31, 250–266.e9. [Google Scholar] [CrossRef] [PubMed]
- Tamanna, N.; Mayengbam, S.; House, J.D.; Treberg, J.R. Methionine restriction leads to hyperhomocysteinemia and alters hepatic H2S production capacity in Fischer-344 rats. Mech. Ageing Dev. 2018, 176, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lü, S.; Ding, Y.; Zheng, M.; Wang, X. Homocysteine activates T cells by enhancing endoplasmic reticulum-mitochondria coupling and increasing mitochondrial respiration. Protein Cell 2016, 7, 391–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, M.; Xu, X.; Xu, Q.; Xu, X.; Azcarate-Peril, M.A.; Wu, X.; Liu, J.; Locasale, J.W.; Li, J.; Shats, I.; et al. Dietary methionine restriction impairs anti-tumor immunity through gut microbiota. biRxiv 2021. [Google Scholar] [CrossRef]
- Carbonero, F.; Benefiel, A.C.; Alizadeh-Ghamsari, A.H.; Gaskins, H.R. Microbial pathways in colonic sulfur metabolism and links with health and disease. Front. Physiol. 2012, 3, 448. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, K.; Martin, F.; Huang, G.; Tumas, D.; Diehl, L.; Chan, A.C. PEST domain-enriched tyrosine phosphatase (PEP) regulation of effector/memory T cells. Science 2004, 303, 685–689. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Garcia, C.; Ravasz, M.; Zehn, D.; Salmond, R.J.; Zamoyska, R. Resistance to TGFβ suppression and improved anti-tumor responses in CD8+ T cells lacking PTPN22. Nat. Commun. 2017, 8, 1343. [Google Scholar] [CrossRef]
- Brownlie, R.J.; Wright, D.; Zamoyska, R.; Salmond, R.J. Deletion of PTPN22 improves effector and memory CD8+ T cell responses to tumors. JCI Insight 2019, 4, e127847. [Google Scholar] [CrossRef]
- Wiede, F.; Lu, K.H.; Du, X.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Liang, S.; Hochheiser, K.; Dodd, G.T.; Goh, P.K.; et al. PTPN2 phosphatase deletion in T cells promotes anti-tumour immunity and CAR T-cell efficacy in solid tumours. EMBO J. 2020, 39, 1–26. [Google Scholar] [CrossRef]
- Ho, W.J.; Croessmann, S.; Lin, J.; Phyo, Z.H.; Charmsaz, S.; Danilova, L.; Mohan, A.A.; Gross, N.E.; Chen, F.; Dong, J.; et al. Systemic inhibition of PTPN22 augments anticancer immunity. J. Clin. Investig. 2021, 131, e146950. [Google Scholar] [CrossRef]
- Cubas, R.; Khan, Z.; Gong, Q.; Moskalenko, M.; Xiong, H.; Ou, Q.; Pai, C.; Rodriguez, R.; Cheung, J.; Chan, A.C. Autoimmunity linked protein phosphatase PTPN22 as a target for cancer immunotherapy. J. Immunother. Cancer 2020, 8, e001439. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Zhang, Q.; Palen, K.; Wang, L.; Qiao, L.; Johnson, B.; Sei, S.; Shoemaker, R.H.; Lubet, R.A.; Wang, Y.; et al. Potentiation of Kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine 2019, 49, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebeisen, M.; Baitsch, L.; Presotto, D.; Baumgaertner, P.; Romero, P.; Michielin, O.; Speiser, D.E.; Rufer, N. SHP-1 phosphatase activity counteracts increased T cell receptor affinity. J. Clin. Investig. 2013, 123, 1044–1056. [Google Scholar] [CrossRef]
- Snook, J.P.; Soedel, A.J.; Ekiz, H.A.; O’Connell, R.M.; Williams, M.A. Inhibition of SHP-1 Expands the Repertoire of Antitumor T Cells Available to Respond to Immune Checkpoint Blockade. Cancer Immunol. Res. 2020, 8, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Kuehm, L.M.; Wolf, K.; Zahour, J.; DiPaolo, R.J.; Teague, R.M. Checkpoint blockade immunotherapy enhances the frequency and effector function of murine tumor-infiltrating T cells but does not alter TCRβ diversity. Cancer Immunol. Immunother. 2019, 68, 1095–1106. [Google Scholar] [CrossRef]
- Castro-Sanchez, P.; Teagle, A.R.; Prade, S.; Zamoyska, R. Modulation of TCR signaling by tyrosine phosphatases: From autoimmunity to immunotherapy. Front. Cell Dev. Biol. 2020, 8, 608747. [Google Scholar] [CrossRef]
