
mRNA-Based Vaccines

 , , ,
, , , 
Abstract
:1. Introduction
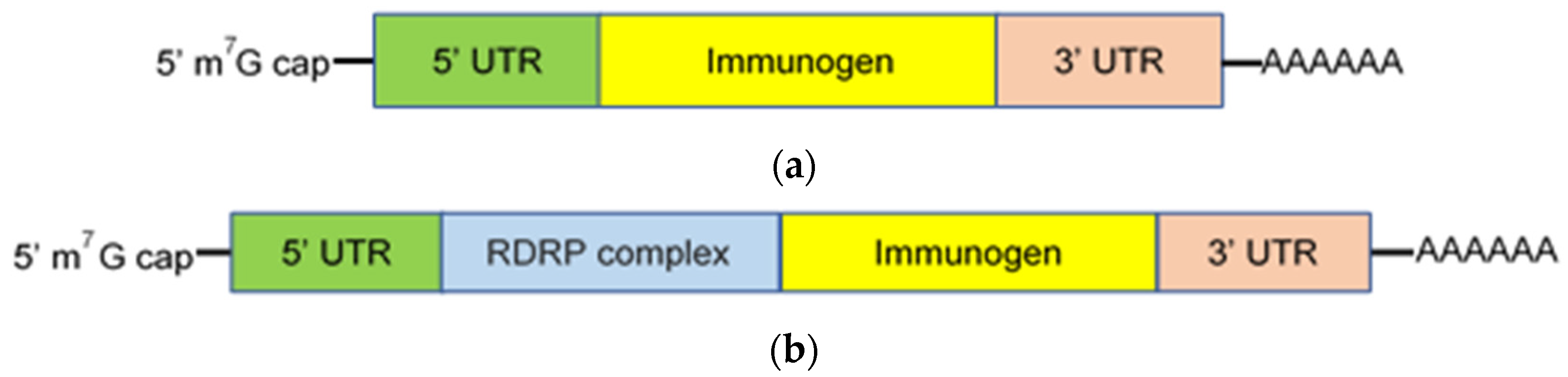
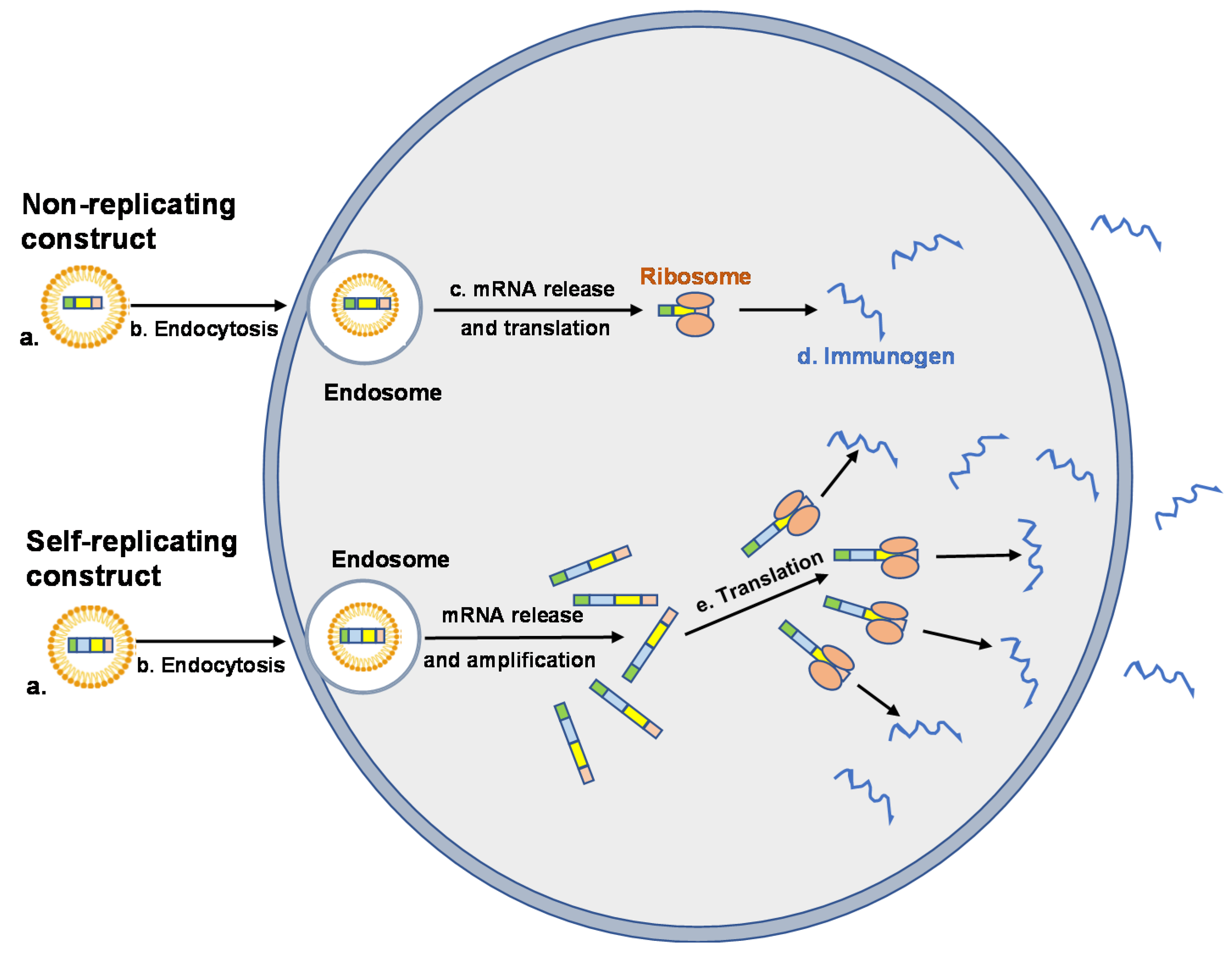
2. mRNA Vaccines
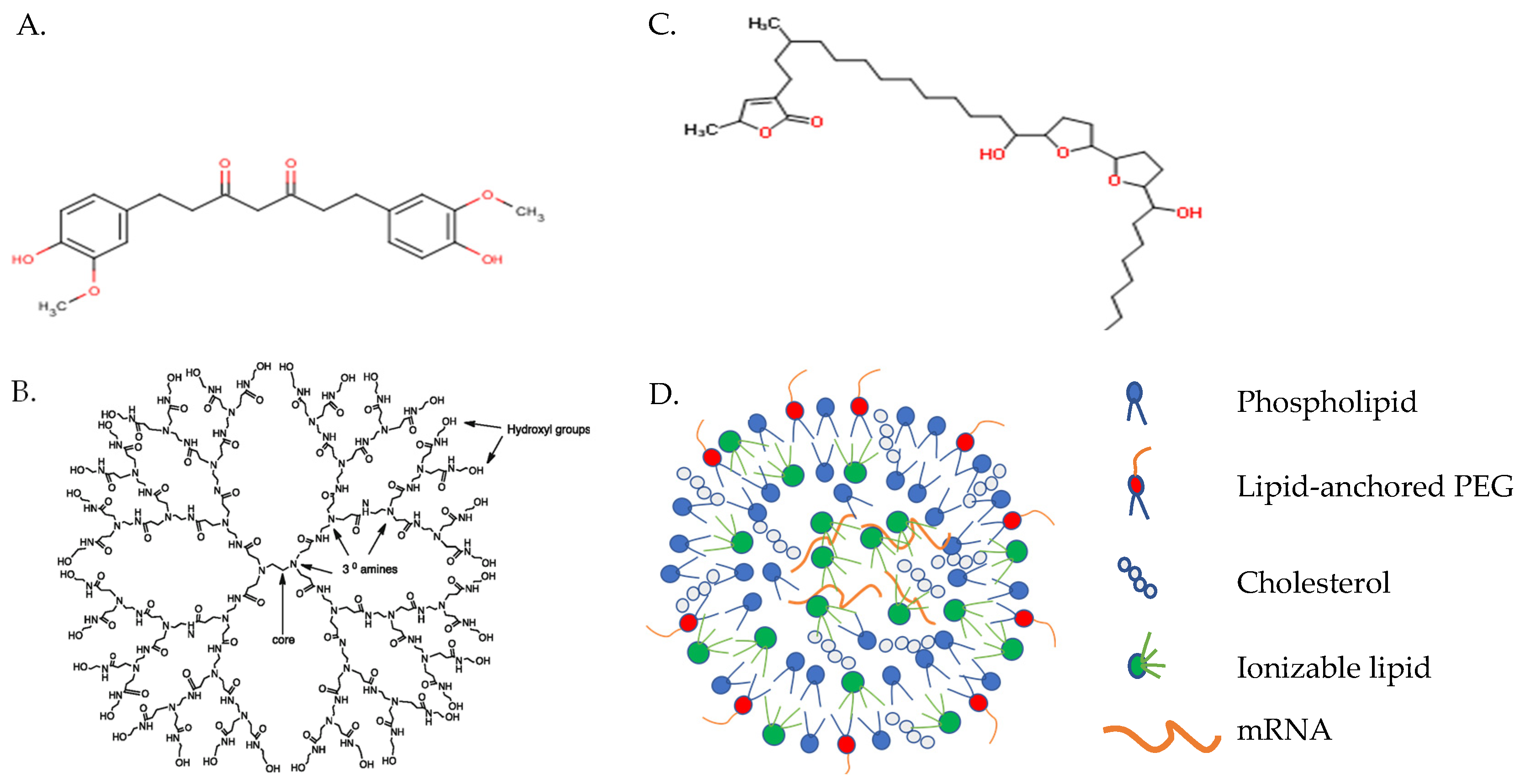
3. Delivery Systems
4. Clinical Applications
4.1. Cancer
4.2. Infectious Diseases
4.3. Allergies and Autoimmune Diseases
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronker, E.S.; Weenen, T.C.; Commandeur, H.; Claassen, E.H.; Osterhaus, A.D. Risk in vaccine research and development quantified. PLoS ONE 2013, 8, e57755. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.; Robinson, J.M.; Cunningham, G.; Iqbal, R.; Larsen, S. The complexity and cost of vaccine manufacturing—An over-view. Vaccine 2017, 35, 4064–4071. [Google Scholar] [CrossRef] [PubMed]
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2020, 21, 1–18. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural basis for m7G recognition and 2′-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Tanguay, R.L.; Gallie, D.R. Translational efficiency is regulated by the length of the 3’ untranslated region. Mol. Cell. Biol. 1996, 16, 146–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstrohm, A.C.; Wickens, M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008, 9, 337–344. [Google Scholar] [CrossRef]
- Lima, S.A.; Chipman, L.B.; Nicholson, A.L.; Chen, Y.-H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milligan, J.F.; Groebe, D.R.; Witherell, G.W.; Uhlenbeck, O.C. Oligoribonucleotide synthesis using T7 RNA polymerase and syn-thetic DNA templates. Nucleic Acids Res. 1987, 15, 8783–8798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Alonso, F.J.; Dabrowski, M.; Wadzack, J.; Nierhaus, K.H. Self-coded 3′-Extension of Run-off Transcripts Produces Aberrant Products during in Vitro Transcription with T7 RNA Polymerase. J. Biol. Chem. 1995, 270, 6298–6307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a transformative technology for vaccine development to control infectious diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Ljungberg, K.; Liljestrom, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2015, 14, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Vogel, A.B.; Lambert, L.; Kinnear, E.; Busse, D.; Erbar, S.; Reuter, K.C.; Wicke, L.; Perkovic, M.; Beissert, T.; Haas, H.; et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol. Ther. 2018, 26, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.W.; Mayerbarber, K.D.