
Multiple Levels of Immunological Memory and Their Association with Vaccination

 ,
, 
Abstract
:1. Introduction
2. Trained Immunity
Consequences on Vaccination
3. Natural Memory
Consequences on Vaccination
4. Overview of B Cell-Mediated Immune Response
5. MZ B Cells and T-Independent B Cell Memory
Consequences on Vaccination
6. T-Dependent, Extrafollicular B Cell Activation
Consequences on Vaccination
7. T Dependent B Cell Memory, Intrafollicular B Cell Activation
Consequences on Vaccination
8. Overview of T Cell-Mediated Immune Response
9. The Effector Cells in T Cell Memory
9.1. Tissue Resident Memory T Cells
9.2. Effector Memory T Cells
10. The Anticipatory Memory Cells in T Cell Memory
10.1. Central Memory T Cells
10.2. Memory Stem T Cells
10.3. Consequences on Vaccination
11. Age-Dependence of Immunological Memory
12. Neonatal Immunity
Consequences on Vaccination
13. Immunity in the Elderly
Consequences on Vaccination
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dominguez-Andres, J.; Netea, M.G. The specifics of innate immune memory. Science 2020, 368, 1052–1053. [Google Scholar] [CrossRef]
- Boraschi, D.; Italiani, P. Innate Immune Memory: Time for Adopting a Correct Terminology. Front. Immunol. 2018, 9, 799. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Latz, E.; Mills, K.H.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.; Xavier, R.J. Trained immunity: A program of innate immune memory in health and disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riksen, N.P.; Netea, M.G. Be aware, innate immune cells remember. Aging 2018, 10, 2218–2219. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Benn, C.S.; Joosten, L.A.; Jacobs, C.; van Loenhout, J.; Xavier, R.J.; Aaby, P.; van der Meer, J.W.; et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. J. Innate Immun. 2014, 6, 152–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netea, M.G.; Latz, E.; Mills, K.H.; O’Neill, L.A. Innate immune memory: A paradigm shift in understanding host defense. Nat. Immunol. 2015, 16, 675–679. [Google Scholar] [CrossRef]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Mitroulis, I.; Ruppova, K.; Wang, B.; Chen, L.S.; Grzybek, M.; Grinenko, T.; Eugster, A.; Troullinaki, M.; Palladini, A.; Kourtzelis, I.; et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell 2018, 172, 147–161.e12. [Google Scholar] [CrossRef] [Green Version]
- Garly, M.L.; Martins, C.L.; Bale, C.; Balde, M.A.; Hedegaard, K.L.; Gustafson, P.; Lisse, I.M.; Whittle, H.C.; Aaby, P. BCG scar and positive tuberculin reaction associated with reduced child mortality in West Africa. A non-specific beneficial effect of BCG? Vaccine 2003, 21, 2782–2790. [Google Scholar] [CrossRef]
- Pollard, A.J.; Finn, A.; Curtis, N. Non-specific effects of vaccines: Plausible and potentially important, but implications uncertain. Arch. Dis. Child. 2017, 102, 1077–1081. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Ramon, S.; Conejero, L.; Netea, M.G.; Sancho, D.; Palomares, O.; Subiza, J.L. Trained Immunity-Based Vaccines: A New Paradigm for the Development of Broad-Spectrum Anti-infectious Formulations. Front. Immunol. 2018, 9, 2936. [Google Scholar] [CrossRef]
- Haas, K.M.; Poe, J.C.; Steeber, D.A.; Tedder, T.F. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 2005, 23, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Alugupalli, K.R.; Leong, J.M.; Woodland, R.T.; Muramatsu, M.; Honjo, T.; Gerstein, R.M. B1b lymphocytes confer T cell-independent long-lasting immunity. Immunity 2004, 21, 379–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taillardet, M.; Haffar, G.; Mondiere, P.; Asensio, M.J.; Gheit, H.; Burdin, N.; Defrance, T.; Genestier, L. The thymus-independent immunity conferred by a pneumococcal polysaccharide is mediated by long-lived plasma cells. Blood 2009, 114, 4432–4440. [Google Scholar] [CrossRef] [Green Version]
- Parker, M.E.; Ciofani, M. Regulation of gammadelta T Cell Effector Diversification in the Thymus. Front. Immunol. 2020, 11, 42. [Google Scholar] [CrossRef]
- Deusch, K.; Luling, F.; Reich, K.; Classen, M.; Wagner, H.; Pfeffer, K. A major fraction of human intraepithelial lymphocytes simultaneously expresses the gamma/delta T cell receptor, the CD8 accessory molecule and preferentially uses the V delta 1 gene segment. Eur. J. Immunol. 1991, 21, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.C.; Andrus, J.P.; Perfetto, S.P.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A.; Roederer, M. Ontogeny of gamma delta T cells in humans. J. Immunol. 2004, 172, 1637–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtmeier, W.; Witthoft, T.; Hennemann, A.; Winter, H.S.; Kagnoff, M.F. The TCR-delta repertoire in human intestine undergoes characteristic changes during fetal to adult development. J. Immunol. 1997, 158, 5632–5641. [Google Scholar] [PubMed]
- Ochsenbein, A.F.; Fehr, T.; Lutz, C.; Suter, M.; Brombacher, F.; Hengartner, H.; Zinkernagel, R.M. Control of early viral and bacterial distribution and disease by natural antibodies. Science 1999, 286, 2156–2159. [Google Scholar] [CrossRef] [PubMed]