- Cheng, Y.; Tian, H. Current development status of MEK inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comin-Anduix, B.; Chodon, T.; Sazegar, H.; Matsunaga, D.; Mock, S.; Jalil, J.; Escuin-Ordinas, H.; Chmielowski, B.; Koya, R.C.; Ribas, A. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin. Cancer Res. 2010, 16, 6040–6048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapkota, B.; Hill, C.E.; Pollack, B.P. Vemurafenib enhances MHC induction in BRAF homozygous melanoma cells. Oncoimmunology 2013, 2, e22890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooijkaas, A.; Gadiot, J.; Morrow, M.; Stewart, R.; Schumacher, T.; Blank, C.U. Selective BRAF inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012, 1, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Dushyanthen, S.; Beavis, P.A.; Salgado, R.; Denkert, C.; Savas, P.; Combs, S.; Rimm, D.L.; Giltnane, J.M.; Estrada, M.V.; et al. RAS/MAPK activation is associated with reduced tumor-infiltrating lymphocytes in triple-negative breast cancer: Therapeutic cooperation between MEK and PD-1/PD-L1 immune checkpoint inhibitors. Clin. Cancer Res. 2015, 22, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Mayes, P.A.; Eastman, S.; Shi, H.; Yadavilli, S.; Zhang, T.; Yang, J.; Seestaller-Wehr, L.; Zhang, S.Y.; Hopson, C.; et al. The BRAF and MEK inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin. Cancer. Res. 2015, 21, 1639–1651. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAF(V600E) melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dushyanthen, S.; Teo, Z.L.; Caramia, F.; Savas, P.; Mintoff, C.P.; Virassamy, B.; Henderson, M.A.; Luen, S.J.; Mansour, M.; Kershaw, M.H.; et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat. Commun. 2017, 8, 606. [Google Scholar] [CrossRef] [PubMed]
- Allegrezza, M.J.; Rutkowski, M.R.; Stephen, T.L.; Svoronos, N.; Tesone, A.J.; Perales-Puchalt, A.; Nguyen, J.M.; Sarmin, F.; Sheen, M.R.; Jeng, E.K.; et al. IL15 Agonists Overcome the Immunosuppressive Effects of MEK Inhibitors. Cancer Res. 2016, 76, 2561–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; van Gool, M.; Kroon, P.; Pineda, C.; Geukes Foppen, M.H.; Scolyer, R.; Song, J.-Y.; et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 2016, 5, e1238557. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Dillon, T.J.; Carey, K.D.; Wetzel, S.A.; Parker, D.C.; Stork, P.J. Regulation of the small GTPase Rap1 and extracellular signal-regulated kinases by the costimulatory molecule CTLA-4. Mol. Cell. Biol. 2005, 25, 4117–4128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohnishi, H.; Takeda, K.; Domenico, J.; Lucas, J.J.; Miyahara, N.; Swasey, C.H.; Dakhama, A.; Gelfand, E.W. Mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2-dependent pathways are essential for CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J. Allergy Clin. Immunol. 2009, 123, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Damasio, M.P.; Marchingo, J.M.; Spinelli, L.; Hukelmann, J.L.; Cantrell, D.A.; Howden, A.J.M. Extracellular signal-regulated kinase (ERK) pathway control of CD8+ T cell differentiation. Biochem. J. 2021, 478, 79–98. [Google Scholar] [CrossRef]
- Cannons, J.L.; Villarino, A.V.; Kapnick, S.M.; Preite, S.; Shih, H.Y.; Gomez-Rodriguez, J.; Kaul, Z.; Shibata, H.; Reilley, J.M.; Huang, B.; et al. PI3Kδ coordinates transcriptional, chromatin, and metabolic changes to promote effector CD8+ T cells at the expense of central memory. Cell Rep. 2021, 37, 109804. [Google Scholar] [CrossRef]
- Ohkusu-Tsukada, K.; Toda, M.; Udono, H.; Kawakami, Y.; Takahashi, K. Targeted inhibition of IL-10-secreting CD25- Treg via p38 MAPK suppression in cancer immunotherapy. Eur. J. Immunol. 2010, 40, 1011–1021. [Google Scholar] [CrossRef]
- Li, C.; Beavis, P.; Palfreeman, A.C.; Amjadi, P.; Kennedy, A.; Brennan, F.M. Activation of p38 mitogen-activated protein kinase is critical step for acquisition of effector function in cytokine-activated T cells, but acts as a negative regulator in T cells activated through the T-cell receptor. Immunology 2011, 132, 104–110. [Google Scholar] [CrossRef]
