
Cancer Vaccines: Promising Therapeutics or an Unattainable Dream

Abstract
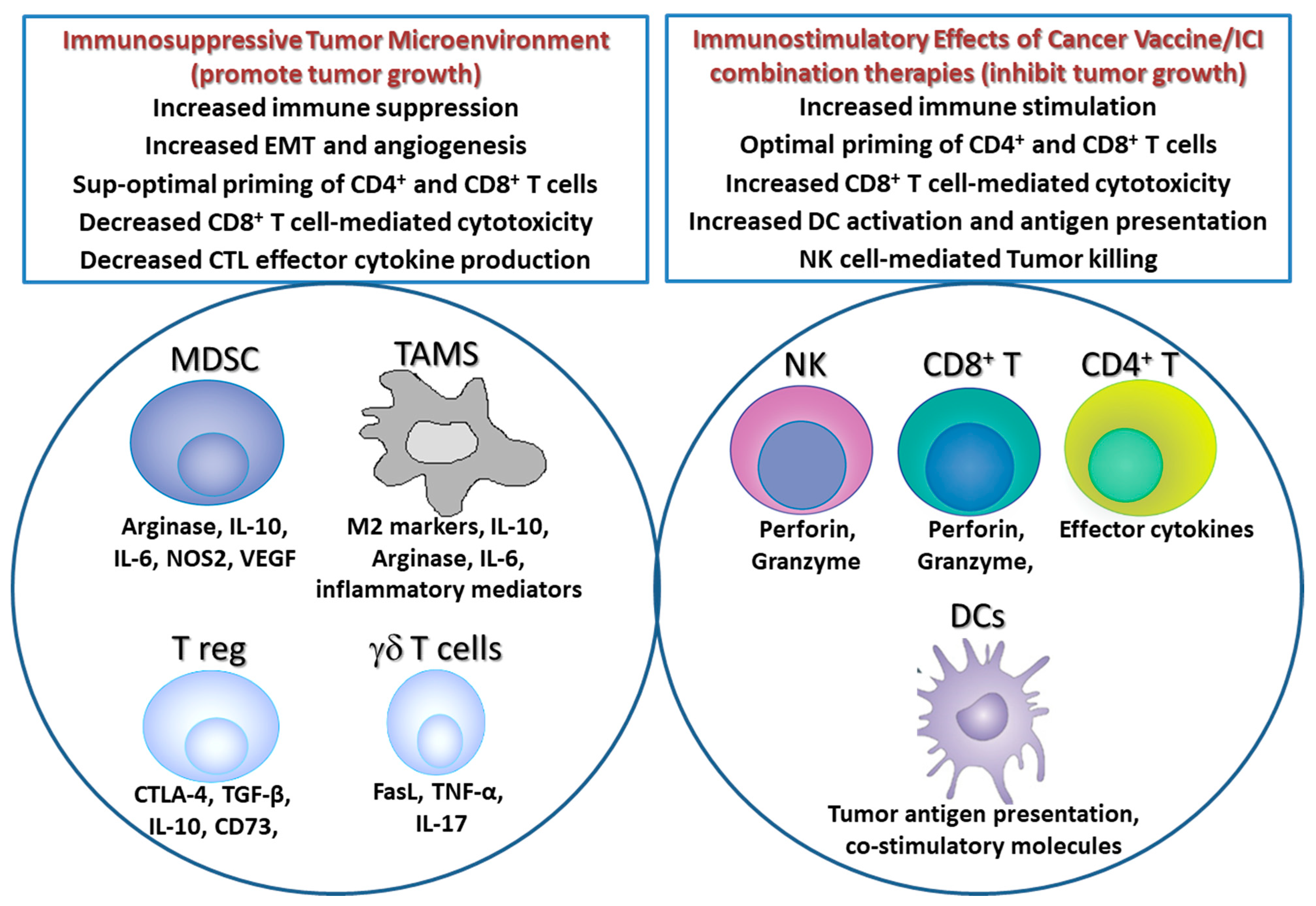
:1. Introduction
2. Preventive and Therapeutic Cancer Vaccines
3. Adjuvants
4. Route of Administration
5. Delivery Strategies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-associated macrophages: Recent insights and therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Gardner, T.A.; Elzey, B.D.; Hahn, N.M. Sipuleucel-T (Provenge) autologous vaccine approved for treatment of men with asymptomatic or minimally symptomatic castrate-resistant metastatic prostate cancer. Hum. Vaccines Immunother. 2012, 8, 534–539. [Google Scholar] [CrossRef]
- Chang, M.H.; Chen, C.J.; Lai, M.S.; Hsu, H.M.; Wu, T.C.; Kong, M.S.; Liang, D.C.; Shau, W.Y.; Chen, D.S. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N. Engl. J. Med. 1997, 336, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Castle, P.E.; Maza, M. Prophylactic HPV vaccination: Past, present, and future. Epidemiol. Infect. 2016, 144, 449–468. [Google Scholar] [CrossRef] [Green Version]
- Maver, P.J.; Poljak, M. Progress in prophylactic human papillomavirus (HPV) vaccination in 2016: A literature review. Vaccine 2018, 36, 5416–5423. [Google Scholar] [CrossRef]
- Frazer, I.H.; Lowy, D.R.; Schiller, J.T. Prevention of cancer through immunization: Prospects and challenges for the 21st century. Eur. J. Immunol. 2007, 37, S148–S155. [Google Scholar] [CrossRef]
- Cutts, F.T.; Hall, A.J. Vaccines for neonatal viral infections: Hepatitis B vaccine. Expert Rev. Vaccines 2004, 3, 349–352. [Google Scholar] [CrossRef]
- Doorbar, J. Molecular biology of human papillomavirus infection and cervical cancer. Clin. Sci. 2006, 110, 525–541. [Google Scholar] [CrossRef] [Green Version]
- Lowy, D.R.; Schiller, J.T. Prophylactic human papillomavirus vaccines. J. Clin. Investig. 2006, 116, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuohy, V.K.; Johnson, J.M.; Mazumder, S. Primary immunoprevention of adult onset cancers by vaccinating against retired tissue-specific self-proteins. Semin. Immunol. 2020, 47, 101392. [Google Scholar] [CrossRef]
- Mazumder, S.; Johnson, J.M.; Swank, V.; Dvorina, N.; Martelli, E.; Ko, J.; Tuohy, V.K. Primary immunoprevention of epithelial ovarian carcinoma by vaccination against the extracellular domain of anti-Mullerian hormone receptor II. Cancer Prev. Res. 2017, 10, 612–624. [Google Scholar] [CrossRef] [Green Version]
- Jaini, R.; Kesaraju, P.; Johnson, J.M.; Altuntas, C.Z.; Jane-Wit, D.; Tuohy, V.K. An autoimmune-mediated strategy for prophylactic breast cancer vaccination. Nat. Med. 2010, 16, 799–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuohy, V.K.; Jaini, R.; Johnson, J.M.; Loya, M.G.; Wilk, D.; Downs-Kelly, E.; Mazumder, S. Targeted vaccination against human α-lactalbumin for immunotherapy and primary immunoprevention of triple negative breast cancer. Cancers 2016, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Bos, R.; van Duikeren, S.; van Hall, T.; Kaaijk, P.; Taubert, R.; Kyewski, B.; Klein, L.; Melief, C.J.; Offringa, R. Expression of a natural tumor antigen by thymic epithelial cells impairs the tumor-protective CD4+ T-cell repertoire. Cancer Res. 2005, 65, 6443–6449. [Google Scholar] [CrossRef] [Green Version]
- Engelhard, V.H.; Bullock, T.N.; Colella, T.A.; Sheasley, S.L.; Mullins, D.W. Antigens derived from melanocyte differentiation proteins: Self-tolerance, autoimmunity, and use for cancer immunotherapy. Immunol. Rev. 2002, 188, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, S.R.; Sorensen, M.R.; Buus, S.; Christensen, J.P.; Thomsen, A.R. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: Implications for cancer immunotherapy. J. Immunol. 2013, 191, 3955–3967. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844. [Google Scholar] [CrossRef]
- Makkouk, A.; Weiner, G.J. Cancer immunotherapy and breaking immune tolerance: New approaches to an old challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Le, D.T.; Pardoll, D.M.; Jaffee, E.M. Cellular vaccine approaches. Cancer J. 2010, 16, 304–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabado, R.L.; Balan, S.; Bhardwaj, N. Dendritic cell-based immunotherapy. Cell Res. 2017, 27, 74–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.A.; Botella, R.; Galloway, T.H.; Murray, N.; Kramp, J.M.; Song, I.S.; Ansel, J.C. Antitumor effects of granulocyte-macrophage colony-stimulating factor production by melanoma cells. Cancer Res. 1996, 56, 2191–2198. [Google Scholar] [PubMed]
- Sanda, M.G.; Ayyagari, S.R.; Jaffee, E.M.; Epstein, J.I.; Clift, S.L.; Cohen, L.K.; Dranoff, G.; Pardoll, D.M.; Mulligan, R.C.; Simons, J.W. Demonstration of a rational strategy for human prostate cancer gene therapy. J. Urol. 1994, 151, 622–628. [Google Scholar] [CrossRef]
- Dunussi-Joannopoulos, K.; Dranoff, G.; Weinstein, H.J.; Ferrara, J.L.; Bierer, B.E.; Croop, J.M. Gene immunotherapy in murine acute myeloid leukemia: Granulocyte-macrophage colony-stimulating factor tumor cell vaccines elicit more potent antitumor immunity compared with B7 family and other cytokine vaccines. Blood 1998, 91, 222–230. [Google Scholar] [CrossRef]
