
Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States


 ,
,  ,
, 
 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
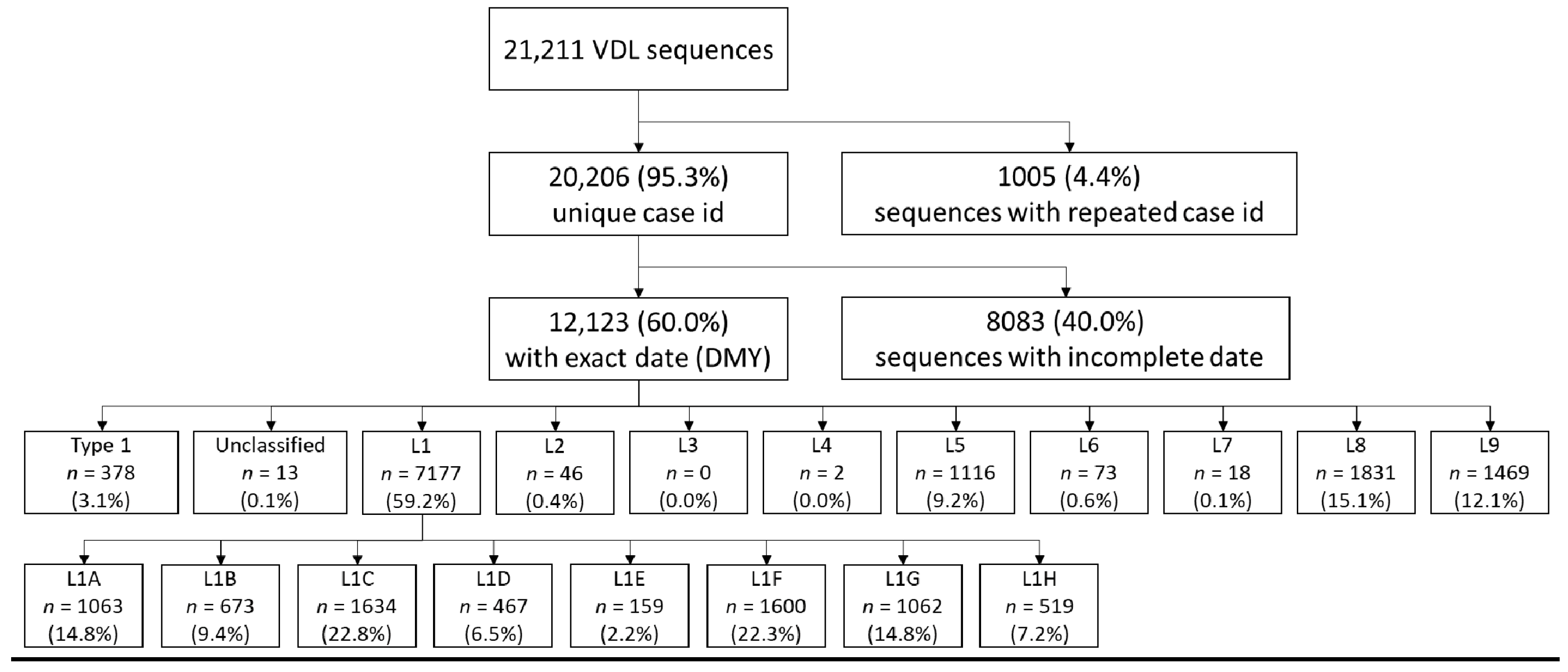
2.1. Source of Sequences
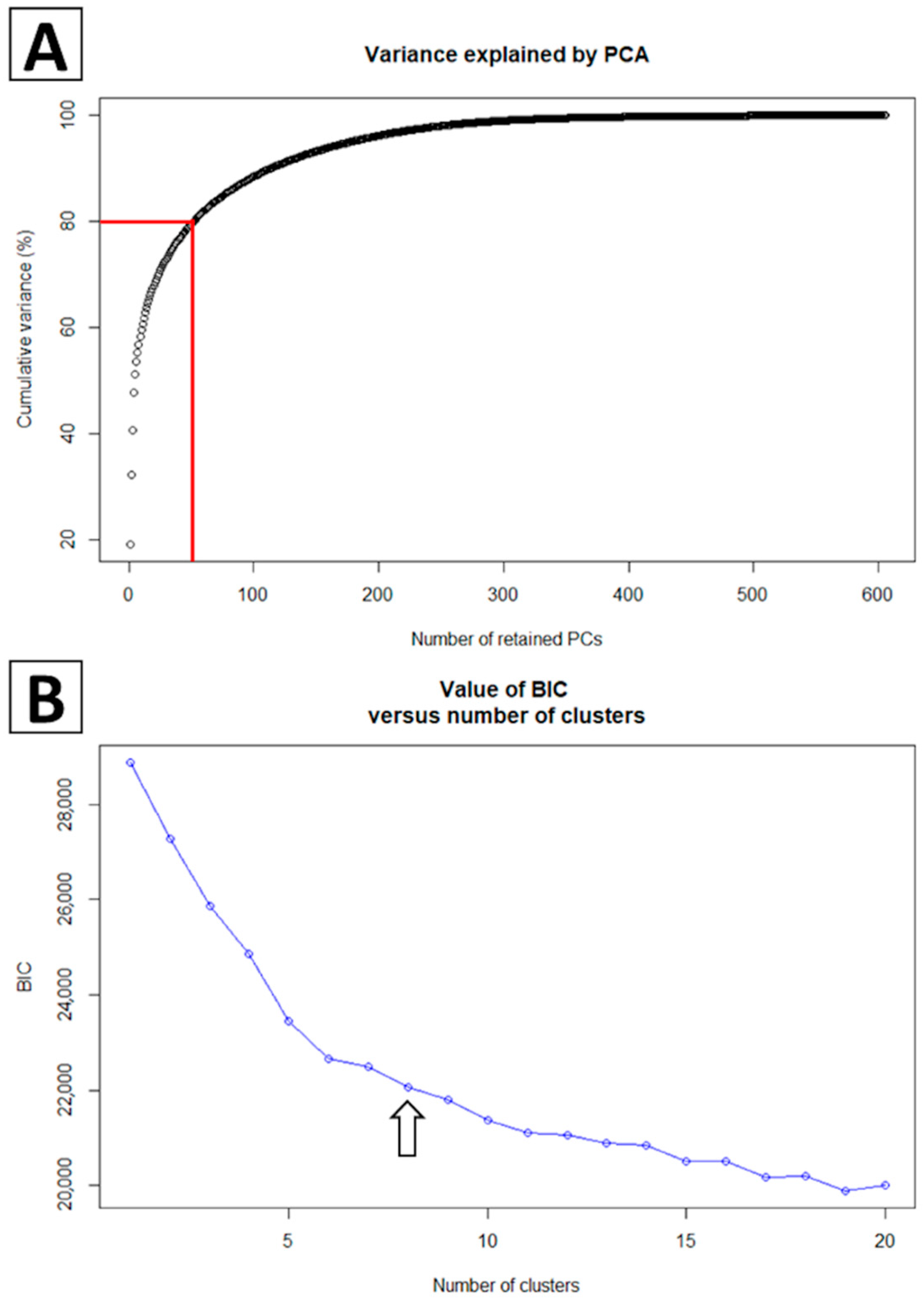
2.2. Phylogenetic Classification Using Discriminant Analysis of Principal Components
2.3. Effective Population Size through Time
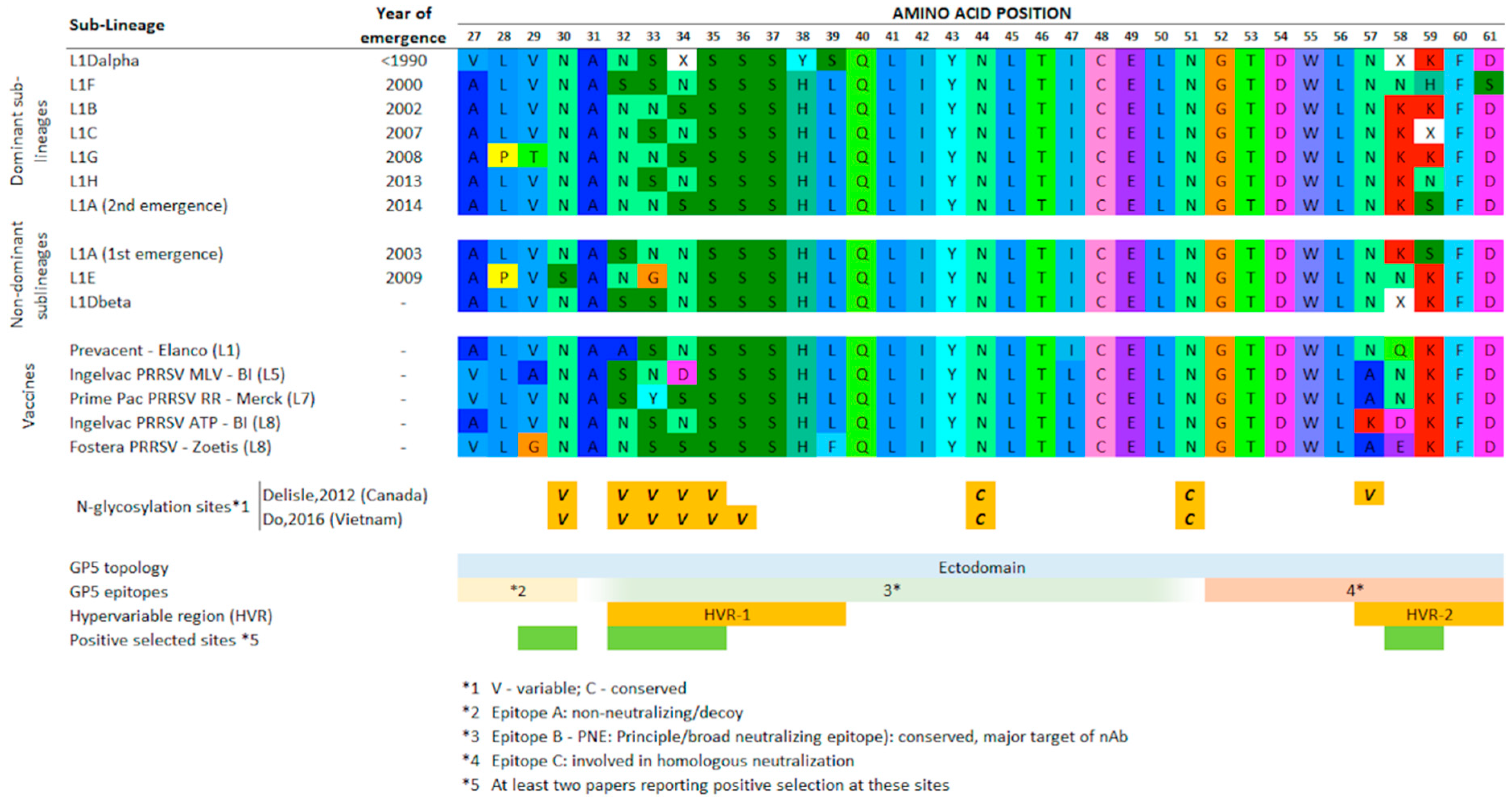
2.4. Clade-Prevalent Mutations
2.5. Comparison of Databases: UMN VDL & GenBank
3. Results
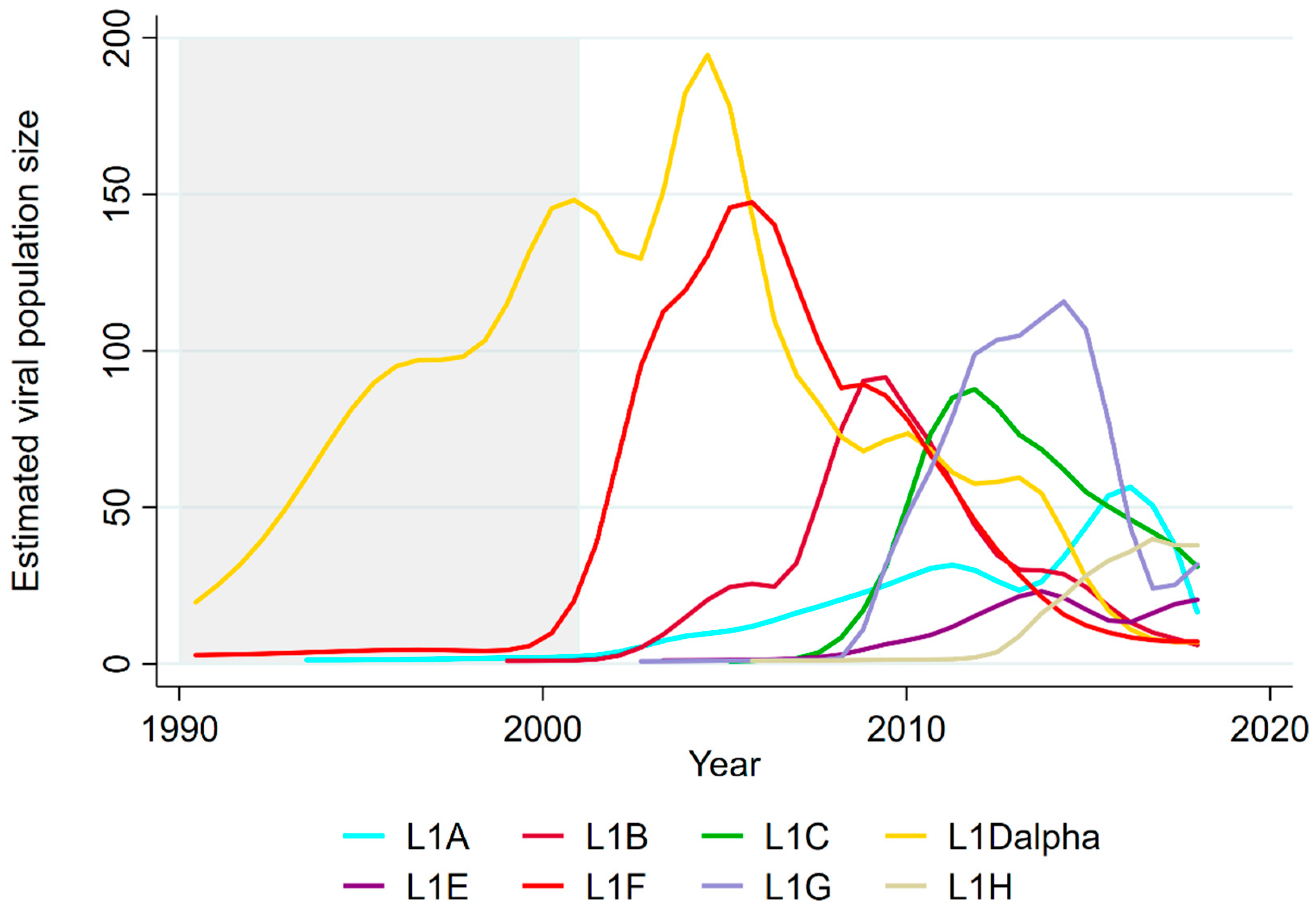
3.1. Sequential Dominance of Sub-Lineages through Time
3.2. Clade-Prevalent Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stadejek, T.; Stankevicius, A.; Murtaugh, M.P.; Oleksiewicz, M.B. Molecular evolution of PRRSV in Europe: Current state of play. Vet. Microbiol. 2013, 165, 21–28. [Google Scholar] [CrossRef] [PubMed]
- VanderWaal, K.; Deen, J. Global trends in infectious diseases of swine. Proc. Natl. Acad. Sci. USA 2018, 115, 11495–11500. [Google Scholar] [CrossRef] [Green Version]
- Tian, K.; Yu, X.; Zhao, T.; Feng, Y.; Cao, Z.; Wang, C.; Hu, Y.; Chen, X.; Hu, D.; Tian, X.; et al. Emergence of Fatal PRRSV Variants: Unparalleled Outbreaks of Atypical PRRS in China and Molecular Dissection of the Unique Hallmark. PLoS ONE 2007, 2, e526. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, H. Porcine reproductive and respiratory syndrome in China. Virus Res. 2010, 154, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Holtkamp, D.J.; Kliebenstein, J.B.; Neumann, E.J.; Zimmerman, J.J.; Rotto, H.F.; Yoder, T.K.; Wang, C.; Yeske, P.E.; Mowrer, C.L.; Haley, C. Assessment of the economic impact of porcine reproductive and respiratory syndrome virus on United States pork producers. J. Swine Health Prod. 2013, 21, 72–84. [Google Scholar]