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by li-pid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Broos, K.; Van der Jeught, K.; Puttemans, J.; Goyvaerts, C.; Heirman, C.; Dewitte, H.; Verbeke, R.; Lentacker, I.; Thielemans, K.; Breckpot, K. Particle-mediated Intravenous Delivery of Antigen mRNA Results in Strong Antigen-specific T-cell Responses Despite the Induction of Type I Interferon. Mol. Ther. Nucleic Acids 2016, 5, e326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Wesselhoeft, R.A.; Kowalski, P.S.; Anderson, D.G. Engineering circular RNA for potent and stable translation in eukaryotic cells. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesselhoeft, R.A.; Kowalski, P.S.; Parker-Hale, F.C.; Huang, Y.; Bisaria, N.; Anderson, D.G. RNA circularization diminishes immu-nogenicity and can extend translation duration in vivo. Mol. Cell 2019, 74, 508–520. [Google Scholar] [CrossRef]
- Feng, Z.; Meng, S.; Zhou, H.; Xu, Z.; Tang, Y.; Li, P.; Liu, C.; Huang, Y.; Wu, M. Functions and Potential Applications of Circular RNAs in Cancer Stem Cells. Front. Oncol. 2019, 9, 500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseley, J.; Tollervey, D. The Many Pathways of RNA Degradation. Cell 2009, 136, 763–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, U.; Kariko, K.; Tureci, O. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Broderick, K.E.; Humeau, L.M. Enhanced Delivery of DNA or RNA Vaccines by Electroporation. Methods Mol. Biol. 2017, 1499, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Benteyn, D.; Heirman, C.; Bonehill, A.; Thielemans, K.; Breckpot, K. mRNA-based dendritic cell vaccines. Expert Rev. Vaccines 2015, 14, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M.; Diken, M.; Boegel, S.; Kreiter, S.; Türeci, Ÿ.; Sahin, U.; Türeci, Ö. Mutanome directed cancer immunotherapy. Curr. Opin. Immunol. 2016, 39, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Smits, E.L.; Anguille, S.; Cools, N.; Berneman, Z.N.; Van Tendeloo, V.F. Dendritic Cell-Based Cancer Gene Therapy. Hum. Gene Ther. 2009, 20, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Stanton, M.G. Current Status of Messenger RNA Delivery Systems. Nucleic Acid Ther. 2018, 28, 158–165. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immuno-therapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Alfagih, I.M.; Aldosari, B.; AlQuadeib, B.; Almurshedi, A.; Alfagih, M.M. Nanoparticles as adjuvants and nanodelivery systems for mRNA-based vaccines. Pharmaceutics 2020, 13, 45. [Google Scholar] [CrossRef]