- Simons, J.W.; Sacks, N. Granulocyte-macrophage colony-stimulating factor-transduced allogeneic cancer cellular immunotherapy: The GVAX vaccine for prostate cancer. Urol. Oncol. 2006, 24, 419–424. [Google Scholar] [CrossRef]
- Small, E.J.; Sacks, N.; Nemunaitis, J.; Urba, W.J.; Dula, E.; Centeno, A.S.; Nelson, W.G.; Ando, D.; Howard, C.; Borellini, F.; et al. Granulocyte macrophage colony-stimulating factor--secreting allogeneic cellular immunotherapy for hormone-refractory prostate cancer. Clin. Cancer Res. 2007, 13, 3883–3891. [Google Scholar] [CrossRef] [Green Version]
- Michael, A.; Ball, G.; Quatan, N.; Wushishi, F.; Russell, N.; Whelan, J.; Chakraborty, P.; Leader, D.; Whelan, M.; Pandha, H. Delayed disease progression after allogeneic cell vaccination in hormone-resistant prostate cancer and correlation with immunologic variables. Clin. Cancer Res. 2005, 11, 4469–4478. [Google Scholar] [CrossRef] [Green Version]
- Reimers, M.A.; Slane, K.E.; Pachynski, R.K. Immunotherapy in metastatic castration-resistant prostate cancer: Past and future strategies for optimization. Curr. Urol. Rep. 2019, 20, 64. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Jaroslawski, S.; Toumi, M. Sipuleucel-T (Provenge(®))—Autopsy of an innovative paradigm change in cancer treatment: Why a single-product biotech company failed to capitalize on its breakthrough invention. BioDrugs 2015, 29, 301–307. [Google Scholar] [CrossRef]
- Chi, J.; Patel, R.; Rehman, H.; Goyal, S.; Saif, M.W. Recent advances in immunotherapy for pancreatic cancer. J. Cancer Metastasis Treat. 2020, 6. [Google Scholar] [CrossRef]
- Sahin, I.H.; Askan, G.; Hu, Z.I.; O’Reilly, E.M. Immunotherapy in pancreatic ductal adenocarcinoma: An emerging entity? Ann. Oncol. 2017, 28, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Jaffee, E.M.; Hruban, R.H.; Biedrzycki, B.; Laheru, D.; Schepers, K.; Sauter, P.R.; Goemann, M.; Coleman, J.; Grochow, L.; Donehower, R.C.; et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: A phase I trial of safety and immune activation. J. Clin. Oncol. 2001, 19, 145–156. [Google Scholar] [CrossRef]
- Laheru, D.; Lutz, E.; Burke, J.; Biedrzycki, B.; Solt, S.; Onners, B.; Tartakovsky, I.; Nemunaitis, J.; Le, D.; Sugar, E.; et al. Allogeneic granulocyte macrophage colony-stimulating factor-secreting tumor immunotherapy alone or in sequence with cyclophosphamide for metastatic pancreatic cancer: A pilot study of safety, feasibility, and immune activation. Clin. Cancer Res. 2008, 14, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, E.; Yeo, C.J.; Lillemoe, K.D.; Biedrzycki, B.; Kobrin, B.; Herman, J.; Sugar, E.; Piantadosi, S.; Cameron, J.L.; Solt, S.; et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma: A phase II trial of safety, efficacy, and immune activation. Ann. Surg. 2011, 253, 328–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardacre, J.M.; Mulcahy, M.; Small, W.; Talamonti, M.; Obel, J.; Krishnamurthi, S.; Rocha-Lima, C.S.; Safran, H.; Lenz, H.J.; Chiorean, E.G. Addition of algenpantucel-L immunotherapy to standard adjuvant therapy for pancreatic cancer: A phase 2 study. J. Gastrointest. Surg. 2013, 17, 94–100. [Google Scholar] [CrossRef]
- Macher, B.A.; Galili, U. The Galalpha1,3Galbeta1,4GlcNAc-R (alpha-Gal) epitope: A carbohydrate of unique evolution and clinical relevance. Biochim. Biophys. Acta 2008, 1780, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galili, U.; Anaraki, F.; Thall, A.; Hillblack, C.; Radic, M. One percent of human circulating B-lymphocytes are capable of producing the natural anti-Gal antibody. Blood 1993, 82, 2485–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, C.J.; Seregina, T.; Atchison, R.; Hall, A.; Muldoon, R.; Levy, J.P. Eliciting hyperacute xenograft response to treat human cancer: Alpha(1,3)galactosyltransferase gene therapy. Anticancer Res. 1998, 18, 2301–2308. [Google Scholar] [PubMed]
- Coveler, A.L.; Rossi, G.R.; Vahanian, N.N.; Link, C.; Chiorean, E.G. Algenpantucel-L immunotherapy in pancreatic adenocarcinoma. Immunotherapy 2016, 8, 117–125. [Google Scholar] [CrossRef]
- LaTemple, D.C.; Abrams, J.T.; Zhang, S.Y.; Galili, U. Increased immunogenicity of tumor vaccines complexed with anti-Gal: Studies in knockout mice for alpha1,3galactosyltransferase. Cancer Res. 1999, 59, 3417–3423. [Google Scholar] [PubMed]
- Dillman, R.O. An update on GM-CSF and its potential role in melanoma management. Melanoma Manag. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Soiffer, R.; Lynch, T.; Mihm, M.; Jung, K.; Rhuda, C.; Schmollinger, J.C.; Hodi, F.S.; Liebster, L.; Lam, P.; Mentzer, S.; et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 1998, 95, 13141–13146. [Google Scholar] [CrossRef] [Green Version]
- Soiffer, R.; Hodi, F.S.; Haluska, F.; Jung, K.; Gillessen, S.; Singer, S.; Tanabe, K.; Duda, R.; Mentzer, S.; Jaklitsch, M.; et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J. Clin. Oncol. 2003, 21, 3343–3350. [Google Scholar] [CrossRef]
- Luiten, R.M.; Kueter, E.W.; Mooi, W.; Gallee, M.P.; Rankin, E.M.; Gerritsen, W.R.; Clift, S.M.; Nooijen, W.J.; Weder, P.; van de Kasteele, W.F.; et al. Immunogenicity, including vitiligo, and feasibility of vaccination with autologous GM-CSF-transduced tumor cells in metastatic melanoma patients. J. Clin. Oncol. 2005, 23, 8978–8991. [Google Scholar] [CrossRef] [Green Version]