- Jayasekera, J.P.; Moseman, E.A.; Carroll, M.C. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 2007, 81, 3487–3494. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.G.; Hewitson, T.D.; Nossal, G.J.; Tarlinton, D.M. The phenotype and fate of the antibody-forming cells of the splenic foci. Eur. J. Immunol. 1996, 26, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Beham-Schmid, C.; Dillon, N.; Sabbattini, P. The life span of short-lived plasma cells is partly determined by a block on activation of apoptotic caspases acting in combination with endoplasmic reticulum stress. Blood 2010, 116, 3445–3455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, S.P.; Ma, E.G.M.; Aranda, C.J.; Curotto de Lafaille, M.A. Non-classical B Cell Memory of Allergic IgE Responses. Front. Immunol. 2019, 10, 715. [Google Scholar] [CrossRef]
- Hammarlund, E.; Thomas, A.; Amanna, I.J.; Holden, L.A.; Slayden, O.D.; Park, B.; Gao, L.; Slifka, M.K. Plasma cell survival in the absence of B cell memory. Nat. Commun. 2017, 8, 1781. [Google Scholar] [CrossRef] [Green Version]
- Slifka, M.K.; Antia, R.; Whitmire, J.K.; Ahmed, R. Humoral immunity due to long-lived plasma cells. Immunity 1998, 8, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Bortnick, A.; Allman, D. What is and what should always have been: Long-lived plasma cells induced by T cell-independent antigens. J. Immunol. 2013, 190, 5913–5918. [Google Scholar] [CrossRef] [Green Version]
- Defrance, T.; Taillardet, M.; Genestier, L. T cell-independent B cell memory. Curr. Opin. Immunol. 2011, 23, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Obukhanych, T.V.; Nussenzweig, M.C. T-independent type II immune responses generate memory B cells. J. Exp. Med. 2006, 203, 305–310. [Google Scholar] [CrossRef] [Green Version]
- Hsu, M.C.; Toellner, K.M.; Vinuesa, C.G.; Maclennan, I.C. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. USA 2006, 103, 5905–5910. [Google Scholar] [CrossRef] [Green Version]
- Bortnick, A.; Chernova, I.; Quinn, W.J., 3rd; Mugnier, M.; Cancro, M.P.; Allman, D. Long-lived bone marrow plasma cells are induced early in response to T cell-independent or T cell-dependent antigens. J. Immunol. 2012, 188, 5389–5396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, K.L.; Hochman, M.; Goldblatt, D. Combined schedules of pneumococcal conjugate and polysaccharide vaccines: Is hyporesponsiveness an issue? Lancet Infect. Dis. 2007, 7, 597–606. [Google Scholar] [CrossRef]
- Daniels, C.C.; Rogers, P.D.; Shelton, C.M. A Review of Pneumococcal Vaccines: Current Polysaccharide Vaccine Recommendations and Future Protein Antigens. J. Pediatr. Pharmacol. Ther. 2016, 21, 27–35. [Google Scholar] [CrossRef] [Green Version]
- Kayhty, H.; Peltola, H.; Karanko, V.; Makela, P.H. The protective level of serum antibodies to the capsular polysaccharide of Haemophilus influenzae type b. J. Infect. Dis. 1983, 147, 1100. [Google Scholar] [CrossRef]
- Weller, S.; Braun, M.C.; Tan, B.K.; Rosenwald, A.; Cordier, C.; Conley, M.E.; Plebani, A.; Kumararatne, D.S.; Bonnet, D.; Tournilhac, O.; et al. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654. [Google Scholar] [CrossRef]
- Zouali, M.; Richard, Y. Marginal zone B-cells, a gatekeeper of innate immunity. Front. Immunol. 2011, 2, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaminski, D.A.; Stavnezer, J. Enhanced IgA class switching in marginal zone and B1 B cells relative to follicular/B2 B cells. J. Immunol. 2006, 177, 6025–6029. [Google Scholar] [CrossRef] [PubMed]
- Breukels, M.A.; Zandvoort, A.; van Den Dobbelsteen, G.P.; van Den Muijsenberg, A.; Lodewijk, M.E.; Beurret, M.; Klok, P.A.; Timens, W.; Rijkers, G.T. Pneumococcal conjugate vaccines overcome splenic dependency of antibody response to pneumococcal polysaccharides. Infect. Immun. 2001, 69, 7583–7587. [Google Scholar] [CrossRef] [Green Version]
- Moberley, S.A.; Holden, J.; Tatham, D.P.; Andrews, R.M. Vaccines for preventing pneumococcal infection in adults. Cochrane Database Syst. Rev. 2008, CD000422. [Google Scholar] [CrossRef]
- Patel, M.; Lee, C.K. Polysaccharide vaccines for preventing serogroup A meningococcal meningitis. Cochrane Database Syst. Rev. 2001, CD001093. [Google Scholar] [CrossRef]
- Papadatou, I.; Tzovara, I.; Licciardi, P.V. The Role of Serotype-Specific Immunological Memory in Pneumococcal Vaccination: Current Knowledge and Future Prospects. Vaccines 2019, 7, 13. [Google Scholar] [CrossRef] [Green Version]
- Turner, V.M.; Mabbott, N.A. Influence of ageing on the microarchitecture of the spleen and lymph nodes. Biogerontology 2017, 18, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Cortegano, I.; Rodriguez, M.; Martin, I.; Prado, M.C.; Ruiz, C.; Hortiguela, R.; Alia, M.; Vilar, M.; Mira, H.; Cano, E.; et al. Altered marginal zone and innate-like B cells in aged senescence-accelerated SAMP8 mice with defective IgG1 responses. Cell Death Dis. 2017, 8, e3000. [Google Scholar] [CrossRef] [PubMed]