- Christianson, W.T.; Joo, H.S. Porcine reproductive and respiratory syndrome: A review. J. Swine Health Prod. 1994, 2, 10–28. [Google Scholar]
- Pejsak, Z.; Stadejek, T.; Markowska-Daniel, I. Clinical signs and economic losses caused by porcine reproductive and respiratory syndrome virus in a large breeding farm. Vet. Microbiol. 1997, 55, 317–322. [Google Scholar] [CrossRef]
- Solano, G.I.; Segalés, J.; Collins, J.E.; Molitor, T.W.; Pijoan, C. Porcine reproductive and respiratory syndrome virus (PRRSv) interac-tion with Haemophilus parasuis. Vet. Microbiol. 1997, 55, 247–257. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Li, L.; Lei, L.; Liu, Y.; Shi, W.; Wu, J.; Li, L.; Rong, F.; Xu, M.; et al. Secondary infection with Streptococcus suis serotype 7 increases the virulence of highly pathogenic porcine reproductive and respiratory syndrome virus in pigs. Virol. J. 2010, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousignant, S.J.; Perez, A.; Morrison, R. Comparison between the 2013–2014 and 2009–2012 annual porcine reproductive and respiratory syndrome virus epidemics in a cohort of sow herds in the United States. Can. Vet. J. 2015, 56, 1087–1089. [Google Scholar]
- Wang, X.; Marthaler, D.; Rovira, A.; Rossow, S.; Murtaugh, M.P. Emergence of a virulent porcine reproductive and respiratory syndrome virus in vaccinated herds in the United States. Virus Res. 2015, 210, 34–41. [Google Scholar] [CrossRef]