- Lipson, E.J.; Sharfman, W.H.; Chen, S.; McMiller, T.L.; Pritchard, T.S.; Salas, J.T.; Sartorius-Mergenthaler, S.; Freed, I.; Ravi, S.; Wang, H.; et al. Safety and immunologic correlates of Melanoma GVAX, a GM-CSF secreting allogeneic melanoma cell vaccine administered in the adjuvant setting. J. Transl. Med. 2015, 13, 214. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Lynch, T.; Skarin, A.; Lucca, J.; Lynch, C.; Jung, K.; Hodi, F.S.; Jaklitsch, M.; Mentzer, S.; Swanson, S.; et al. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J. Clin. Oncol. 2003, 21, 624–630. [Google Scholar] [CrossRef]
- Kelly, R.J.; Giaccone, G. Lung cancer vaccines. Cancer J. 2011, 17, 302–308. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Dillman, R.O.; Schwarzenberger, P.O.; Senzer, N.; Cunningham, C.; Cutler, J.; Tong, A.; Kumar, P.; Pappen, B.; Hamilton, C.; et al. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J. Clin. Oncol. 2006, 24, 4721–4730. [Google Scholar] [CrossRef] [PubMed]
- Nemunaitis, J.; Nemunaitis, M.; Senzer, N.; Snitz, P.; Bedell, C.; Kumar, P.; Pappen, B.; Maples, P.B.; Shawler, D.; Fakhrai, H. Phase II trial of Belagenpumatucel-L, a TGF-β2 antisense gene modified allogeneic tumor vaccine in advanced non small cell lung cancer (NSCLC) patients. Cancer Gene Ther. 2009, 16, 620–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhasz, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.H.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Kanne, D.B.; Leong, M.; Glickman, L.H.; McWhirter, S.M.; Lemmens, E.; Mechette, K.; Leong, J.J.; Lauer, P.; Liu, W.; et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 2015, 7, 283ra252. [Google Scholar] [CrossRef] [Green Version]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Hulett, T.W.; Jensen, S.M.; Wilmarth, P.A.; Reddy, A.P.; Ballesteros-Merino, C.; Afentoulis, M.E.; Dubay, C.; David, L.L.; Fox, B.A. Coordinated responses to individual tumor antigens by IgG antibody and CD8+ T cells following cancer vaccination. J. Immunother. Cancer 2018, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Schrump, D.S.; Gildersleeve, J.C. Whole-cell cancer vaccines induce large antibody responses to carbohydrates and glycoproteins. Cell Chem. Biol. 2016, 23, 1515–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.P.; You, Y.H.; Besaratinia, A. Mutations induced by ultraviolet light. Mutat. Res. 2005, 571, 19–31. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunological mechanisms underneath the efficacy of cancer therapy. Cancer Immunol. Res. 2016, 4, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Baumgaertner, P.; Costa Nunes, C.; Cachot, A.; Maby-El Hajjami, H.; Cagnon, L.; Braun, M.; Derre, L.; Rivals, J.P.; Rimoldi, D.; Gnjatic, S.; et al. Vaccination of stage III/IV melanoma patients with long NY-ESO-1 peptide and CpG-B elicits robust CD8+and CD4+ T-cell responses with multiple specificities including a novel DR7-restricted epitope. Oncoimmunology 2016, 5, e1216290. [Google Scholar] [CrossRef] [Green Version]
- Takeoka, T.; Nagase, H.; Kurose, K.; Ohue, Y.; Yamasaki, M.; Takiguchi, S.; Sato, E.; Isobe, M.; Kanazawa, T.; Matsumoto, M.; et al. NY-ESO-1 protein cancer vaccine with poly-ICLC and OK-432: Rapid and strong induction of NY-ESO-1-specific immune responses by poly-ICLC. J. Immunother. 2017, 40, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zeng, G.; Wang, X.; Robbins, P.F.; Rosenberg, S.A.; Wang, R.F. CD4+ T cell recognition of MHC class II-restricted epitopes from NY-ESO-1 presented by a prevalent HLA DP4 allele: Association with NY-ESO-1 antibody production. Proc. Natl. Acad. Sci. USA 2001, 98, 3964–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Slingluff, C.L., Jr.; Petroni, G.R.; Olson, W.C.; Smolkin, M.E.; Chianese-Bullock, K.A.; Mauldin, I.S.; Smith, K.T.; Deacon, D.H.; Varhegyi, N.E.; Donnelly, S.B.; et al. A randomized pilot trial testing the safety and immunologic effects of a MAGE-A3 protein plus AS15 immunostimulant administered into muscle or into dermal/subcutaneous sites. Cancer Immunol. Immunother. 2016, 65, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Saiag, P.; Gutzmer, R.; Ascierto, P.A.; Maio, M.; Grob, J.J.; Murawa, P.; Dreno, B.; Ross, M.; Weber, J.; Hauschild, A.; et al. Prospective assessment of a gene signature potentially predictive of clinical benefit in metastatic melanoma patients following MAGE-A3 immunotherapeutic (PREDICT). Ann. Oncol. 2016, 27, 1947–1953. [Google Scholar] [CrossRef]
- Dreno, B.; Thompson, J.F.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.J.; Korovin, S.; et al. MAGE-A3 immunotherapeutic as adjuvant therapy for patients with resected, MAGE-A3-positive, stage III melanoma (DERMA): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef] [Green Version]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Aljahdali, I.; Ling, X. Cancer therapeutics using survivin BIRC5 as a target: What can we do after over two decades of study? J. Exp. Clin. Cancer Res. 2019, 38, 368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onodi, F.; Maherzi-Mechalikh, C.; Mougel, A.; Ben Hamouda, N.; Taboas, C.; Gueugnon, F.; Tran, T.; Nozach, H.; Marcon, E.; Gey, A.; et al. High therapeutic efficacy of a new survivin LSP-cancer vaccine containing CD4+ and CD8+ T-cell epitopes. Front. Oncol. 2018, 8, 517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef] [Green Version]