- Turner, V.M.; Mabbott, N.A. Ageing adversely affects the migration and function of marginal zone B cells. Immunology 2017, 151, 349–362. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Yamazaki, T.; Okubo, Y.; Uehara, Y.; Sugane, K.; Agematsu, K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J. Immunol. 2005, 175, 3262–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takemori, T.; Kaji, T.; Takahashi, Y.; Shimoda, M.; Rajewsky, K. Generation of memory B cells inside and outside germinal centers. Eur. J. Immunol. 2014, 44, 1258–1264. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- McHeyzer-Williams, M.; Okitsu, S.; Wang, N.; McHeyzer-Williams, L. Molecular programming of B cell memory. Nat. Rev. Immunol. 2011, 12, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsner, R.A.; Shlomchik, M.J. Germinal Center and Extrafollicular B Cell Responses in Vaccination, Immunity, and Autoimmunity. Immunity 2020, 53, 1136–1150. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.D.; Gatto, D.; Wood, K.; Camidge, T.; Basten, A.; Brink, R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 2009, 183, 3139–3149. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, B.P.; Vogel, L.A.; Zhang, W.; Loo, W.; Shnider, D.; Lind, E.F.; Ratliff, M.; Noelle, R.J.; Erickson, L.D. Imprinting the fate of antigen-reactive B cells through the affinity of the B cell receptor. J. Immunol. 2006, 177, 7723–7732. [Google Scholar] [CrossRef] [Green Version]
- Paus, D.; Phan, T.G.; Chan, T.D.; Gardam, S.; Basten, A.; Brink, R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006, 203, 1081–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco, Y.R.; Batista, F.D. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity 2007, 27, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuber, E.; Strober, W. The T cell-B cell interaction via OX40-OX40L is necessary for the T cell-dependent humoral immune response. J. Exp. Med. 1996, 183, 979–989. [Google Scholar] [CrossRef] [Green Version]
- MacLennan, I.C.; Toellner, K.M.; Cunningham, A.F.; Serre, K.; Sze, D.M.; Zuniga, E.; Cook, M.C.; Vinuesa, C.G. Extrafollicular antibody responses. Immunol. Rev. 2003, 194, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pape, K.A.; Kouskoff, V.; Nemazee, D.; Tang, H.L.; Cyster, J.G.; Tze, L.E.; Hippen, K.L.; Behrens, T.W.; Jenkins, M.K. Visualization of the genesis and fate of isotype-switched B cells during a primary immune response. J. Exp. Med. 2003, 197, 1677–1687. [Google Scholar] [CrossRef]
- Toyama, H.; Okada, S.; Hatano, M.; Takahashi, Y.; Takeda, N.; Ichii, H.; Takemori, T.; Kuroda, Y.; Tokuhisa, T. Memory B cells without somatic hypermutation are generated from Bcl6-deficient B cells. Immunity 2002, 17, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef]
- Kaji, T.; Ishige, A.; Hikida, M.; Taka, J.; Hijikata, A.; Kubo, M.; Nagashima, T.; Takahashi, Y.; Kurosaki, T.; Okada, M.; et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J. Exp. Med. 2012, 209, 2079–2097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamine, A.; Takahashi, Y.; Baba, N.; Miyake, K.; Tokuhisa, T.; Takemori, T.; Abe, R. Two waves of memory B-cell generation in the primary immune response. Int. Immunol. 2005, 17, 581–589. [Google Scholar] [CrossRef]
- Vieira, P.; Rajewsky, K. Persistence of memory B cells in mice deprived of T cell help. Int. Immunol. 1990, 2, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, M.; Lam, K.P.; Rajewsky, K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature 2000, 407, 636–642. [Google Scholar] [CrossRef]
- Kaneko, N.; Kuo, H.H.; Boucau, J.; Farmer, J.R.; Allard-Chamard, H.; Mahajan, V.S.; Piechocka-Trocha, A.; Lefteri, K.; Osborn, M.; Bals, J.; et al. Loss of Bcl-6-Expressing T Follicular Helper Cells and Germinal Centers in COVID-19. Cell 2020, 183, 143–157.e13. [Google Scholar] [CrossRef] [PubMed]
- Finney, J.; Yeh, C.H.; Kelsoe, G.; Kuraoka, M. Germinal center responses to complex antigens. Immunol. Rev. 2018, 284, 42–50. [Google Scholar] [CrossRef]
- Dal Porto, J.M.; Haberman, A.M.; Kelsoe, G.; Shlomchik, M.J. Very low affinity B cells form germinal centers, become memory B cells, and participate in secondary immune responses when higher affinity competition is reduced. J. Exp. Med. 2002, 195, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Schwickert, T.A.; Lindquist, R.L.; Shakhar, G.; Livshits, G.; Skokos, D.; Kosco-Vilbois, M.H.; Dustin, M.L.; Nussenzweig, M.C. In vivo imaging of germinal centres reveals a dynamic open structure. Nature 2007, 446, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.D.; Okada, T.; Tang, H.L.; Cyster, J.G. Imaging of germinal center selection events during affinity maturation. Science 2007, 315, 528–531. [Google Scholar] [CrossRef]
- Batista, F.D.; Harwood, N.E. The who, how and where of antigen presentation to B cells. Nat. Rev. Immunol. 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Good-Jacobson, K.L.; Szumilas, C.G.; Chen, L.; Sharpe, A.H.; Tomayko, M.M.; Shlomchik, M.J. PD-1 regulates germinal center B cell survival and the formation and affinity of long-lived plasma cells. Nat. Immunol. 2010, 11, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Victora, G.D.; Schwickert, T.A.; Fooksman, D.R.; Kamphorst, A.O.; Meyer-Hermann, M.; Dustin, M.L.; Nussenzweig, M.C. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell 2010, 143, 592–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, J.S.; Ke, F.; Grigorova, I.L. B Cell Receptor Crosslinking Augments Germinal Center B Cell Selection when T Cell Help Is Limiting. Cell Rep. 2018, 25, 1395–1403.e4. [Google Scholar] [CrossRef] [Green Version]