- Durymanov, M.; Reineke, J. Nonviral delivery of nucleic acids: Insight into mechanisms of overcoming intracellular barriers. Front. Pharmacol. 2018, 9, 971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauch, S.; Lutz, J.; Kowalczyk, A.; Schlake, T.; Heidenreich, R. RNActive(R) technology: Generation and testing of stable and immunogenic mRNA vaccines. Methods Mol. Biol. 2017, 1499, 89–107. [Google Scholar] [PubMed]
- Kallen, K.J.; Heidenreich, R.; Schnee, M.; Petsch, B.; Schlake, T.; Thess, A.; Baumhof, P.; Scheel, B.; Koch, S.D.; Fotin-Mleczek, M. A novel, disruptive vaccination technology: Self-adjuvanted RNActive vaccines. Hum. Vaccin. Immunother. 2013, 9, 2263–2276. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armbruster, N.; Jasny, E.; Petsch, B. Advances in RNA Vaccines for Preventive Indications: A Case Study of a Vaccine Against Rabies. Vaccines 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, M.; Carrasco, M.; Alishetty, S.; Paige, M.; Alameh, M.; Weissman, D. Nanomaterial Delivery Systems for mRNA Vaccines. Vaccines 2021, 9, 65. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepini, T.; Pulichino, A.M.; Carsillo, T.; Carlson, A.L.; Sari-Sarraf, F.; Ramsauer, K.; Debasitis, J.C.; Maruggi, G.; Otten, G.R.; Geall, A.J.; et al. Induction of an IFN-mediated antiviral response by a self-amplifying RNA vac-cine: Implications for vaccine design. J. Immunol. 2017, 198, 4012–4024. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, G.; Thoryk, E.A.; Cox, K.S.; Meschino, S.; Dubey, S.A.; Vora, K.A.; Celano, R.; Gindy, M.; Casimiro, D.R.; Bett, A.J. A novel lipid nanoparticle adjuvant significantly enhances B cell and T cell responses to sub-unit vaccine antigens. Vaccine 2016, 34, 110–119. [Google Scholar] [CrossRef]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.A.; Midoux, P. Enhancement of Dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with Mannosylated Histidylated lipopolyplexes loaded with tumor antigen mRNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef]
- Tacken, P.J.; Torensma, R.; Figdor, C.G. Targeting antigens to dendritic cells in vivo. Immunobiology 2006, 211, 599–608. [Google Scholar] [CrossRef]