- Ohno, S.; Okuyama, R.; Aruga, A.; Sugiyama, H.; Yamamoto, M. Phase I trial of Wilms’ Tumor 1 (WT1) peptide vaccine with GM-CSF or CpG in patients with solid malignancy. Anticancer Res. 2012, 32, 2263–2269. [Google Scholar]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef]
- Calvo Tardon, M.; Allard, M.; Dutoit, V.; Dietrich, P.Y.; Walker, P.R. Peptides as cancer vaccines. Curr. Opin. Pharmacol. 2019, 47, 20–26. [Google Scholar] [CrossRef]
- Kruit, W.H.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.J.; et al. Selection of immunostimulant AS15 for active immunization with MAGE-A3 protein: Results of a randomized phase II study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Zielinski, M.; Linder, A.; Dahabreh, J.; Gonzalez, E.E.; Malinowski, W.; Lopez-Brea, M.; Vanakesa, T.; Jassem, J.; Kalofonos, H.; et al. Adjuvant MAGE-A3 immunotherapy in resected non-small-cell lung cancer: Phase II randomized study results. J. Clin. Oncol. 2013, 31, 2396–2403. [Google Scholar] [CrossRef]
- Wurz, G.T.; Kao, C.J.; Wolf, M.; DeGregorio, M.W. Tecemotide: An antigen-specific cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 3383–3393. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.; Thatcher, N.; Socinski, M.A.; Wasilewska-Tesluk, E.; Horwood, K.; Szczesna, A.; Martin, C.; Ragulin, Y.; Zukin, M.; Helwig, C.; et al. Tecemotide in unresectable stage III non-small-cell lung cancer in the phase III START study: Updated overall survival and biomarker analyses. Ann. Oncol. 2015, 26, 1134–1142. [Google Scholar] [CrossRef]
- Chianese-Bullock, K.A.; Pressley, J.; Garbee, C.; Hibbitts, S.; Murphy, C.; Yamshchikov, G.; Petroni, G.R.; Bissonette, E.A.; Neese, P.Y.; Grosh, W.W.; et al. MAGE-A1-, MAGE-A10-, and gp100-derived peptides are immunogenic when combined with granulocyte-macrophage colony-stimulating factor and montanide ISA-51 adjuvant and administered as part of a multipeptide vaccine for melanoma. J. Immunol. 2005, 174, 3080–3086. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, E.; Liu, H.; Huckriede, A.; Hak, E. Safety and tolerability evaluation of the use of Montanide ISA™51 as vaccine adjuvant: A systematic review. Hum. Vaccines Immunother. 2016, 12, 159–169. [Google Scholar] [CrossRef] [Green Version]
- Pavlick, A.; Blazquez, A.B.; Meseck, M.; Lattanzi, M.; Ott, P.A.; Marron, T.U.; Holman, R.M.; Mandeli, J.; Salazar, A.M.; McClain, C.B.; et al. Combined vaccination with NY-ESO-1 protein, poly-ICLC, and montanide improves humoral and cellular immune responses in patients with high-risk melanoma. Cancer Immunol. Res. 2020, 8, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapenta, C.; Donati, S.; Spadaro, F.; Lattanzi, L.; Urbani, F.; Macchia, I.; Sestili, P.; Spada, M.; Cox, M.C.; Belardelli, F.; et al. Lenalidomide improves the therapeutic effect of an interferon-alpha-dendritic cell-based lymphoma vaccine. Cancer Immunol. Immunother. 2019, 68, 1791–1804. [Google Scholar] [CrossRef]
- Sakamaki, I.; Kwak, L.W.; Cha, S.C.; Yi, Q.; Lerman, B.; Chen, J.; Surapaneni, S.; Bateman, S.; Qin, H. Lenalidomide enhances the protective effect of a therapeutic vaccine and reverses immune suppression in mice bearing established lymphomas. Leukemia 2014, 28, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyzzer, E.E. Tumor immunity. J. Cancer Res. 1916, 1, 125–155. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Foley, E.J. Antigenic properties of methylcholanthrene-induced tumors in mice of the strain of origin. Cancer Res. 1953, 13, 835–837. [Google Scholar]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- van Rooij, N.; van Buuren, M.M.; Philips, D.; Velds, A.; Toebes, M.; Heemskerk, B.; van Dijk, L.J.; Behjati, S.; Hilkmann, H.; El Atmioui, D.; et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 2013, 31, e439–e442. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gros, A.; Parkhurst, M.R.; Tran, E.; Pasetto, A.; Robbins, P.F.; Ilyas, S.; Prickett, T.D.; Gartner, J.J.; Crystal, J.S.; Roberts, I.M.; et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 2016, 22, 433–438. [Google Scholar] [CrossRef]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Lower, M.; Diekmann, J.; Boegel, S.; Schrors, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubin, M.M.; Zhang, X.; Schuster, H.; Caron, E.; Ward, J.P.; Noguchi, T.; Ivanova, Y.; Hundal, J.; Arthur, C.D.; Krebber, W.J.; et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014, 515, 577–581. [Google Scholar] [CrossRef]