- Akkaya, M.; Kwak, K.; Pierce, S.K. B cell memory: Building two walls of protection against pathogens. Nat. Rev. Immunol. 2020, 20, 229–238. [Google Scholar] [CrossRef]
- Inoue, T.; Moran, I.; Shinnakasu, R.; Phan, T.G.; Kurosaki, T. Generation of memory B cells and their reactivation. Immunol. Rev. 2018, 283, 138–149. [Google Scholar] [CrossRef]
- Pape, K.A.; Maul, R.W.; Dileepan, T.; Paustian, A.S.; Gearhart, P.J.; Jenkins, M.K. Naive B Cells with High-Avidity Germline-Encoded Antigen Receptors Produce Persistent IgM(+) and Transient IgG(+) Memory B Cells. Immunity 2018, 48, 1135–1143.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, A.K.; Desikan, R.; Dixit, N.M. Preferential Presentation of High-Affinity Immune Complexes in Germinal Centers Can Explain How Passive Immunization Improves the Humoral Response. Cell Rep. 2019, 29, 3946–3957.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosaki, T.; Kometani, K.; Ise, W. Memory B cells. Nat. Rev. Immunol. 2015, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Pape, K.A.; Taylor, J.J.; Maul, R.W.; Gearhart, P.J.; Jenkins, M.K. Different B cell populations mediate early and late memory during an endogenous immune response. Science 2011, 331, 1203–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogan, I.; Bertocci, B.; Vilmont, V.; Delbos, F.; Megret, J.; Storck, S.; Reynaud, C.A.; Weill, J.C. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 2009, 10, 1292–1299. [Google Scholar] [CrossRef]
- Plotkin, S.A. Correlates of protection induced by vaccination. Clin. Vaccine Immunol. 2010, 17, 1055–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A. Updates on immunologic correlates of vaccine-induced protection. Vaccine 2020, 38, 2250–2257. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.A. Vaccines: Correlates of vaccine-induced immunity. Clin. Infect. Dis. 2008, 47, 401–409. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.; Foo, S.S.; Bruzzone, R.; Dinh, L.V.; King, N.J.; Mahalingam, S. Fc receptors in antibody-dependent enhancement of viral infections. Immunol. Rev. 2015, 268, 340–364. [Google Scholar] [CrossRef] [Green Version]
- Winarski, K.L.; Tang, J.; Klenow, L.; Lee, J.; Coyle, E.M.; Manischewitz, J.; Turner, H.L.; Takeda, K.; Ward, A.B.; Golding, H.; et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc. Natl. Acad. Sci. USA 2019, 116, 15194–15199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arvin, A.M.; Fink, K.; Schmid, M.A.; Cathcart, A.; Spreafico, R.; Havenar-Daughton, C.; Lanzavecchia, A.; Corti, D.; Virgin, H.W. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature 2020, 584, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2020, 21, 83–100. [Google Scholar] [CrossRef]
- Phan, T.G.; Paus, D.; Chan, T.D.; Turner, M.L.; Nutt, S.L.; Basten, A.; Brink, R. High affinity germinal center B cells are actively selected into the plasma cell compartment. J. Exp. Med. 2006, 203, 2419–2424. [Google Scholar] [CrossRef]
- Blanchard-Rohner, G.; Pulickal, A.S.; Jol-van der Zijde, C.M.; Snape, M.D.; Pollard, A.J. Appearance of peripheral blood plasma cells and memory B cells in a primary and secondary immune response in humans. Blood 2009, 114, 4998–5002. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Garcia, I.; Rodriguez-Bayona, B.; Mora-Lopez, F.; Campos-Caro, A.; Brieva, J.A. Increased survival is a selective feature of human circulating antigen-induced plasma cells synthesizing high-affinity antibodies. Blood 2008, 111, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Castiglione, F.; Mantile, F.; De Berardinis, P.; Prisco, A. How the interval between prime and boost injection affects the immune response in a computational model of the immune system. Comput. Math. Methods Med. 2012, 2012, 842329. [Google Scholar] [CrossRef] [Green Version]
- Tam, H.H.; Melo, M.B.; Kang, M.; Pelet, J.M.; Ruda, V.M.; Foley, M.H.; Hu, J.K.; Kumari, S.; Crampton, J.; Baldeon, A.D.; et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl. Acad. Sci. USA 2016, 113, E6639–E6648. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.; Palm, A.E.; Krammer, F.; Wilson, P.C. From Original Antigenic Sin to the Universal Influenza Virus Vaccine. Trends Immunol. 2018, 39, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Ma, C.S.; Brink, R.; Deenick, E.K. The good, the bad and the ugly—TFH cells in human health and disease. Nat. Rev. Immunol. 2013, 13, 412–426. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Kartashov, A.V.; Liu, C.; Imamichi, H.; Yang, W.; Peng, W.; Lane, H.C.; Zhao, K. Rapid Recall Ability of Memory T cells is Encoded in their Epigenome. Sci. Rep. 2017, 7, 39785. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muruganandah, V.; Sathkumara, H.D.; Navarro, S.; Kupz, A. A Systematic Review: The Role of Resident Memory T Cells in Infectious Diseases and Their Relevance for Vaccine Development. Front. Immunol. 2018, 9, 1574. [Google Scholar] [CrossRef]