- Rowland, R.R.; Steffen, M.; Ackerman, T.; Benfield, D.A. The Evolution of Porcine Reproductive and Respiratory Syndrome Virus: Quasispecies and Emergence of a Virus Subpopulation during Infection of Pigs with VR-2332. Virology 1999, 259, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Cano, J.P.; Dee, S.A.; Murtaugh, M.P.; Trincado, C.A.; Pijoan, C.B. Effect of vaccination with a modified-live porcine reproductive and respiratory syndrome virus vaccine on dynamics of homologous viral infection in pigs. Am. J. Vet. Res. 2007, 68, 565–571. [Google Scholar] [CrossRef]
- Díaz, I.; Gimeno, M.; Darwich, L.; Navarro, N.; Kuzemtseva, L.; López, S.; Galindo, I.; Segalés, J.; Martín, M.; Pujols, J.; et al. Characterization of homologous and heterologous adaptive immune responses in porcine reproductive and respiratory syndrome virus infection. Vet. Res. 2012, 43, 30. [Google Scholar] [CrossRef] [Green Version]
- Geldhof, M.F.; Vanhee, M.; Van Breedam, W.; Van Doorsselaere, J.; Karniychuk, U.U.; Nauwynck, H.J. Comparison of the efficacy of autogenous inactivated Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) vaccines with that of commercial vaccines against homologous and heterologous challenges. BMC Vet. Res. 2012, 8, 182. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Galliher-Beckley, A.; Pappan, L.; Trible, B.; Kerrigan, M.; Beck, A.; Hesse, R.; Blecha, F.; Nietfeld, J.C.; Rowland, R.R.; et al. Comparison of Host Immune Responses to Homolo-gous and Heterologous Type II Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Challenge in Vaccinated and Unvaccinated Pigs. Biomed Res. Int. 2014, 2014, 1–10. [Google Scholar]