- Duan, F.; Duitama, J.; Al Seesi, S.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Lower, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrors, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Carreno, B.M.; Magrini, V.; Becker-Hapak, M.; Kaabinejadian, S.; Hundal, J.; Petti, A.A.; Ly, A.; Lie, W.R.; Hildebrand, W.H.; Mardis, E.R.; et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 2015, 348, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsen, M.O.; Vogel, M.; Riether, C.; Muller, J.; Salatino, S.; Ternette, N.; Gomes, A.C.; Cabral-Miranda, G.; El-Turabi, A.; Ruedl, C.; et al. Targeting mutated plus germline epitopes confers pre-clinical efficacy of an instantly formulated cancer nano-vaccine. Front. Immunol. 2019, 10, 1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1+ CD38hi cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A phase II study of allogeneic GM-CSF-transfected pancreatic tumor vaccine (GVAX) with ipilimumab as maintenance treatment for metastatic pancreatic cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Scholz, M.; Yep, S.; Chancey, M.; Kelly, C.; Chau, K.; Turner, J.; Lam, R.; Drake, C.G. Phase I clinical trial of sipuleucel-T combined with escalating doses of ipilimumab in progressive metastatic castrate-resistant prostate cancer. Immunotargets Ther. 2017, 6, 11–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II Study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef]
- Attia, P.; Phan, G.Q.; Maker, A.V.; Robinson, M.R.; Quezado, M.M.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. 2005, 23, 6043–6053. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Sanderson, K.; Scotland, R.; Lee, P.; Liu, D.; Groshen, S.; Snively, J.; Sian, S.; Nichol, G.; Davis, T.; Keler, T.; et al. Autoimmunity in a phase I trial of a fully human anti-cytotoxic T-lymphocyte antigen-4 monoclonal antibody with multiple melanoma peptides and Montanide ISA 51 for patients with resected stages III and IV melanoma. J. Clin. Oncol. 2005, 23, 741–750. [Google Scholar] [CrossRef]
- Sarnaik, A.A.; Yu, B.; Yu, D.; Morelli, D.; Hall, M.; Bogle, D.; Yan, L.; Targan, S.; Solomon, J.; Nichol, G.; et al. Extended dose ipilimumab with a peptide vaccine: Immune correlates associated with clinical benefit in patients with resected high-risk stage IIIc/IV melanoma. Clin. Cancer Res. 2011, 17, 896–906. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef] [Green Version]
- Madan, R.A.; Arlen, P.M.; Mohebtash, M.; Hodge, J.W.; Gulley, J.L. Prostvac-VF: A vector-based vaccine targeting PSA in prostate cancer. Expert Opin. Investig. Drugs 2009, 18, 1001–1011. [Google Scholar] [CrossRef] [Green Version]
- Arlen, P.M.; Skarupa, L.; Pazdur, M.; Seetharam, M.; Tsang, K.Y.; Grosenbach, D.W.; Feldman, J.; Poole, D.J.; Litzinger, M.; Steinberg, S.M.; et al. Clinical safety of a viral vector based prostate cancer vaccine strategy. J. Urol. 2007, 178, 1515–1520. [Google Scholar] [CrossRef]
- Kantoff, P.W.; Schuetz, T.J.; Blumenstein, B.A.; Glode, L.M.; Bilhartz, D.L.; Wyand, M.; Manson, K.; Panicali, D.L.; Laus, R.; Schlom, J.; et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2010, 28, 1099–1105. [Google Scholar] [CrossRef]
- Gulley, J.L.; Borre, M.; Vogelzang, N.J.; Ng, S.; Agarwal, N.; Parker, C.C.; Pook, D.W.; Rathenborg, P.; Flaig, T.W.; Carles, J.; et al. Phase III trial of PROSTVAC in asymptomatic or minimally symptomatic metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019, 37, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Mohebtash, M.; Arlen, P.M.; Vergati, M.; Rauckhorst, M.; Steinberg, S.M.; Tsang, K.Y.; Poole, D.J.; Parnes, H.L.; Wright, J.J.; et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 501–508. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zeng, H.; Xu, R.H.; Liu, B.; Li, Z. Vaccination with human pluripotent stem cells generates a broad spectrum of immunological and clinical responses against colon cancer. Stem Cells 2009, 27, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Du, J.; Shen, H.; Gao, D.; Li, Z.; Wang, G.; Mu, X.; Liu, Q. Administration of embryonic stem cells generates effective antitumor immunity in mice with minor and heavy tumor load. Cancer Immunol. Immunother. 2010, 59, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, K.; Mitchell, R.A.; Putty, K.; Willer, S.; Sharma, R.K.; Yan, J.; Bodduluri, H.; Eaton, J.W. Vaccination with embryonic stem cells protects against lung cancer: Is a broad-spectrum prophylactic vaccine against cancer possible? PLoS ONE 2012, 7, e42289. [Google Scholar] [CrossRef]
- Brewer, B.G.; Mitchell, R.A.; Harandi, A.; Eaton, J.W. Embryonic vaccines against cancer: An early history. Exp. Mol. Pathol. 2009, 86, 192–197. [Google Scholar] [CrossRef]
- Gold, P.; Freedman, S.O. Specific carcinoembryonic antigens of the human digestive system. J. Exp. Med. 1965, 122, 467–481. [Google Scholar] [CrossRef]
- Alexander, P.; Fairley, G.H. Cellular resistance to tumours. Br. Med. Bull. 1967, 23, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Laurence, D.J.; Neville, A.M. Foetal antigens and their role in the diagnosis and clinical management of human neoplasms: A review. Br. J. Cancer. 1972, 26, 335–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, P. Foetal “antigens” in cancer. Nature 1972, 235, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, R.W.; Glaves, D.; Vose, B.M. Embryonic antigen expression in chemically induced rat hepatomas and sarcomas. Int. J. Cancer 1972, 10, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Stonehill, E.H.; Bendich, A. Retrogenetic expression: The reappearance of embryonal antigens in cancer cells. Nature 1970, 228, 370–372. [Google Scholar] [CrossRef]