- Veiga, N.; Diesendruck, Y.; Peer, D. Targeted lipid nanoparticles for RNA therapeutics and immunomodulation in leukocytes. Adv. Drug Deliv. Rev. 2020, 159, 364–376. [Google Scholar] [CrossRef]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Lindgren, G.; Lin, A.; Thompson, E.A.; Ols, S.; Röhss, J.; John, S.; Hassett, K.; Yuzhakov, O.; Bahl, K.; et al. Efficient Targeting and Activation of Antigen-Presenting Cells In Vivo after Modified mRNA Vaccine Administration in Rhesus Macaques. Mol. Ther. 2017, 25, 2635–2647. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, M.F.; Jennings, G.T. Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 2010, 10, 787–796. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, C.; Walker, P.G.; Dong, Y. Formulation and Delivery Technologies for mRNA Vaccines. In Current Topics in Microbiology and Immunology; Springer Nature: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Rausch, S.; Schwentner, C.; Stenzl, A.; Bedke, J. mRNA vaccine CV9103 and CV9104 for the treatment of prostate cancer. Hum. Vaccines Immunother. 2014, 10, 3146–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiedler, K.; Lazzaro, S.; Lütz, J.; Rauch, S.; Heidenreich, R. mRNA Cancer Vaccines. Adv. Struct. Saf. Stud. 2016, 209, 61–85. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bordon, Y. An RNA vaccine for advanced melanoma. Nat. Rev. Immun. 2020, 20, 571. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA vaccines protect against Zika virus infection. Cell 2017, 168, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Aldrich, C.; Leroux-Roels, I.; Huang, K.B.; Bica, M.A.; Loeliger, E.; Schoenborn-Kellenberger, O.; Walz, L.; Leroux-Roels, G.; von Sonnenburg, F.; Oostvogels, L. Proof-of-concept of a low-dose unmodified mRNA-based rabies vaccine formulated with lipid nanoparticles in human volunteers: A phase 1 trial. Vaccine 2021, 39, 1310–1318. [Google Scholar] [CrossRef]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Shimabukuro, T.; Nair, N. Allergic reactions including anaphylaxis after receipt of the first dose of Pfizer-BioNTech COVID-19 vaccine. JAMA 2021, 325, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2020, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.A.; Roberts, P.C.; Graham, B.S. A SARS-CoV-2 mRNA vaccine—Preliminary report. reply. N. Engl. J. Med. 2020, 383, 1191–1192. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Gooch, K.; Hall, Y.; Salguero, F.J.; Dennis, M.J.; Gleeson, F.V.; Harris, D.; Ho, C.; Humphries, H.E.; Longet, S.; et al. mRNA vaccine CVnCoV protects non-human primates from SARS-CoV-2 challenge infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kremsner, P.; Mann, P.; Oostvogels, L.; Kreidenweiss, A.; Leroux-Roels, I.; Leroux-Roels, G.; Kroidl, A.; Schunk, M.; Schindler, C.; Fendel, R.; et al. Phase 1 Assessment of the safety and immunogenicity of an mRNA-lipid nanoparticle vaccine candidate against SARS-CoV-2 in human volunteers. medRxiv 2020. [Google Scholar] [CrossRef]