- Kogkopoulou, O.; Tzakos, E.; Mavrothalassitis, G.; Baldari, C.T.; Paliogianni, F.; Young, H.A.; Thyphronitis, G. Conditional up-regulation of IL-2 production by p38 MAPK inactivation is mediated by increased Erk1/2 activity. J. Leukoc. Biol. 2006, 79, 1052–1060. [Google Scholar] [CrossRef]
- Gurusamy, D.; Henning, A.N.; Yamamoto, T.N.; Yu, Z.; Zacharakis, N.; Krishna, S.; Kishton, R.J.; Vodnala, S.K.; Eidizadeh, A.; Jia, L.; et al. Multi-phenotype CRISPR-Cas9 screen identifies p38 kinase as a target for adoptive immunotherapies. Cancer Cell 2020, 37, 818–833.e9. [Google Scholar] [CrossRef]
- Henson, S.M.; Macaulay, R.; Riddell, N.E.; Nunn, C.J.; Akbar, A.N. Blockade of PD-1 or p38 MAP kinase signaling enhances senescent human CD8(+) T-cell proliferation by distinct pathways. Eur. J. Immunol. 2015, 45, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Liu, S.; Chen, S.; Ye, J.; Wu, X.; Bian, Z.; Chen, X. The p38 MAPK Inhibitor SB203580 Abrogates Tumor Necrosis Factor-Induced Proliferative Expansion of Mouse CD4+Foxp3+ Regulatory T Cells. Front. Immunol. 2018, 9, 1556. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Limón, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From biology to cancer therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arranz-Nicolás, J.; Martin-Salgado, M.; Adán-Barrientos, I.; Liébana, R.; Del Carmen Moreno-Ortíz, M.; Leitner, J.; Steinberger, P.; Ávila-Flores, A.; Merida, I. Diacylglycerol kinase α inhibition cooperates with PD-1-targeted therapies to restore the T cell activation program. Cancer Immunol. Immunother. 2021, 70, 3277–3289. [Google Scholar] [CrossRef]
- Tang, K.H.; Li, S.; Khodadadi-Jamayran, A.; Jen, J.; Han, H.; Guidry, K.; Chen, T.; Hao, Y.; Fedele, C.; Zebala, J.A.; et al. Combined inhibition of SHP2 and CXCR1/2 promotes anti-tumor T cell response in NSCLC. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR Signalling Controls Tumour Cell Growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Pollizzi, N.; Patel, H.; Sun, I.H.; Oh, M.H.; Waickman, A.T.; Wen, J.; Delgoffe, G.M.; Powell, J.D. Mtorc1 and mtorc2 selectively regulate cd8(+) t cell differentiation. J. Clin. Investig. 2015, 125, 2090–2108. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.T.; Hassan, M.; Morris, A.B.; Jeffery, J.; Lee, K.; Jagirdar, N.; Staton, A.D.; Raikar, S.S.; Spencer, H.T.; Sulchek, T.; et al. Improving T-cell expansion and function for adoptive T-cell therapy using ex vivo treatment with PI3Kdelta inhibitors and VIP antagonists. Blood Adv. 2018, 2, 210–223. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Kato, K.; Jiang, J.; Wahl, D.R.; Mineishi, S.; Fisher, E.M.; Murasko, D.M.; Glick, G.D.; Zhang, Y. Characterization of the metabolic phenotype of rapamycin-treated CD8+ T cells with augmented ability to generate long-lasting memory cells. PLoS ONE 2011, 6, e20107. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, X.Y.; Subjeck, J.R.; Shrikant, P.A.; Kim, H.L. Temsirolimus, an mtor inhibitor, enhances anti-tumour effects of heat shock protein cancer vaccines. Br. J. Cancer 2011, 104, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Beziaud, L.; Boullerot, L.; Tran, T.; Mansi, L.; Marie-Joseph, E.L.; Ravel, P.; Johannes, L.; Bayry, J.; Tartour, E.; Adotévi, O. Rapalog combined with ccr4 antagonist improves anticancer vaccines efficacy. Int. J. Cancer 2018, 143, 3008–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Sparwasser, T.; Figlin, R.; Kim, H.L. Foxp3+ t cells inhibit antitumor immune memory modulated by mtor inhibition. Cancer Res. 2014, 74, 2217–2228. [Google Scholar] [CrossRef] [Green Version]
- Chaoul, N.; Fayolle, C.; Desrues, B.; Oberkampf, M.; Tang, A.; Ladant, D.; Leclerc, C. Rapamycin impairs anti-tumour CD8+ t-cell responses and vaccine-induced tumor eradication. Cancer Res. 2015, 75, 3279–3291. [Google Scholar] [CrossRef] [Green Version]
- Diken, M.; Kreiter, S.; Vascotto, F.; Selmi, A.; Attig, S.; Diekmann, J.; Huber, C.; Türeci, Ö.; Sahin, U. mTOR inhibition improves antitumor effects of vaccination with antigen-encoding RNA. Cancer Immunol. Res. 2013, 1, 386–392. [Google Scholar] [CrossRef] [Green Version]