- Mueller, S.N.; Mackay, L.K. Tissue-resident memory T cells: Local specialists in immune defence. Nat. Rev. Immunol. 2016, 16, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Skon, C.N.; Lee, J.Y.; Anderson, K.G.; Masopust, D.; Hogquist, K.A.; Jameson, S.C. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol. 2013, 14, 1285–1293. [Google Scholar] [CrossRef] [Green Version]
- Mackay, L.K.; Rahimpour, A.; Ma, J.Z.; Collins, N.; Stock, A.T.; Hafon, M.L.; Vega-Ramos, J.; Lauzurica, P.; Mueller, S.N.; Stefanovic, T.; et al. The developmental pathway for CD103(+)CD8+ tissue-resident memory T cells of skin. Nat. Immunol. 2013, 14, 1294–1301. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Jameson, S.C.; Masopust, D. Understanding Subset Diversity in T Cell Memory. Immunity 2018, 48, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Park, S.L.; Zaid, A.; Hor, J.L.; Christo, S.N.; Prier, J.E.; Davies, B.; Alexandre, Y.O.; Gregory, J.L.; Russell, T.A.; Gebhardt, T.; et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol. 2018, 19, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Mitchell, J.S.; Thompson, E.A.; Schenkel, J.M.; Mohammed, J.; Wijeyesinghe, S.; Fonseca, R.; Burbach, B.J.; Hickman, H.D.; Vezys, V.; et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat. Immunol. 2018, 19, 173–182. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Fraser, K.A.; Beura, L.K.; Pauken, K.E.; Vezys, V.; Masopust, D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 2014, 346, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Masopust, D.; Vezys, V.; Marzo, A.L.; Lefrancois, L. Preferential localization of effector memory cells in nonlymphoid tissue. Science 2001, 291, 2413–2417. [Google Scholar] [CrossRef] [Green Version]
- Bouneaud, C.; Garcia, Z.; Kourilsky, P.; Pannetier, C. Lineage relationships, homeostasis, and recall capacities of central- and effector-memory CD8 T cells in vivo. J. Exp. Med. 2005, 201, 579–590. [Google Scholar] [CrossRef]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central memory and effector memory T cell subsets: Function, generation, and maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef]
- Youngblood, B.; Hale, J.S.; Kissick, H.T.; Ahn, E.; Xu, X.; Wieland, A.; Araki, K.; West, E.E.; Ghoneim, H.E.; Fan, Y.; et al. Effector CD8 T cells dedifferentiate into long-lived memory cells. Nature 2017, 552, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Ahmed, R. Memory T follicular helper CD4 T cells. Front. Immunol. 2015, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luthje, K.; Kallies, A.; Shimohakamada, Y.; Belz, G.T.; Light, A.; Tarlinton, D.M.; Nutt, S.L. The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat. Immunol. 2012, 13, 491–498. [Google Scholar] [CrossRef]
- Kunzli, M.; Schreiner, D.; Pereboom, T.C.; Swarnalekha, N.; Litzler, L.C.; Lotscher, J.; Ertuna, Y.I.; Roux, J.; Geier, F.; Jakob, R.P.; et al. Long-lived T follicular helper cells retain plasticity and help sustain humoral immunity. Sci. Immunol. 2020, 5, eaay5552. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Craft, J. T follicular helper cell heterogeneity: Time, space, and function. Immunol. Rev. 2019, 288, 85–96. [Google Scholar] [CrossRef]
- Asrir, A.; Aloulou, M.; Gador, M.; Perals, C.; Fazilleau, N. Interconnected subsets of memory follicular helper T cells have different effector functions. Nat. Commun. 2017, 8, 847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.T.; Kanno, Y.; Cannons, J.L.; Handon, R.; Bible, P.; Elkahloun, A.G.; Anderson, S.M.; Wei, L.; Sun, H.; O’Shea, J.J.; et al. Functional and epigenetic studies reveal multistep differentiation and plasticity of in vitro-generated and in vivo-derived follicular T helper cells. Immunity 2011, 35, 622–632. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Speiser, D.E.; Lichterfeld, M.; Bonini, C. T memory stem cells in health and disease. Nat. Med. 2017, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Lugli, E.; Ji, Y.; Pos, Z.; Paulos, C.M.; Quigley, M.F.; Almeida, J.R.; Gostick, E.; Yu, Z.; Carpenito, C.; et al. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011, 17, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Lugli, E.; Dominguez, M.H.; Gattinoni, L.; Chattopadhyay, P.K.; Bolton, D.L.; Song, K.; Klatt, N.R.; Brenchley, J.M.; Vaccari, M.; Gostick, E.; et al. Superior T memory stem cell persistence supports long-lived T cell memory. J. Clin. Investig. 2013, 123, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuertes Marraco, S.A.; Soneson, C.; Delorenzi, M.; Speiser, D.E. Genome-wide RNA profiling of long-lasting stem cell-like memory CD8 T cells induced by Yellow Fever vaccination in humans. Genom. Data 2015, 5, 297–301. [Google Scholar] [CrossRef] [Green Version]
- McMahan, K.; Yu, J.; Mercado, N.B.; Loos, C.; Tostanoski, L.H.; Chandrashekar, A.; Liu, J.; Peter, L.; Atyeo, C.; Zhu, A.; et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 2020. [Google Scholar] [CrossRef]
- Billeskov, R.; Beikzadeh, B.; Berzofsky, J.A. The effect of antigen dose on T cell-targeting vaccine outcome. Hum. Vaccine Immunother. 2019, 15, 407–411. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, C.E.; Kim, C.; Weyand, C.M.; Goronzy, J.J. Influence of immune aging on vaccine responses. J. Allergy Clin. Immunol. 2020, 145, 1309–1321. [Google Scholar] [CrossRef]