- Baldwin, R.W.; Glaves, D.; Pimm, M.V.; Vose, B.M. Tumour specific and embryonic antigen expression of chemically induced rat tumours. Ann. Inst. Pasteur (Paris) 1972, 122, 715–728. [Google Scholar]
- Brawn, R.J. Possible association of embryonal antigen(s) with several primary 3-methylcholanthrene-induced murine sarcomas. Int. J. Cancer 1970, 6, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Klavins, J.V.; Mesa-Tejada, R.; Weiss, M. Human carcinoma antigens cross reacting with anti-embryonic antibodies. Nat. New Biol. 1971, 234, 153–154. [Google Scholar] [CrossRef]
- Menard, S.; Colnaghi, M.I.; Della Porta, G. In vitro demonstration of tumor-specific common antigens and embryonal antigens in murine fibrosarcomas induced by 7,12-dimethylbenz(a)anthracene. Cancer Res. 1973, 33, 478–481. [Google Scholar] [PubMed]
- Colnaghi, M.I.; Della Porta, G. Evidence for virus-related and unrelated antigens on murine lymphomas induced by chemical carcinogens. J. Natl. Cancer Inst. 1973, 50, 173–180. [Google Scholar] [CrossRef] [PubMed]
- LeMevel, B.P.; Wells, S.A., Jr. Foetal antigens cross-reactive with tumour-specific transplantation antigens. Nat. New Biol. 1973, 244, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Bendich, A.; Borenfreund, E.; Stonehill, E.H. Protection of adult mice against tumor challenge by immunization with irradiated adult skin or embryo cells. J. Immunol. 1973, 111, 284–285. [Google Scholar]
- Pearson, G.; Freeman, G. Evidence suggesting a relationship between polyoma virus-induced transplantation antigen and normal embryonic antigen. Cancer Res. 1968, 28, 1665–1673. [Google Scholar]
- Ambrose, K.R.; Candler, E.L.; Coggin, J.H., Jr. Characterization of tumor-specific transplantation immunity reactions in immunodiffusion chambers in vivo. Proc. Soc. Exp. Biol. Med. 1969, 132, 1013–1020. [Google Scholar] [CrossRef]
- Buttle, G.A.; Frayn, A. Effect of previous injection of homologous embryonic tissue on the growth of certain transplantable mouse tumours. Nature 1967, 215, 1495–1497. [Google Scholar] [CrossRef]
- Coggin, J.H., Jr.; Ambrose, K.R. A rapid in vivo assay for SV40 tumor immunity in hamsters. Proc. Soc. Exp. Biol. Med. 1969, 130, 246–252. [Google Scholar] [CrossRef]
- Grant, J.P.; Wells, S.A., Jr. Tumor resistance in rats immunized to fetal tissues. J. Surg. Res. 1974, 16, 533–540. [Google Scholar] [CrossRef]
- Rees, R.C.; Shah, L.P.; Baldwin, R.W. Inhibition of pulmonary tumour development in rats sensitised to rat embryonic tissue. Nature 1975, 255, 329–330. [Google Scholar] [CrossRef]
- Ambrose, K.R.; Anderson, N.G.; Coggin, J.H. Interruption of SV40 oncogenesis with human foetal antigen. Nature 1971, 233, 194–195. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dranoff, G. GM-CSF-based cancer vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaddanapudi, K.; Meng, S.; Whitt, A.G.; Al Rayyan, N.; Richie, J.; Tu, A.; Eaton, J.W.; Li, C. Exosomes from GM-CSF expressing embryonic stem cells are an effective prophylactic vaccine for cancer prevention. Oncoimmunology 2019, 8, 1561119. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Taieb, J.; Andre, F.; Zitvogel, L. The potential of exosomes in immunotherapy. Expert Opin. Biol. Ther. 2005, 5, 737–747. [Google Scholar] [CrossRef]
- Thery, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Kooreman, N.G.; Kim, Y.; de Almeida, P.E.; Termglinchan, V.; Diecke, S.; Shao, N.Y.; Wei, T.T.; Yi, H.; Dey, D.; Nelakanti, R.; et al. Autologous iPSC-based vaccines elicit anti-tumor responses in vivo. Cell Stem Cell 2018, 22, 501–513.e507. [Google Scholar] [CrossRef] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Xin, L.; Lawson, D.A.; Witte, O.N. The Sca-1 cell surface marker enriches for a prostate-regenerating cell subpopulation that can initiate prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 6942–6947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.C.; Hsu, H.S.; Chen, Y.W.; Tsai, T.H.; How, C.K.; Wang, C.Y.; Hung, S.C.; Chang, Y.L.; Tsai, M.L.; Lee, Y.Y.; et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS ONE 2008, 3, e2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, S.H.; Yu, C.C.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Tsai, T.H.; Chou, S.H.; Chien, C.S.; Ku, H.H.; Lo, J.F. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin. Cancer Res. 2008, 14, 4085–4095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovey, J.S.; Zacharek, S.J.; Kim, C.F.; Lees, J.A. Bmi1 is critical for lung tumorigenesis and bronchioalveolar stem cell expansion. Proc. Natl. Acad. Sci. USA 2008, 105, 11857–11862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [Green Version]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer vaccines: Adjuvant potency, importance of age, lifestyle, and treatments. Front. Immunol. 2020, 11, 615240. [Google Scholar] [CrossRef]