- Esfahani, K.; Al-Aubodah, T.A.; Thebault, P.; Lapointe, R.; Hudson, M.; Johnson, N.A.; Baran, D.; Bhulaiga, N.; Takano, T.; Cailhier, J.F.; et al. Targeting the mTOR pathway uncouples the efficacy and toxicity of PD-1 blockade in renal transplantation. Nat. Commun. 2019, 10, 4712. [Google Scholar] [CrossRef] [Green Version]
- Beziaud, L.; Mansi, L.; Ravel, P.; Marie-Joseph, E.L.; Laheurte, C.; Rangan, L.; Bonnefoy, F.; Pallandre, J.R.; Boullerot, L.; Gamonet, C.; et al. Rapalogs efficacy relies on the modulation of antitumor t-cell immunity. Cancer Res. 2016, 76, 4100–4112. [Google Scholar] [CrossRef] [Green Version]
- Berezhnoy, A.; Castro, I.; Levay, A.; Malek, T.R.; Gilboa, E. Aptamer-targeted inhibition of mtor in t cells enhances antitumor immunity. J. Clin. Investig. 2014, 124, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Essen, M.R.; Kongsbak, M.; Schjerling, P.; Olgaard, K.; Odum, N.; Geisler, C. Vitamin D controls T cell antigen receptor signaling and activation of human T cells. Nat. Immunol. 2010, 11, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Joseph, R.W.; Bayraktar, U.D.; Kim, T.K.; St John, L.S.; Popat, U.; Khalili, J.; Molldrem, J.J.; Wieder, E.D.; Komanduri, K.V. Vitamin D receptor upregulation in alloreactive human T cells. Hum. Immunol 2012, 73, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, A.; Reddy, G.S.; Kobayashi, T.; Okano, T.; Park, J.; Sharma, S. Nuclear factor of activated T cells (NFAT) as a molecular target for 1alpha,25-dihydroxyvitamin D3-mediated effects. J. Immunol. 1998, 160, 209–218. [Google Scholar]
- Murayama, A.; Takeyama, K.; Kitanaka, S.; Kodera, Y.; Kawaguchi, Y.; Hosoya, T.; Kato, S. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology 1999, 140, 2224–2231. [Google Scholar] [CrossRef]
- Kim, S.; Shevde, N.K.; Pike, J.W. 1,25-Dihydroxyvitamin D3 stimulates cyclic vitamin D receptor/retinoid X receptor DNA-binding, co-activator recruitment, and histone acetylation in intact osteoblasts. J. Bone Miner. Res. 2005, 20, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Ramana, C.V.; Dusso, A.S. Stat1-vitamin D receptor interactions antagonize 1,25-dihydroxyvitamin D transcriptional activity and enhance stat1-mediated transcription. Mol. Cell. Biol. 2002, 22, 2777–2787. [Google Scholar] [CrossRef] [Green Version]
- Aguirre, M.; Manzano, N.; Salas, Y.; Angel, M.; Díaz-Couselo, F.A.; Zylberman, M. Vitamin D deficiency in patients admitted to the general ward with breast, lung, and colorectal cancer in Buenos Aires, Argentina. Arch. Osteoporos. 2016, 11, 4. [Google Scholar] [CrossRef]
- Rossini, M.; Maddali, B.S.; La, M.G.; La Montagna, G.; Minisola, G.; Malavolta, N.; Bernini, L.; Cacace, E.; Sinigaglia, L.; Di Munno, O.; et al. Vitamin D deficiency in rheumatoid arthritis: Prevalence, determinants and associations with disease activity and disability. Arthritis Res. Ther. 2010, 12, R216. [Google Scholar] [CrossRef] [Green Version]
- Greer, R.M.; Portelli, S.L.; Hung, B.S.; Cleghorn, G.J.; McMahon, S.K.; Batch, J.A.; Conwell, L.S. Serum vitamin D levels are lower in Australian children an adolescents with Type 1 diabetes than in children without diabetes. Pediatric Diabetes 2012, 14, 31–41. [Google Scholar] [CrossRef]
- Ginde, A.A.; Mansbach, J.M.; Camargo, C.A., Jr. Association between serum 25- hydroxyvitamin D level and upper respiratory tract infection in the Third National Health and Nutrition Examination Survey. Arch. Intern. Med. 2009, 169, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannell, J.J.; Vieth, R.; Umhau, J.C.; Holick, M.F.; Grant, W.B.; Madronich, S.; Garland, C.F.; Giovannucci, E. Epidemic influenza and vitamin D. Epidemiol. Infect. 2006, 134, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Grant, W.B. Variations in vitamin D production could possibly explain the seasonality of childhood respiratory infections in Hawaii. Pediatric Infect. Dis. J. 2008, 27, 853. [Google Scholar] [CrossRef] [PubMed]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin D and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fassò, M.; Wang, Y.V.; et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: Long-term safety, clinical activity, and immune correlates from a Phase Ia study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef]