- Keck, S.; Schmaler, M.; Ganter, S.; Wyss, L.; Oberle, S.; Huseby, E.S.; Zehn, D.; King, C.G. Antigen affinity and antigen dose exert distinct influences on CD4 T-cell differentiation. Proc. Natl. Acad. Sci. USA 2014, 111, 14852–14857. [Google Scholar] [CrossRef] [Green Version]
- Fazilleau, N.; McHeyzer-Williams, L.J.; Rosen, H.; McHeyzer-Williams, M.G. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat. Immunol. 2009, 10, 375–384. [Google Scholar] [CrossRef]
- Ise, W.; Inoue, T.; McLachlan, J.B.; Kometani, K.; Kubo, M.; Okada, T.; Kurosaki, T. Memory B cells contribute to rapid Bcl6 expression by memory follicular helper T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11792–11797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warfel, J.M.; Edwards, K.M. Pertussis vaccines and the challenge of inducing durable immunity. Curr. Opin. Immunol. 2015, 35, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Auladell, M.; Jia, X.; Hensen, L.; Chua, B.; Fox, A.; Nguyen, T.H.O.; Doherty, P.C.; Kedzierska, K. Recalling the Future: Immunological Memory Toward Unpredictable Influenza Viruses. Front. Immunol. 2019, 10, 1400. [Google Scholar] [CrossRef] [PubMed]
- Huisman, W.; Martina, B.E.; Rimmelzwaan, G.F.; Gruters, R.A.; Osterhaus, A.D. Vaccine-induced enhancement of viral infections. Vaccine 2009, 27, 505–512. [Google Scholar] [CrossRef]
- Su, S.; Du, L.; Jiang, S. Learning from the past: Development of safe and effective COVID-19 vaccines. Nat. Rev. Microbiol. 2020, 16, 1–9. [Google Scholar] [CrossRef]
- Walker, J.C.; Smolders, M.A.; Gemen, E.F.; Antonius, T.A.; Leuvenink, J.; de Vries, E. Development of lymphocyte subpopulations in preterm infants. Scand. J. Immunol. 2011, 73, 53–58. [Google Scholar] [CrossRef]
- Kollmann, T.R.; Kampmann, B.; Mazmanian, S.K.; Marchant, A.; Levy, O. Protecting the Newborn and Young Infant from Infectious Diseases: Lessons from Immune Ontogeny. Immunity 2017, 46, 350–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vono, M.; Eberhardt, C.S.; Auderset, F.; Mastelic-Gavillet, B.; Lemeille, S.; Christensen, D.; Andersen, P.; Lambert, P.H.; Siegrist, C.A. Maternal Antibodies Inhibit Neonatal and Infant Responses to Vaccination by Shaping the Early-Life B Cell Repertoire within Germinal Centers. Cell Rep. 2019, 28, 1773–1784.e5. [Google Scholar] [CrossRef] [Green Version]
- Ygberg, S.; Nilsson, A. The developing immune system—From foetus to toddler. Acta Paediatr. 2012, 101, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Zandvoort, A.; Timens, W. The dual function of the splenic marginal zone: Essential for initiation of anti-TI-2 responses but also vital in the general first-line defense against blood-borne antigens. Clin. Exp. Immunol. 2002, 130, 4–11. [Google Scholar] [CrossRef]
- Siegrist, C.A.; Aspinall, R. B-cell responses to vaccination at the extremes of age. Nat. Rev. Immunol. 2009, 9, 185–194. [Google Scholar] [CrossRef]
- Mastelic, B.; Kamath, A.T.; Fontannaz, P.; Tougne, C.; Rochat, A.F.; Belnoue, E.; Combescure, C.; Auderset, F.; Lambert, P.H.; Tacchini-Cottier, F.; et al. Environmental and T cell-intrinsic factors limit the expansion of neonatal follicular T helper cells but may be circumvented by specific adjuvants. J. Immunol. 2012, 189, 5764–5772. [Google Scholar] [CrossRef]
- Morbach, H.; Eichhorn, E.M.; Liese, J.G.; Girschick, H.J. Reference values for B cell subpopulations from infancy to adulthood. Clin. Exp. Immunol. 2010, 162, 271–279. [Google Scholar] [CrossRef]
- Adkins, B.; Leclerc, C.; Marshall-Clarke, S. Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 2004, 4, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Huygens, A.; Lecomte, S.; Tackoen, M.; Olislagers, V.; Delmarcelle, Y.; Burny, W.; Van Rysselberge, M.; Liesnard, C.; Larsen, M.; Appay, V.; et al. Functional Exhaustion Limits CD4+ and CD8+ T-Cell Responses to Congenital Cytomegalovirus Infection. J. Infect. Dis. 2015, 212, 484–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risdon, G.; Gaddy, J.; Horie, M.; Broxmeyer, H.E. Alloantigen priming induces a state of unresponsiveness in human umbilical cord blood T cells. Proc. Natl. Acad. Sci. USA 1995, 92, 2413–2417. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Surendran, N. Neonatal Vaccination: Challenges and Intervention Strategies. Neonatology 2016, 109, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Burt, T.D. Fetal regulatory T cells and peripheral immune tolerance in utero: Implications for development and disease. Am. J. Reprod. Immunol. 2013, 69, 346–358. [Google Scholar] [CrossRef] [Green Version]
- Boer, M.C.; Joosten, S.A.; Ottenhoff, T.H. Regulatory T-Cells at the Interface between Human Host and Pathogens in Infectious Diseases and Vaccination. Front. Immunol. 2015, 6, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sautois, B.; Fillet, G.; Beguin, Y. Comparative cytokine production by in vitro stimulated mononucleated cells from cord blood and adult blood. Exp. Hematol 1997, 25, 103–108. [Google Scholar]