- Mateu, E.; Diaz, I. The challenge of PRRS immunology. Vet. J. 2008, 177, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Xu, H.; Wang, K.; He, F. Emergence of Two different recombinant PRRSV strains with low neutralizing antibody sus-ceptibility in China. Sci. Rep. 2019, 9, 2490. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, J.; Yang, J.; Zeng, H.; Guo, L.; Ren, S.; Sun, W.; Chen, Z.; Cong, X.; Shi, J.; et al. Emergence of Different Recombinant Porcine Reproductive and Respiratory Syndrome Viruses, China. Sci. Rep. 2018, 8, 4118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Hui, R.K.-H.; Faaberg, K.S.; Wennblom, T.; Murtaugh, M.P.; Stadejek, T.; Leung, F.C.-C. Molecular epidemiology of PRRSV: A phylogenetic perspective. Virus Res. 2010, 154, 7–17. [Google Scholar] [CrossRef]
- Kapur, V.; Elam, M.R.; Pawlovich, T.M.; Murtaugh, M.P. Genetic variation in porcine reproductive and respiratory syndrome virus isolates in the midwestern United States. J. Gen. Virol. 1996, 77, 1271–1276. [Google Scholar] [CrossRef]
- Wesley, R.D.; Mengeling, W.L.; Lager, K.M.; Clouser, D.F.; Landgraf, J.G.; Frey, M.L. Differentiation of a porcine reproductive and respiratory syndrome virus vaccine strain from North American field strains by restriction fragment length polymorphism analysis of ORF 5. J. Vet. Diagn. Investig. 1998, 10, 140–144. [Google Scholar] [CrossRef] [Green Version]
- Paploski, I.A.D.; Corzo, C.; Rovira, A.; Murtaugh, M.P.; Sanhueza, J.M.; Vilalta, C.; Schroeder , D.C.; VanderWaal, K. Temporal Dynamics of Co-circulating Line-ages of Porcine Reproductive and Respiratory Syndrome Virus. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.-H.; Chang, C.-C.; Yoon, K.-J. Instability of the Restriction Fragment Length Polymorphism Pattern of Open Reading Frame 5 of Porcine Reproductive and Respiratory Syndrome Virus during Sequential Pig-to-Pig Passages. J. Clin. Microbiol. 2004, 42, 4462–4467. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lam, T.T.-Y.; Hon, C.-C.; Murtaugh, M.P.; Davies, P.R.; Hui, R.K.-H.; Li, J.; Wong, L.T.-W.; Yip, C.-W.; Jiang, J.-W.; et al. Phylogeny-Based Evolutionary, Demographical, and Geographical Dissection of North American Type 2 Porcine Reproductive and Respiratory Syndrome Viruses. J. Virol. 2010, 84, 8700–8711. [Google Scholar] [CrossRef] [Green Version]
- VanderWaal, K.; Paploski, I.A.; Makau, D.N.; Corzo, C.A. Contrasting animal movement and spatial connectivity networks in shaping transmission pathways of a genetically diverse virus. Prev. Vet. Med. 2020, 178, 104977. [Google Scholar] [CrossRef]
- Makau, D.N.; Paploski, I.A.D.; Corzo, C.A.; VanderWaal, K. Dynamic network connectivity influences the spread of a sub-lineage of porcine reproductive and respiratory syndrome virus. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Plat-forms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- StataCorp. Stata Statistical Software: Release 15; StataCorp, LLC: College Station, TX, USA, 2017. [Google Scholar]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing Computer Program, Version 3.6.1; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, V.; Baele, G. Bayesian Estimation of Past Population Dynamics in BEAST 1.10 Using the Skygrid Coalescent Model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhamis, M.A.; Arruda, A.G.; Morrison, R.B.; Perez, A.M. Novel approaches for Spatial and Molecular Surveillance of Porcine Reproductive and Respiratory Syndrome Virus (PRRSv) in the United States. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Geneious Prime. Available online: http://www.geneious.com/ (accessed on 7 April 2020).