- Timerman, D.; McEnery-Stonelake, M.; Nambudiri, V.E.; Hodi, F.S.; Claus, E.B.; Ibrahim, N.; Lin, J.Y. Vitamin D deficiency is associated with a worse prognosis in metastatic melanoma. Oncotarget 2017, 8, 6873–6882. [Google Scholar] [CrossRef] [Green Version]
- Grover, S.; Dougan, M.; Tyan, K.; Giobbie-Hurder, A.; Blum, S.M.; Ishizuka, J.; Qazi, T.; Elias, R.; Vora, K.B.; Ruan, A.B.; et al. Vitamin D intake is associated with decreased risk of immune checkpoint inhibitor-induced colitis. Cancer 2020, 126, 3758–3767. [Google Scholar] [CrossRef]
- Toti, E.; Chen, C.O.; Palmery, M.; Villaño Valencia, D.; Peluso, I. Non-Provitamin A and Provitamin A Carotenoids as Immunomodulators: Recommended Dietary Allowance, Therapeutic Index, or Personalized Nutrition? Oxid Med. Cell Longev. 2018, 2018, 4637861. [Google Scholar] [CrossRef]
- Erkelens, N.; Mebius, E. Retinoic acid and immune homeostasis: A balancing act. Trends Immunol. 2017, 38, 168–180. [Google Scholar] [CrossRef]
- Brown, C.C.; Esterhazy, D.; Sarde, A.; London, M.; Pullabhatla, V.; Osma-Garcia, I.; Al-Bader, R.; Ortiz, C.; Elgueta, R.; Arno, M.; et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 2015, 42, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Cannons, J.L.; Grainger, J.R.; Dos Santos, L.M.; Hand, T.W.; Naik, S.; Wohlfert, E.A.; Chou, D.B.; Oldenhove, G.; Robinson, M.; et al. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 2011, 34, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, H.; Kanno, T.; Nakayamada, S.; Hirahara, K.; Sciumè, G.; Muljo, S.A.; Kuchen, S.; Casellas, R.; Wei, L.; Kanno, Y.; et al. TGF-β and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 2012, 13, 587–595. [Google Scholar] [CrossRef]
- Takeuchi, H.; Yokota, A.; Ohoka, Y.; Iwata, M. Cyp26b1 regulates retinoic acid-dependent signals in T cells and its expression is inhibited by transforming growth factor-β. PLoS ONE 2011, 6, e16089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, N.; Yuan, R.; Prestwood, T.R.; Penny, H.L.; DiMaio, M.A.; Reticker-Flynn, N.E.; Krois, C.R.; Kenkel, J.A.; Pham, T.D.; Carmi, Y.; et al. Normalizing microbiota-induced retinoic acid deficiency stimulates protective CD8(+) T cell-mediated immunity in colorectal cancer. Immunity 2016, 45, 641–655. [Google Scholar] [CrossRef] [Green Version]
- McCarter, M.; Tobin, R.P.; Cogswell, D.T.; Vorwald, V.M.; Davis, D.; Gulick, J.V.R.; Couts, K.L.; Jordan, K.R.; Nuanes, V.; Gao, D.; et al. Pembrolizumab and all-trans retinoic acid combination treatment of advanced melanoma. J. Clin. Oncol. 2021, 39, 9536. [Google Scholar] [CrossRef]
- Tan, X.; Sande, J.L.; Pufnock, J.S.; Blattman, J.N.; Greenberg, P.D. Retinoic acid as a vaccine adjuvant enhances CD8+ T cell response and mucosal protection from viral challenge. J. Virol. 2011, 85, 8316–8327. [Google Scholar] [CrossRef] [Green Version]
- Devalaraja, S.; To, T.K.J.; Folkert, I.W.; Folkert, I.W.; Natesan, R.; Alam, M.Z.; Li, M.; Tada, Y.; Budagyan, K.; Dang, M.T.; et al. Tumor-Derived Retinoic Acid Regulates Intratumoral Monocyte Differentiation to Promote Immune Suppression. Cell 2020, 180, 1098–1114.e16. [Google Scholar] [CrossRef]