- Bowen, W.S.; Svrivastava, A.K.; Batra, L.; Barsoumian, H.; Shirwan, H. Current challenges for cancer vaccine adjuvant development. Expert Rev. Vaccines 2018, 17, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Temizoz, B.; Kuroda, E.; Ishii, K.J. Vaccine adjuvants as potential cancer immunotherapeutics. Int. Immunol. 2016, 28, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.B.; Xu, J. Better adjuvants for better vaccines: Progress in adjuvant delivery systems, modifications, and adjuvant-antigen codelivery. Vaccines 2020, 8, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogenesch, H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front. Immunol. 2012, 3, 406. [Google Scholar] [CrossRef] [Green Version]
- Hoeller, C.; Michielin, O.; Ascierto, P.A.; Szabo, Z.; Blank, C.U. Systematic review of the use of granulocyte-macrophage colony-stimulating factor in patients with advanced melanoma. Cancer Immunol. Immunother. 2016, 65, 1015–1034. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.L.; Shen, K.Y.; Tien, C.Y.; Chen, Y.A.; Liu, S.J. Recent progress in GM-CSF-based cancer immunotherapy. Immunotherapy 2017, 9, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Moynihan, K.D.; Zheng, Y.; Szeto, G.L.; Li, A.V.; Huang, B.; Van Egeren, D.S.; Park, C.; Irvine, D.J. Structure-based programming of lymph-node targeting in molecular vaccines. Nature 2014, 507, 519–522. [Google Scholar] [CrossRef] [Green Version]
- Fifis, T.; Gamvrellis, A.; Crimeen-Irwin, B.; Pietersz, G.A.; Li, J.; Mottram, P.L.; McKenzie, I.F.; Plebanski, M. Size-dependent immunogenicity: Therapeutic and protective properties of nano-vaccines against tumors. J. Immunol. 2004, 173, 3148–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbach, J.; Neumann, A.; Atmaca, A.; Wahle, C.; Brand, K.; von Boehmer, L.; Knuth, A.; Bender, A.; Ritter, G.; Old, L.J.; et al. Efficient in vivo priming by vaccination with recombinant NY-ESO-1 protein and CpG in antigen naive prostate cancer patients. Clin. Cancer Res. 2011, 17, 861–870. [Google Scholar] [CrossRef] [Green Version]
- Stebbing, J.; Dalgleish, A.; Gifford-Moore, A.; Martin, A.; Gleeson, C.; Wilson, G.; Brunet, L.R.; Grange, J.; Mudan, S. An intra-patient placebo-controlled phase I trial to evaluate the safety and tolerability of intradermal IMM-101 in melanoma. Ann. Oncol. 2012, 23, 1314–1319. [Google Scholar] [CrossRef]
- Rittig, S.M.; Haentschel, M.; Weimer, K.J.; Heine, A.; Muller, M.R.; Brugger, W.; Horger, M.S.; Maksimovic, O.; Stenzl, A.; Hoerr, I.; et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol. Ther. 2011, 19, 990–999. [Google Scholar] [CrossRef]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef]
- Welters, M.J.; Kenter, G.G.; de Vos van Steenwijk, P.J.; Lowik, M.J.; Berends-van der Meer, D.M.; Essahsah, F.; Stynenbosch, L.F.; Vloon, A.P.; Ramwadhdoebe, T.H.; Piersma, S.J.; et al. Success or failure of vaccination for HPV16-positive vulvar lesions correlates with kinetics and phenotype of induced T-cell responses. Proc. Natl. Acad. Sci. USA 2010, 107, 11895–11899. [Google Scholar] [CrossRef] [Green Version]
- van den Hende, M.; van Poelgeest, M.I.; van der Hulst, J.M.; de Jong, J.; Drijfhout, J.W.; Fleuren, G.J.; Valentijn, A.R.; Wafelman, A.R.; Slappendel, G.M.; Melief, C.J.; et al. Skin reactions to human papillomavirus (HPV) 16 specific antigens intradermally injected in healthy subjects and patients with cervical neoplasia. Int. J. Cancer 2008, 123, 146–152. [Google Scholar] [CrossRef]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Marroquin, C.E.; Westwood, J.A.; Lapointe, R.; Mixon, A.; Wunderlich, J.R.; Caron, D.; Rosenberg, S.A.; Hwu, P. Mobilization of dendritic cell precursors in patients with cancer by flt3 ligand allows the generation of higher yields of cultured dendritic cells. J. Immunother. 2002, 25, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Nair, S.; Fernandez-Casal, M.; Deng, Y.; St Peter, M.; Williams, R.; Hobeika, A.; Mosca, P.; Clay, T.; Cumming, R.I.; et al. Preoperative mobilization of circulating dendritic cells by Flt3 ligand administration to patients with metastatic colon cancer. J. Clin. Oncol. 2000, 18, 3883–3893. [Google Scholar] [CrossRef] [PubMed]
- de Vos van Steenwijk, P.J.; van Poelgeest, M.I.; Ramwadhdoebe, T.H.; Lowik, M.J.; Berends-van der Meer, D.M.; van der Minne, C.E.; Loof, N.M.; Stynenbosch, L.F.; Fathers, L.M.; Valentijn, A.R.; et al. The long-term immune response after HPV16 peptide vaccination in women with low-grade pre-malignant disorders of the uterine cervix: A placebo-controlled phase II study. Cancer Immunol. Immunother. 2014, 63, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Sultan, H.; Kumai, T.; Nagato, T.; Wu, J.; Salazar, A.M.; Celis, E. The route of administration dictates the immunogenicity of peptide-based cancer vaccines in mice. Cancer Immunol. Immunother. 2019, 68, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.I.; Barrios, K.; Lee, Y.R.; Linowski, A.K.; Celis, E. BiVax: A peptide/poly-IC subunit vaccine that mimics an acute infection elicits vast and effective anti-tumor CD8 T-cell responses. Cancer Immunol. Immunother. 2013, 62, 787–799. [Google Scholar] [CrossRef]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S. Talimogene laherparepvec for the treatment of advanced melanoma. Clin. Cancer Res. 2016, 22, 3127–3131. [Google Scholar] [CrossRef] [Green Version]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef]