- Novel Lipids and Lipid Nanoparticle Formulations for Delivery of Nucleic Acids. Available online: https://patents.google.com/patent/WO2017075531A1/en (accessed on 8 April 2021).
- Burki, T. Understanding variants of SARS-CoV-2. Lancet 2021, 397, 462. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021, 1–2. [Google Scholar] [CrossRef]
- Muik, A.; Chen, A.; Wallish, A.K.; Sänger, B.; Swanson, K.A.; Mühl, J.; Chen, W.; Cai, H.; Maurus, D.; Sarkar, R.; et al. Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera. Science 2021, 371, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Scheiblhofer, S.; Thalhamer, J.; Weiss, R. DNA and mRNA vaccination against allergies. Pediatr. Allergy Immunol. 2018, 29, 679–688. [Google Scholar] [CrossRef]
- Hattinger, E.; Scheiblhofer, S.; Roesler, E.; Thalhamer, T.; Weiss, R. Prophylactic mRNA Vaccination against Allergy Confers Long-Term Memory Responses and Persistent Protection in Mice. J. Immunol. Res. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Company | Vaccine | mRNA | Immunogen | LNP (Probable Ionizable Lipid) | Dose (μg mRNA, Twice) |
|---|---|---|---|---|---|
| Pfizer-BioNTech | BNT162b2 | modified nucleoside | prefusion stabilized spike protein | Acuitas ALC-0315 [81] | 30 |
| Moderna | mRNA-1273 | modified nucleoside | prefusion stabilized spike protein | Lipid H [52] | 100 |
| CureVac | CVnCoV | unmodified | prefusion stabilized spike protein | Acuitas ALC-0315 [81] | 12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kowalzik, F.; Schreiner, D.; Jensen, C.; Teschner, D.; Gehring, S.; Zepp, F. mRNA-Based Vaccines. Vaccines 2021, 9, 390. https://doi.org/10.3390/vaccines9040390
Kowalzik F, Schreiner D, Jensen C, Teschner D, Gehring S, Zepp F. mRNA-Based Vaccines. Vaccines. 2021; 9(4):390. https://doi.org/10.3390/vaccines9040390
Chicago/Turabian StyleKowalzik, Frank, Daniel Schreiner, Christian Jensen, Daniel Teschner, Stephan Gehring, and Fred Zepp. 2021. "mRNA-Based Vaccines" Vaccines 9, no. 4: 390. https://doi.org/10.3390/vaccines9040390
APA StyleKowalzik, F., Schreiner, D., Jensen, C., Teschner, D., Gehring, S., & Zepp, F. (2021). mRNA-Based Vaccines. Vaccines, 9(4), 390. https://doi.org/10.3390/vaccines9040390