- Larange, A.; Cheroutre, H. Retinoic acid and retinoic acid receptors as pleiotropic modulators of the immune system. Annu. Rev. Immunol. 2016, 34, 369–394. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016, 45, 374–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Shangguan, X.; Zhou, W.; Cao, Y.; Zheng, Q.; Tu, J.; Hu, G.; Liang, Z.; Jiang, C.; Deng, L.; et al. Glucose limitation activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T cell memory development. Nat. Commun. 2021, 12, 4371. [Google Scholar] [CrossRef] [PubMed]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.V.; Melo, A.C.L.; Low, J.S.; de Castro, Í.A.; Braga, T.T.; Almeida, D.C.; Batista de Lima, A.G.U.; Hiyane, M.I.; Correa-Costa, M.; Andrade-Oliveira, V.; et al. Metformin exerts antitumor activity via induction of multiple death pathways in tumor cells and activation of a protective immune response. Oncotarget 2018, 9, 25808–25825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, F.; Tian, Y.; Cao, L.; Gao, Q.; Zhang, C.; Zhang, K.; Shen, C.; Ping, Y.; Maimela, N.R.; et al. Metformin enhances the antitumor activity of CD8+ T lymphocytes via the AMPK-miR-107-eomes-PD-1 pathway. J. Immunol. 2020, 204, 2575–2588. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, F.; Wang, L.; Qiu, S.; Yao, Y.; Xiong, X.; Chen, X.; Ji, Q.; Cao, J.; Li, D.; et al. Potentiating the anti-tumor response of tumor infiltrated T cells by NAD+ supplementation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Choi, H.J.; Jang, S.Y.; Hwang, E.S. High-dose nicotinamide suppresses ROS generation and augments population expansion during CD8(+) T cell activation. Mol. Cells 2015, 38, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabalirajan, U.; Ahmad, T.; Leishangthem, G.D.; Dinda, A.K.; Agrawal, A.; Ghosh, B. L-arginine reduces mitochondrial dysfunction and airway injury in murine allergic airway inflammation. Int. Immunopharmacol. 2010, 10, 1514–1519. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, X.; Cheng, C.; Yu, W.; Yi, P. Metabolism of amino acids in cancer. Front. Cell Dev. Biol. 2021, 8, 603837. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino acid depletion therapies: Starving cancer cells to death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef]
- Wang, T.; Gnanaprakasam, J.N.R.; Chen, X.; Kang, S.; Xu, X.; Sun, H.; Liu, L.; Rodgers, H.; Miller, E.; Cassel, T.A.; et al. Inosine is an alternative carbon source for CD8+-T-cell function under glucose restriction. Nat. Metab. 2020, 2, 635–647. [Google Scholar] [CrossRef]
- Trompette, A.; Gollwitzer, E.S.; Pattaroni, C.; Lopez-Mejia, I.C.; Riva, E.; Pernot, J.; Ubags, N.; Fajas, L.; Nicod, L.P.; Marsland, B.J.; et al. Dietary fiber confers protection against flu by shaping Ly6c- patrolling monocyte hematopoiesis and CD8+ T cell metabolism. Immunity 2018, 48, 992–1005.e8. [Google Scholar] [CrossRef] [Green Version]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar] [CrossRef] [Green Version]



| Setting | Effect | Mechanism |
|---|---|---|
| Cancer | Exhaustion | Chronic antigenic exposure, altered differentiation, expression of checkpoints, and loss of effector function. |
| Tolerance induction | Inhibition within the TME including T-regulatory cells MDSC and hypoxia. Tumour antigen with low affinity for cognate TCR resulting in weak T-cell activation. | |
| Chronic inflammation | Cytokine induced dysfunction | Loss of TCR responsiveness and effector functions, bystander activation, apoptosis |
| Senescence | Unresponsive T-cells | Reduced production of naïve T-cells, lower expression of costimulatory receptors. Uncoupling of TCR signalling pathways and skewed T-cell differentiation. |
| Microbiota | Effect on T-Cells | Questions | Refs |
|---|---|---|---|
| FMT | Can recapitulate ICB responsiveness in non-responder patients. | Nonstandardised and possibility of toxicity | [126,127] |
| VE800 | An consortium of 11 bacteria which enhances IFN-γ expression and CD8+ T-cell activation in mice. | Is the constorium diverse enough for human use? A trial of VE800 and nivolumab is underway (NCT04208958) | [132] |
| EDP1503 | Inducing systemic anti-tumour immunity by activating both innate and adaptive immunity characterised by increased cytokine expression including IFN-γ and CXCL10, activation of CD8+ T-cells. | What is the best combination and schedule in combination with other immunotherapies? Overall response rate of 14% across 29 patients—is a single bacterial species sufficient NCT03775850? | [133,134] |