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; A Neher, R. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Hanada, K.; Suzuki, Y.; Nakane, T.; Hirose, O.; Gojobori, T. The Origin and Evolution of Porcine Reproductive and Respiratory Syndrome Viruses. Mol. Biol. Evol. 2005, 22, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Delisle, B.; Gagnon, C.A.; Lambert, M.-È.; D’Allaire, S. Porcine reproductive and respiratory syndrome virus diversity of Eastern Canada swine herds in a large sequence dataset reveals two hypervariable regions under positive selection. Infect. Genet. Evol. 2012, 12, 1111–1119. [Google Scholar] [CrossRef]
- Do, H.Q.; Trinh, D.T.; Nguyen, T.L.; Vu, T.T.H.; Than, D.D.; Van Lo, T.; Yeom, M.; Song, D.; Choe, S.; An, D.-J.; et al. Molecular evolution of type 2 porcine reproductive and respiratory syndrome viruses circulating in Vietnam from 2007 to 2015. BMC Vet. Res. 2016, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Li, X.; Zhang, Z.; Shuai, J.; Chen, N.; Liu, G.; Fang, W. Porcine reproductive and respiratory syndrome viruses predominant in southeastern China from 2004 to 2007 were from a common source and underwent further divergence. Arch. Virol. 2009, 154, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, D.; Odogwu, N.; Kevill, J.; Yang, M.; Krishna, V.; Kikuti, M. Genomes of porcine reproductive and respiratory syndrome virus type-2 variants responsible for four distinct disease outbreaks in US swine farms over the past 6 years. Submited Publ.
- Murtaugh, M.P.; Stadejek, T.; Abrahante, J.; Lam, T.T.; Leung, F.C.-C. The ever-expanding diversity of porcine reproductive and respiratory syndrome virus. Virus Res. 2010, 154, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.-I.; Kim, J.-J.; Cha, S.-H.; Wu, W.-H.; Cooper, V.; Evans, R.; Choi, E.-J.; Yoon, K.-J. Significance of genetic variation of PRRSV ORF5 in virus neutralization and molecular determinants corresponding to cross neutralization among PRRS viruses. Vet. Microbiol. 2013, 162, 10–22. [Google Scholar] [CrossRef]
- Correas, I.; Osorio, F.A.; Steffen, D.; Pattnaik, A.K.; Vu, H.L.X. Cross reactivity of immune responses to porcine reproductive and respiratory syndrome virus infection. Vaccine 2017, 35, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, I.; Kwon, B.; Osorio, F.A.; Pattnaik, A.K. Influence of N-Linked Glycosylation of Porcine Reproductive and Respiratory Syndrome Virus GP5 on Virus Infectivity, Antigenicity, and Ability To Induce Neutralizing Antibodies. J. Virol. 2006, 80, 3994–4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, T.; Yoo, S.J.; Lee, D.-U.; Sunwoo, S.Y.; Je, S.H.; Park, J.W.; Kim, M.-H.; Park, C.-K.; Lyoo, Y.S. Differential evolution of antigenic regions of porcine reproduc-tive and respiratory syndrome virus 1 before and after vaccine introduction. Virus Res. 2019, 260, 12–19. [Google Scholar] [CrossRef]
- Dea, S.; Gagnon, C.A.; Mardassi, H.; Pirzadeh, B.; Rogan, D. Current knowledge on the structural proteins of porcine reproductive and respiratory syndrome (PRRS) virus: Comparison of the North American and European isolates. Arch. Virol. 2000, 145, 659–688. [Google Scholar] [CrossRef]
- Popescu, L.N.; Trible, B.R.; Chen, N.; Rowland, R.R. GP5 of porcine reproductive and respiratory syndrome virus (PRRSV) as a target for homologous and broadly neutralizing antibodies. Vet. Microbiol. 2017, 209, 90–96. [Google Scholar] [CrossRef]