- Saso, A.; Kampmann, B. Vaccine responses in newborns. Semin. Immunopathol. 2017, 39, 627–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, N.; Siegrist, C.A. Neonatal immunization: Where do we stand? Curr. Opin. Infect. Dis. 2011, 24, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, K.A.; Liang, M.D.; Andresen, T.K.; Thomas, K.E.; Marty, A.M.; Cuesta, N.; Vogel, S.N.; Fenton, M.J. TLR2 and TLR4 serve distinct roles in the host immune response against Mycobacterium bovis BCG. J. Leukoc. Biol. 2003, 74, 277–286. [Google Scholar] [CrossRef]
- Marchant, A.; Goetghebuer, T.; Ota, M.O.; Wolfe, I.; Ceesay, S.J.; De Groote, D.; Corrah, T.; Bennett, S.; Wheeler, J.; Huygen, K.; et al. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J. Immunol. 1999, 163, 2249–2255. [Google Scholar] [PubMed]
- Rousseau, M.C.; El-Zein, M.; Conus, F.; Legault, L.; Parent, M.E. Bacillus Calmette-Guerin (BCG) Vaccination in Infancy and Risk of Childhood Diabetes. Paediatr. Perinat. Epidemiol. 2016, 30, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Thostesen, L.M.; Kjaergaard, J.; Pihl, G.T.; Birk, N.M.; Nissen, T.N.; Aaby, P.; Jensen, A.K.G.; Olesen, A.W.; Stensballe, L.G.; Jeppesen, D.L.; et al. Neonatal BCG vaccination and atopic dermatitis before 13 months of age: A randomized clinical trial. Allergy 2018, 73, 498–504. [Google Scholar] [CrossRef]
- Morra, M.E.; Kien, N.D.; Elmaraezy, A.; Abdelaziz, O.A.M.; Elsayed, A.L.; Halhouli, O.; Montasr, A.M.; Vu, T.L.; Ho, C.; Foly, A.S.; et al. Early vaccination protects against childhood leukemia: A systematic review and meta-analysis. Sci. Rep. 2017, 7, 15986. [Google Scholar] [CrossRef] [Green Version]
- Demirjian, A.; Levy, O. Safety and efficacy of neonatal vaccination. Eur. J. Immunol. 2009, 39, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Vojtek, I.; Dieussaert, I.; Doherty, T.M.; Franck, V.; Hanssens, L.; Miller, J.; Bekkat-Berkani, R.; Kandeil, W.; Prado-Cohrs, D.; Vyse, A. Maternal immunization: Where are we now and how to move forward? Ann. Med. 2018, 50, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein Klouwenberg, P.; Bont, L. Neonatal and infantile immune responses to encapsulated bacteria and conjugate vaccines. Clin. Dev. Immunol. 2008, 2008, 628963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landers, C.D.; Chelvarajan, R.L.; Bondada, S. The role of B cells and accessory cells in the neonatal response to TI-2 antigens. Immunol. Res. 2005, 31, 25–36. [Google Scholar] [CrossRef]
- Frasca, D.; Blomberg, B.B. Inflammaging decreases adaptive and innate immune responses in mice and humans. Biogerontology 2016, 17, 7–19. [Google Scholar] [CrossRef]
- Fuentes, E.; Fuentes, M.; Alarcon, M.; Palomo, I. Immune System Dysfunction in the Elderly. An. Acad. Bras. Cienc. 2017, 89, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Pera, A.; Campos, C.; Lopez, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Zugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.L.; Wu, Y.C.; Barnett, Y.; Duggan, O.; Vaughan, R.; Kondeatis, E.; Nilsson, B.O.; Wikby, A.; Kipling, D.; Dunn-Walters, D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8, 18–25. [Google Scholar] [CrossRef] [Green Version]
- Ciabattini, A.; Nardini, C.; Santoro, F.; Garagnani, P.; Franceschi, C.; Medaglini, D. Vaccination in the elderly: The challenge of immune changes with aging. Semin. Immunol. 2018, 40, 83–94. [Google Scholar] [CrossRef]
- Frasca, D.; Riley, R.L.; Blomberg, B.B. Aging murine B cells have decreased class switch induced by anti-CD40 or BAFF. Exp. Gerontol. 2007, 42, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritz, T.; Lair, J.; Ban, M.; Keller, M.; Weinberger, B.; Krismer, M.; Grubeck-Loebenstein, B. Plasma cell numbers decrease in bone marrow of old patients. Eur. J. Immunol. 2015, 45, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Howard, W.A.; Gibson, K.L.; Dunn-Walters, D.K. Antibody quality in old age. Rejuvenation Res. 2006, 9, 117–125. [Google Scholar] [CrossRef]
- Frasca, D.; Landin, A.M.; Lechner, S.C.; Ryan, J.G.; Schwartz, R.; Riley, R.L.; Blomberg, B.B. Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. J. Immunol. 2008, 180, 5283–5290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, B.B.; Frasca, D. Age effects on mouse and human B cells. Immunol. Res. 2013, 57, 354–360. [Google Scholar] [CrossRef] [Green Version]
- Frasca, D.; Van der Put, E.; Riley, R.L.; Blomberg, B.B. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J. Immunol. 2004, 172, 2155–2162. [Google Scholar] [CrossRef] [Green Version]
- Colonna-Romano, G.; Bulati, M.; Aquino, A.; Pellicano, M.; Vitello, S.; Lio, D.; Candore, G.; Caruso, C. A double-negative (IgD-CD27-) B cell population is increased in the peripheral blood of elderly people. Mech. Ageing Dev. 2009, 130, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, S.; Yang, K.; Ozen, Z.; Peng, W.; Marinova, E.; Kelsoe, G.; Zheng, B. Enhanced differentiation of splenic plasma cells but diminished long-lived high-affinity bone marrow plasma cells in aged mice. J. Immunol. 2003, 170, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mogling, R.; de Boer, A.B.; Willems, N.; Schrijver, E.H.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef] [Green Version]
- Malek, T.R.; Bayer, A.L. Tolerance, not immunity, crucially depends on IL-2. Nat. Rev. Immunol. 2004, 4, 665–674. [Google Scholar] [CrossRef]