| Enterococcus gallinarum | Associated with ICB responsiveness, Induces anti-tumour responses associated with an increase in the CD8+ T-cell:Treg ratio. | A clinical trial of MRx0518 and Pembrolizumab to treat patients with advanced solid tumours having progressed on anti-PD-1/PDL-1 monotherapy is ongoing (NCT03637803) | [120,136,137] |
| MET4 | A greater number of MET4-associated taxa were detectable in MET4 recipients than controls. | Effect on anti-tumour immune responses not yet determined | [135] |
| Clostridium butyricum MIYAIRI 588 | Associated with ICB responsiveness, Monotherapy with MRX0518 was able to reduce tumour size in syngeneic mouse models of breast, renal and lung carcinoma associated with an increase in the CD8+ T-cell:Treg ratio. | Mechanism of action involves supporting the colonisation of microbes associated with ICB response—is this the best approach? A phase I trial in combination with nivolumab plus ipilimumab in patients with metastatic RCC is underway (NCT03829111). | [138] |
| Ketogenic diet | Induces the induction of 3-hydroxybutyrate mediated reductions in PDL-1 expression on DC | Efficacy in a patient setting yet to be ascertained; issues with compliance? | [145] |
| Methionine restriction | Enhances the effects of chemotherapy in methionine dependent tumours, anti-tumour effect and potential role in T-cell activation | May also inhibit T-cell activation, potentially corrected by homocysteine supplementation however increased homocysteine is implicated in cardiovascular disease | [147,152,156] |
| Butyrate | Modulates T-cell activation via HDAC inhibition, associated with anti-PD-1 ICB responsiveness | Negatively correlated with the efficacy of anti-CTLA-4 ICB responsiveness | [130,140] |
| Inosine | Facilitates T-cell activation via aHR | Not studied in a patient setting | [149] |
| Dietary fibre | Increases the efficacy of anti-PD-1 therapy by altering the gut microbiome and increasing the production of SCFA including butyrate and propionate | Currently studied in murine models of colon cancer and in NSCLC patients administered high-fibre diets. The mechanism(s) of action not yet understood. | [142,143,144] |
| T-Cell Metabolism | Effect on T-Cells | Questions | References |
|---|---|---|---|
| Metformin | Activates AMPK, effecting mitochondrial biogenesis and mTOR inhibition. Inhibits tumour proliferation. | May inhibit effector T-cell function by downregulating glycolysis. Sequence of Metformin administration is important (NCT03800602; NCT03709147). | [247,248,249] |
| Glutamine antagonism | Glutamine blockade using JHU083 inhibits tumour cell viability but results in T-cells oxidative metabolism and adopting a long-lived, highly activated phenotype | Glutamine antagonism is untested in clinical trials | [254] |
| L-arginine | improves anti-tumour T-cell responses by enhancing memory formation and mitochondrial respiration. | Mechanism of action not well understood | [252,253] |
| Butyrate | Butyrate signalling via GPCR41, preferential fuelling of oxidative phosphorylation through sustained glutamine utilization and fatty acid catabolism and ensuring that effector CD8+ T-cells contract in a manner that supports a pool of circulating memory cells with the necessary metabolic adaptations for long-term survival | Butyrate is positively and negatively associated with the efficacy of different ICB immunotherapies. It is unknown whether butyrate supplementation can improve cancer immunotherapy. | [130] |
| NAD+ | Improved mitochondrial fitness upon supplementation with Nicotinamide riboside. Increased presence of TILs and survival in combination with anti-PD-1 ICB in a murine model | Direct effects on tumours are unknown. It is unknown whether supplementation is a useful strategy to improve immunotherapy | [208,250,251] |
| Inosine | Alternative substrate for T-cell growth and function | Some tumour cells are capable of utilising inosine as a carbon source, which may promote tumour growth | [257] |
| Name | Trial/Cancer | Immunotherapies | ClinicalTrials.Gov Identifier |
|---|---|---|---|