- Chen, J.; Liu, T.; Zhu, C.-G.; Jin, Y.-F.; Zhang, Y.-Z. Genetic Variation of Chinese PRRSV Strains Based on ORF5 Sequence. Biochem. Genet. 2006, 44, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Key, K.F.; Haqshenas, G.; Guenette, D.K.; Swenson, S.L.; Toth, T.E.; Meng, X.-J. Genetic variation and phylogenetic analyses of the ORF5 gene of acute porcine reproductive and respiratory syndrome virus isolates. Vet. Microbiol. 2001, 83, 249–263. [Google Scholar] [CrossRef]
- LaRochelle, R.; D’Allaire, S.; Magar, R. Molecular epidemiology of porcine reproductive and respiratory syndrome virus (PRRSV) in Québec. Virus Res. 2003, 96, 3–14. [Google Scholar] [CrossRef]
- Faaberg, K.S.; Hocker, J.D.; Erdman, M.M.; Harris, D.H.; Nelson, E.A.; Torremorell, M.; Plagemann, P.G. Neutralizing Antibody Responses of Pigs Infected with Natural GP5 N-Glycan Mutants of Porcine Reproductive and Respiratory Syndrome Virus. Viral Immunol. 2006, 19, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Tao, S.; Orlando, R.; Murtaugh, M.P. N-glycosylation profiling of porcine reproductive and respiratory syndrome virus en-velope glycoprotein 5. Virology 2015, 478, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.; Galeota, J.A.; Jar, A.M.; Platt, K.B.; Osorio, F.A.; Lopez, O.J. Identification of neutralizing and nonneutralizing epitopes in the porcine reproductive and respiratory syndrome virus GP5 ectodomain. J. Virol. 2002, 76, 4241–4250. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Wei, Y.; Lin, S.; Huang, J.; He, W.; Yao, J.; Chen, Y.; Kang, O.; Huang, W.; Wei, Z. Genetic analysis of porcine productive and respiratory syndrome virus between 2013 and 2014 in Southern parts of China: Identification of several novel strains with amino acid deletions or insertions in nsp2. BMC Vet. Res. 2019, 15, 171. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Li, G.; Yu, L.; Li, L.; Zhang, Y.; Zhou, Y.; Tong, W.; Liu, C.; Gao, F.; Tong, G. Genetic Diversity of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) From 1996 to 2017 in China. Front. Microbiol. 2020, 11, 618. [Google Scholar] [CrossRef]
- Lee, J.-A.; Lee, N.-H.; Lee, J.-B.; Park, S.-Y.; Song, C.-S.; Choi, I.-S.; Lee, S.-W. Genetic diversity of the Korean field strains of porcine repro-ductive and respiratory syndrome virus. Infect. Genet. Evol. 2016, 40, 288–294. [Google Scholar] [CrossRef]
- Brar, M.S.; Murtaugh, M.P.; Shi, M.; Leung, F.C.-C. Evolutionary diversification of type 2 porcine reproductive and respiratory syndrome virus. J. Gen. Virol. 2015, 96, 1570–1580. [Google Scholar] [CrossRef]
- Kikuti, M.; Paploski, I.; Pamornchainavakul, N.; Picasso-Risso, C.; Schwartz, M.; Yeske, P. Outbreak of a Type 2 Lineage 1C variant of Porcine Reproductive and Respiratory Syndrome Virus in the United States. Submited Publ.
- LaLonde, C.; Provost, C.; Gagnon, C.A. Whole-Genome Sequencing of Porcine Reproductive and Respiratory Syndrome Virus from Field Clinical Samples Improves the Genomic Surveillance of the Virus. J. Clin. Microbiol. 2020, 58. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | L1A | L1B | L1C | L1D | L1Dalpha | L1Dbeta | L1E | L1F | L1G | L1H | L2 | L4 | L5 | L6 | L7 | L8 | L9 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 1095 | n = 683 | n = 1663 | n = 469 | n = 405 | n = 64 | n = 161 | n = 1600 | n = 1062 | n = 519 | n = 46 | n = 2 | n = 1116 | n = 73 | n = 18 | n = 1831 | n = 1469 | |
| L1A | 4.0 | ||||||||||||||||
| L1B | 10.1 | 4.0 | |||||||||||||||
| L1C | 13.4 | 14.5 | 6.0 | ||||||||||||||
| L1D | 12.6 | 14.4 | 14.2 | 12.0 | |||||||||||||
| L1Dalpha | 12.7 | 14.6 | 14.7 | 12.0 | 12.0 | ||||||||||||
| L1Dbeta | 11.8 | 13.6 | 11.4 | 12.2 | 13.0 | 7.0 | |||||||||||
| L1E | 14.9 | 15.7 | 16.2 | 14.6 | 14.6 | 14.6 | 5.0 | ||||||||||