- Ahmed, M.; Lanzer, K.G.; Yager, E.J.; Adams, P.S.; Johnson, L.L.; Blackman, M.A. Clonal expansions and loss of receptor diversity in the naive CD8 T cell repertoire of aged mice. J. Immunol. 2009, 182, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Goronzy, J.J.; Fang, F.; Cavanagh, M.M.; Qi, Q.; Weyand, C.M. Naive T cell maintenance and function in human aging. J. Immunol. 2015, 194, 4073–4080. [Google Scholar] [CrossRef] [PubMed]
- Renkema, K.R.; Li, G.; Wu, A.; Smithey, M.J.; Nikolich-Zugich, J. Two separate defects affecting true naive or virtual memory T cell precursors combine to reduce naive T cell responses with aging. J. Immunol. 2014, 192, 151–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virgin, H.W.; Wherry, E.J.; Ahmed, R. Redefining chronic viral infection. Cell 2009, 138, 30–50. [Google Scholar] [CrossRef] [Green Version]
- Naylor, K.; Li, G.; Vallejo, A.N.; Lee, W.W.; Koetz, K.; Bryl, E.; Witkowski, J.; Fulbright, J.; Weyand, C.M.; Goronzy, J.J. The influence of age on T cell generation and TCR diversity. J. Immunol. 2005, 174, 7446–7452. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Human T cell immunosenescence and inflammation in aging. J. Leukoc. Biol. 2017, 102, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef]
- Young, B.; Zhao, X.; Cook, A.R.; Parry, C.M.; Wilder-Smith, A.; MC, I.C. Do antibody responses to the influenza vaccine persist year-round in the elderly? A systematic review and meta-analysis. Vaccine 2017, 35, 212–221. [Google Scholar] [CrossRef]
- Lefebvre, J.S.; Masters, A.R.; Hopkins, J.W.; Haynes, L. Age-related impairment of humoral response to influenza is associated with changes in antigen specific T follicular helper cell responses. Sci. Rep. 2016, 6, 25051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Self, W.H.; Wunderink, R.G.; Fakhran, S.; Balk, R.; Bramley, A.M.; Reed, C.; Grijalva, C.G.; Anderson, E.J.; Courtney, D.M.; et al. Community-Acquired Pneumonia Requiring Hospitalization among U.S. Adults. N. Engl. J. Med. 2015, 373, 415–427. [Google Scholar] [CrossRef] [Green Version]
- Schmader, K.E.; Johnson, G.R.; Saddier, P.; Ciarleglio, M.; Wang, W.W.; Zhang, J.H.; Chan, I.S.; Yeh, S.S.; Levin, M.J.; Harbecke, R.M.; et al. Effect of a zoster vaccine on herpes zoster-related interference with functional status and health-related quality-of-life measures in older adults. J. Am. Geriatr. Soc. 2010, 58, 1634–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, H.F.; Harpaz, R.; Luo, Y.; Hales, C.M.; Sy, L.S.; Tartof, S.Y.; Bialek, S.; Hechter, R.C.; Jacobsen, S.J. Declining Effectiveness of Herpes Zoster Vaccine in Adults Aged ≥60 Years. J. Infect. Dis. 2016, 213, 1872–1875. [Google Scholar] [CrossRef] [Green Version]



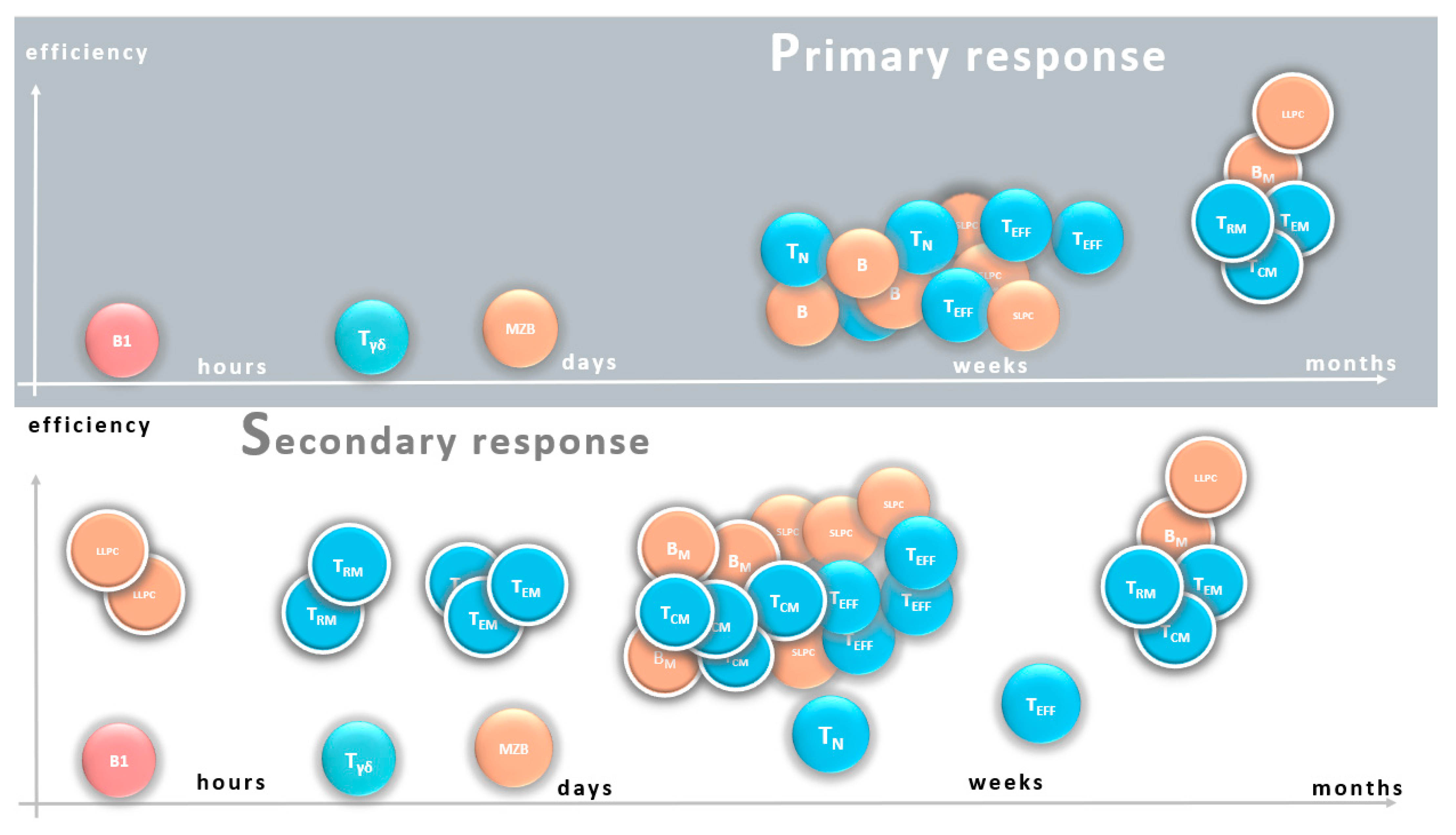{kind=link}
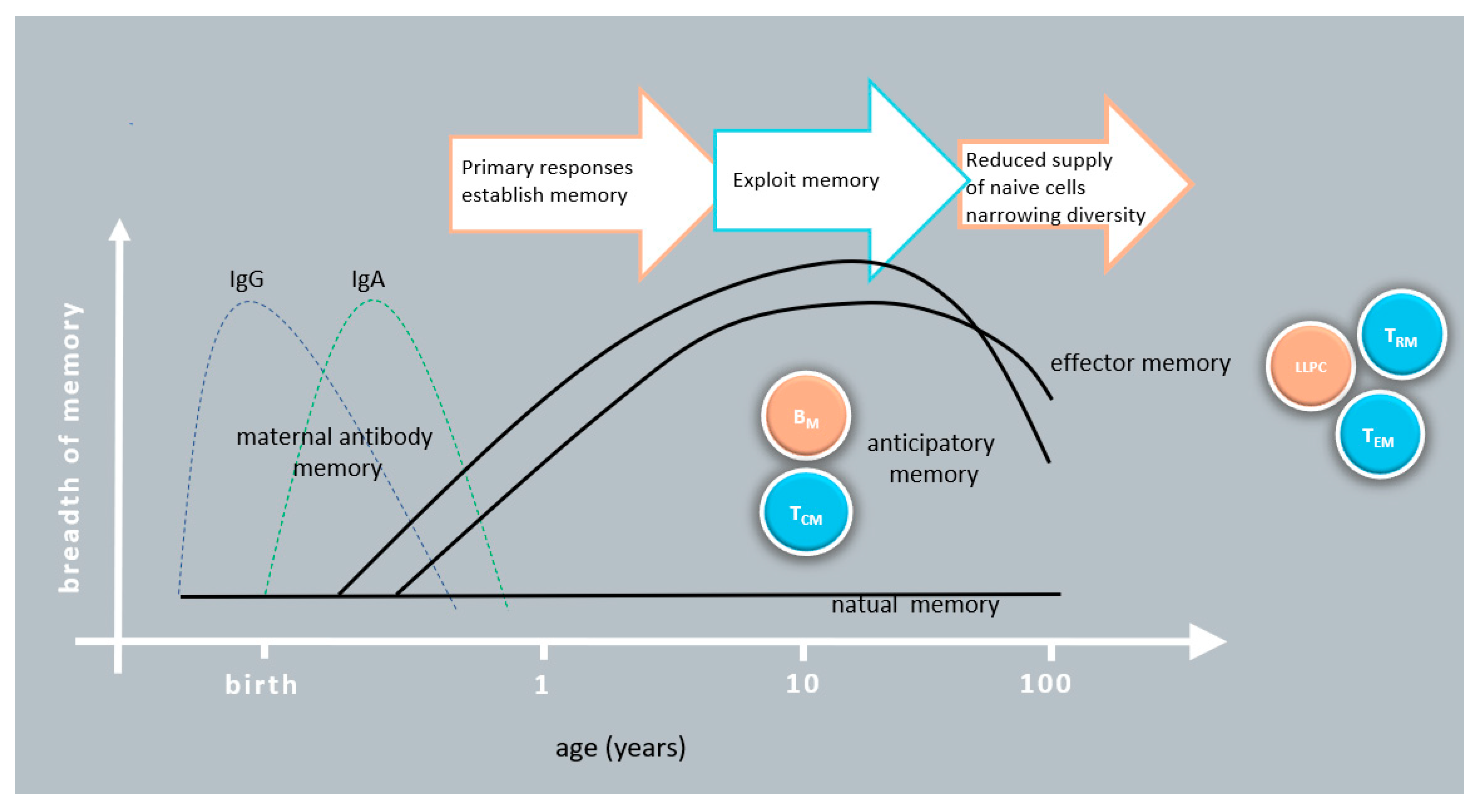{kind=link}
| Name | Memory | Cells | Mechanism | Antigen Specificity | Duration | Targets |
|---|---|---|---|---|---|---|
| Trained innate immunity | Innate | Mo, MF, ILC, NK | Epigenetic | No/limited | Transient | Conserved/personal |
| Natural immunity | Natural adaptive | B1, Tγδ | Epigenetic and genetic | Limited | Lasting | Conserved |
| Immunological memory | Adaptive | B cells, T cells | Epigenetic and genetic | Huge | Lasting | Personal |
| Characteristics | Naive | Natural Memory | Anticipatory Memory | Effector Memory |
|---|---|---|---|---|
| B cell types | B2 | B1 | BM | LLPC |
| T cell types | TN | Tγδ | TCM, TSCM | TEM, TRM |
| Activation state | Resting | Self-renewing | Resting | Active |
| Function | Replenishment, seeding new memory | Natural gatekeeper | Circulating surveillance | Terminally differentiated effector |
| Response type | Primary response | Maintenance | Secondary response | Maintenance |
| Proliferative capacity | High upon activation | Constant | High upon activation | No/limited |
| Time of protection | Slow | Immediate/quick | Medium | Immediate/quick |
| Efficiency | Medium | Low | Medium to high | High |
| Flexibility of repertoire | High | Limited | Medium | No/minimal |
| Vaccine Type | Strength in Memory Response | Weakness in Memory Response |
|---|---|---|
| Attenuated |
| |
| Inactivated |
|
|
| Subunit |
|
|
| Conjugated |
|
|
| Toxoid |
| |
| VLP |
|
|
| RNA |
|
|
| DNA |
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bugya, Z.; Prechl, J.; Szénási, T.; Nemes, É.; Bácsi, A.; Koncz, G. Multiple Levels of Immunological Memory and Their Association with Vaccination. Vaccines 2021, 9, 174. https://doi.org/10.3390/vaccines9020174
Bugya Z, Prechl J, Szénási T, Nemes É, Bácsi A, Koncz G. Multiple Levels of Immunological Memory and Their Association with Vaccination. Vaccines. 2021; 9(2):174. https://doi.org/10.3390/vaccines9020174
Chicago/Turabian StyleBugya, Zsófia, József Prechl, Tibor Szénási, Éva Nemes, Attila Bácsi, and Gábor Koncz. 2021. "Multiple Levels of Immunological Memory and Their Association with Vaccination" Vaccines 9, no. 2: 174. https://doi.org/10.3390/vaccines9020174
APA StyleBugya, Z., Prechl, J., Szénási, T., Nemes, É., Bácsi, A., & Koncz, G. (2021). Multiple Levels of Immunological Memory and Their Association with Vaccination. Vaccines, 9(2), 174. https://doi.org/10.3390/vaccines9020174