| Interleukin-15 and -21 Armored Glypican-3-specific Chimeric Antigen Receptor Expressing Autologous T Cells as an Immunotherapy for Children with Solid Tumors (CARE) | Single group, open label, interventional study in 24 participants with paediatric solid tumours | This study will test T-cells genetically engineered with a GPC3-targeting CAR, IL15 gene and IL21 gene. | NCT04715191 |
| Metformin Plus/Minus Fasting Mimicking Diet to Target the Metabolic Vulnerabilities of LKB1-inactive Lung Adenocarcinoma (FAME) | Interventional, nonrandomized, open-labeled, triple arm, non-comparative phase II trial 64 participants, LKB1-inactive lung adenocarcinoma | A combination of Metformin, anti-PD-1 ICB with Pembrolizumab, platinum-based immunotherapy and fasting-mimicking diet in immune suppressive, metabolically vulnerable LKB1-inactive lung adenocarcinoma. | NCT03709147 |
| Study of GEN-1 With NACT for Treatment of Ovarian Cancer (OVATION 2) | Parallel assignment, open label, interventional study in 130 participants with ovarian cancer | GEN-1 is an IL-12 expressing plasmid being administered along with neoadjuvant chemotherapy using Carboplatin and Paclitaxel +/− subsequent GEN-1. | NCT03393884 |
| The Effect of Diet and Exercise on ImmuNotherapy and the Microbiome (EDEN) | Parallel assignment, open label, interventional study in 80 participants with melanoma | Combination of anti-PD-1 ICB, a high fibre diet, and weekly exercise. The study will measure adherence, changes in the gut microbiome and effects on PFS and OS. | NCT04866810 |
| Docetaxel Chemotherapy and Pembrolizumab Plus Interleukin-12 Gene Therapy and L-NMMA in Triple Negative Breast (INTEGRAL) | Single group, open label, interventional study in 30 participants with triple negative breast cancer | Combination of Interleukin 12 (IL-12) gene therapy, Methylarginine and antiPD-1 immunotherapy alongside neoadjuvant chemotherapy with docitaxel. | NCT04095689 |
| Ketogenic Diet for Patients Receiving First Line Treatment for Metastatic Renal Cell Carcinoma (CETOREIN) | Single group, open label, interventional study in 20 participants with metastatic Renal Cell Carcinoma | Ketogenic diet and vitamin supplementation in patients treated for a metastatic renal cell carcinoma with standard of care treatment including ICB and tyrosine kinase inhibitors. | NCT04316520 |
| All-Trans Retinoic Acid and Atezolizumab for the Treatment of Recurrent or Metastatic Non-Small Cell Lung Cancer | Single group, open label, interventional phase Ib trial in 18 participants with recurrent or metastatic NSCLC | A dose De-Escalation Study of ATRA and atezolizumab | NCT04919369 |
| Dendritic Cell Immunotherapy Plus Standard Treatment of Advanced Renal Cell Carcinoma | Phase 2b open label, Parallel assignment interventional trial in 120 participants with advanced renal cell carcinoma | CMN-001 dendritic cell vaccine plus ICB with VEGFR kinase inhibition with Lenvatinib and mTOR inhibition with Everolimus | NCT04203901 |
| Sirolimus and Durvalumab for the Treatment of Stage I-IIIA Non-small Cell Lung Cancer | Single group, open label, interventional study in 31 participants with NSCLC. | Inhibition of mTOR with Sirolimus for 22 days followed by anti-PDL-1 ICB using Durvalumab | NCT04348292 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, P.L.; Piadel, K.; Dalgleish, A.G. Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy. Vaccines 2021, 9, 1392. https://doi.org/10.3390/vaccines9121392
Smith PL, Piadel K, Dalgleish AG. Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy. Vaccines. 2021; 9(12):1392. https://doi.org/10.3390/vaccines9121392
Chicago/Turabian StyleSmith, Peter Lawrence, Katarzyna Piadel, and Angus George Dalgleish. 2021. "Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy" Vaccines 9, no. 12: 1392. https://doi.org/10.3390/vaccines9121392
APA StyleSmith, P. L., Piadel, K., & Dalgleish, A. G. (2021). Directing T-Cell Immune Responses for Cancer Vaccination and Immunotherapy. Vaccines, 9(12), 1392. https://doi.org/10.3390/vaccines9121392