| L1F | 11.7 | 13.7 | 12.5 | 13.6 | 13.9 | 11.4 | 15.5 | 6.0 | |||||||||
| L1G | 11.0 | 6.2 | 15.0 | 15.2 | 15.4 | 14.0 | 16.3 | 14.0 | 5.0 | ||||||||
| L1H | 14.2 | 15.9 | 14.7 | 14.7 | 15.3 | 11.4 | 17.1 | 13.1 | 15.9 | 5.0 | |||||||
| L2 | 15.4 | 16.6 | 17.5 | 15.4 | 15.5 | 15.4 | 16.3 | 16.2 | 17.2 | 16.8 | 12.0 | ||||||
| L4 | 14.9 | 15.6 | 16.7 | 15.3 | 15.3 | 15.3 | 17.1 | 15.8 | 16.7 | 17.3 | 16.6 | 16.0 | |||||
| L5 | 14.2 | 15.4 | 16.9 | 14.0 | 13.9 | 15.0 | 16.7 | 16.4 | 16.0 | 17.7 | 13.5 | 14.7 | 4.0 | ||||
| L6 | 17.3 | 18.5 | 19.4 | 16.6 | 16.5 | 17.5 | 18.8 | 18.1 | 18.9 | 17.8 | 17.0 | 17.4 | 13.4 | 6.0 | |||
| L7 | 13.8 | 16.0 | 16.0 | 13.6 | 13.5 | 14.1 | 15.8 | 15.8 | 17.0 | 16.9 | 14.2 | 14.8 | 11.3 | 14.7 | 5.0 | ||
| L8 | 15.3 | 16.0 | 17.1 | 14.2 | 14.0 | 15.3 | 16.4 | 16.6 | 17.2 | 17.5 | 14.7 | 15.7 | 12.0 | 15.1 | 12.4 | 6.0 | |
| L9 | 16.8 | 18.0 | 17.9 | 15.0 | 14.9 | 15.7 | 17.7 | 17.3 | 18.7 | 17.7 | 15.8 | 16.9 | 13.7 | 15.0 | 12.2 | 12.5 | 11.0 |
| Prevacent-Elanco (L1) | 12.4 | 15.0 | 12.8 | 13.2 | 14.1 | 7.5 | 15.7 | 12.5 | 15.1 | 12.2 | 16.8 | 16.6 | 15.7 | 19.0 | 14.5 | 16.9 | 16.8 |
| Ingelvac PRRSV MLV-BI (L5) | 13.9 | 15.3 | 16.8 | 13.7 | 13.5 | 14.8 | 16.7 | 16.1 | 15.8 | 17.6 | 13.2 | 14.3 | 2.2 | 12.7 | 10.8 | 11.7 | 13.5 |
| Prime Pac PRRSV RR-Merck (L7) | 13.8 | 16.0 | 16.1 | 13.3 | 13.2 | 13.9 | 16.5 | 15.7 | 17.0 | 16.5 | 13.8 | 14.7 | 10.6 | 14.1 | 3.2 | 11.7 | 11.4 |
| Ingelvac PRRSV ATP-BI (L8) | 14.8 | 15.4 | 16.9 | 13.8 | 13.6 | 15.2 | 15.9 | 16.3 | 16.7 | 17.4 | 14.3 | 15.2 | 11.2 | 14.8 | 12.3 | 4.0 | 12.4 |
| Fostera PRRSV-Zoetis (L8) | 14.6 | 15.9 | 16.2 | 12.6 | 12.4 | 14.3 | 15.4 | 15.9 | 16.7 | 17.1 | 13.0 | 14.1 | 9.8 | 12.4 | 8.8 | 7.3 | 9.3 |
| Sublineage | Year of 1st Emergence | Year of 2nd Emergence | ||||
|---|---|---|---|---|---|---|
| Emergence | Peak | Difference between Emergence and Peak | Emergence | Peak | Difference btween Emergence and Peak | |
| L1Dalpha | <1990 | 2004 | >15 | - | - | - |
| L1F | 2000 | 2005 | 5 | - | - | - |
| L1B | 2002 | 2008 | 6 | - | - | - |
| L1A | 2003 | 2011 | 8 | 2014 | 2016 | 2 |
| L1C | 2007 | 2011 | 4 | - | - | - |
| L1G | 2008 | 2014 | 6 | - | - | - |
| L1E | 2009 | 2014 | 5 | 2016 | 2018 * | 2 |
| L1H | 2013 | 2017 * | 4 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paploski, I.A.D.; Pamornchainavakul, N.; Makau, D.N.; Rovira, A.; Corzo, C.A.; Schroeder, D.C.; Cheeran, M.C.-J.; Doeschl-Wilson, A.; Kao, R.R.; Lycett, S.; et al. Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States. Vaccines 2021, 9, 608. https://doi.org/10.3390/vaccines9060608
Paploski IAD, Pamornchainavakul N, Makau DN, Rovira A, Corzo CA, Schroeder DC, Cheeran MC-J, Doeschl-Wilson A, Kao RR, Lycett S, et al. Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States. Vaccines. 2021; 9(6):608. https://doi.org/10.3390/vaccines9060608
Chicago/Turabian StylePaploski, Igor A. D., Nakarin Pamornchainavakul, Dennis N. Makau, Albert Rovira, Cesar A. Corzo, Declan C. Schroeder, Maxim C-J. Cheeran, Andrea Doeschl-Wilson, Rowland R. Kao, Samantha Lycett, and et al. 2021. "Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States" Vaccines 9, no. 6: 608. https://doi.org/10.3390/vaccines9060608
APA StylePaploski, I. A. D., Pamornchainavakul, N., Makau, D. N., Rovira, A., Corzo, C. A., Schroeder, D. C., Cheeran, M. C. -J., Doeschl-Wilson, A., Kao, R. R., Lycett, S., & VanderWaal, K. (2021). Phylogenetic Structure and Sequential Dominance of Sub-Lineages of PRRSV Type-2 Lineage 1 in the United States. Vaccines, 9(6), 608. https://doi.org/10.3390/vaccines9060608