- Redelman-Sidi, G.; Glickman, M.S.; Bochner, B.H. The mechanism of action of BCG therapy for bladder cancer—A current perspective. Nat. Rev. Urol. 2014, 11, 153–162. [Google Scholar] [CrossRef]
- Jacouton, E.; Torres Maravilla, E.; Boucard, A.S.; Pouderous, N.; Pessoa Vilela, A.P.; Naas, I.; Chain, F.; Azevedo, V.; Langella, P.; Bermudez-Humaran, L.G. Anti-tumoral effects of recombinant Lactococcus lactis strain secreting IL-17A cytokine. Front. Microbiol. 2018, 9, 3355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohseni, A.H.; Taghinezhad, S.S.; Keyvani, H. The first clinical use of a recombinant Lactococcus lactis expressing human papillomavirus type 16 E7 oncogene oral vaccine: A phase I safety and immunogenicity trial in healthy women volunteers. Mol. Cancer Ther. 2020, 19, 717–727. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Luna, M.A.; Luria-Perez, R. Cancer immunotherapy: Priming the host immune response with live attenuated Salmonella enterica. J. Immunol. Res. 2018, 2018, 2984247. [Google Scholar] [CrossRef] [Green Version]
- Flickinger, J.C., Jr.; Rodeck, U.; Snook, A.E. Listeria monocytogenes as a vector for cancer immunotherapy: Current understanding and progress. Vaccines 2018, 6, 48. [Google Scholar] [CrossRef] [Green Version]
- Gunn, G.R.; Zubair, A.; Peters, C.; Pan, Z.K.; Wu, T.C.; Paterson, Y. Two Listeria monocytogenes vaccine vectors that express different molecular forms of human papilloma virus-16 (HPV-16) E7 induce qualitatively different T cell immunity that correlates with their ability to induce regression of established tumors immortalized by HPV-16. J. Immunol. 2001, 167, 6471–6479. [Google Scholar] [CrossRef] [Green Version]
- Sewell, D.A.; Shahabi, V.; Gunn, G.R., 3rd; Pan, Z.K.; Dominiecki, M.E.; Paterson, Y. Recombinant Listeria vaccines containing PEST sequences are potent immune adjuvants for the tumor-associated antigen human papillomavirus-16 E7. Cancer Res. 2004, 64, 8821–8825. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Ho, A.H.; Wainberg, Z.A.; Picozzi, V.J.; Kindler, H.L.; Wang-Gillam, A.; Oberstein, P.E.; Morse, M.; Zeh, H.; Weekes, C.D.; et al. Results from a phase 2b, randomized, multicenter study of GVAX pancreas and CRS-207 compared to chemotherapy in adults with previously-treated metastatic pancreatic adenocarcinoma (ECLIPSE Study). J. Clin. Oncol. 2017, 35, 345. [Google Scholar] [CrossRef]
- Larocca, C.; Schlom, J. Viral vector-based therapeutic cancer vaccines. Cancer J. 2011, 17, 359–371. [Google Scholar] [CrossRef]
- DiPaola, R.S.; Plante, M.; Kaufman, H.; Petrylak, D.P.; Israeli, R.; Lattime, E.; Manson, K.; Schuetz, T. A phase I trial of pox PSA vaccines (PROSTVAC-VF) with B7-1, ICAM-1, and LFA-3 co-stimulatory molecules (TRICOM) in patients with prostate cancer. J. Transl. Med. 2006, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Gulley, J.L.; Pico-Navarro, C. Revised overall survival analysis of a phase II, randomized, double-blind, controlled study of PROSTVAC in men with metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2017, 35, 124–125. [Google Scholar] [CrossRef]
- Spaner, D.E.; Astsaturov, I.; Vogel, T.; Petrella, T.; Elias, I.; Burdett-Radoux, S.; Verma, S.; Iscoe, N.; Hamilton, P.; Berinstein, N.L. Enhanced viral and tumor immunity with intranodal injection of canary pox viruses expressing the melanoma antigen, gp100. Cancer 2006, 106, 890–899. [Google Scholar] [CrossRef]
- von Mehren, M.; Arlen, P.; Gulley, J.; Rogatko, A.; Cooper, H.S.; Meropol, N.J.; Alpaugh, R.K.; Davey, M.; McLaughlin, S.; Beard, M.T.; et al. The influence of granulocyte macrophage colony-stimulating factor and prior chemotherapy on the immunological response to a vaccine (ALVAC-CEA B7.1) in patients with metastatic carcinoma. Clin. Cancer Res. 2001, 7, 1181–1191. [Google Scholar]
- Kaufman, H.L.; Lenz, H.J.; Marshall, J.; Singh, D.; Garett, C.; Cripps, C.; Moore, M.; von Mehren, M.; Dalfen, R.; Heim, W.J.; et al. Combination chemotherapy and ALVAC-CEA/B7.1 vaccine in patients with metastatic colorectal cancer. Clin. Cancer Res. 2008, 14, 4843–4849. [Google Scholar] [CrossRef] [Green Version]
- Chodon, T.; Lugade, A.A.; Battaglia, S.; Odunsi, K. Emerging role and future directions of immunotherapy in advanced ovarian cancer. Hematol. Oncol. Clin. N. Am. 2018, 32, 1025–1039. [Google Scholar] [CrossRef]
- Cho, H.; Cockle, P.; Joe, B.; Risini, W.; White, P.; Jooss, K. Vaccine based immunotherapy regimen (VBIR) for the treatment of prostate cancer. [Abstract]. Cancer Res. 2016, 76, LB-093. [Google Scholar]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New viral vectors for infectious diseases and cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef]
- Schuler, G.; Schuler-Thurner, B.; Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. [Google Scholar] [CrossRef]
- Schreibelt, G.; Bol, K.F.; Westdorp, H.; Wimmers, F.; Aarntzen, E.H.; Duiveman-de Boer, T.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Effective clinical responses in metastatic melanoma patients after vaccination with primary myeloid dendritic cells. Clin. Cancer Res. 2016, 22, 2155–2166. [Google Scholar] [CrossRef] [Green Version]
- Westdorp, H.; Creemers, J.H.A.; van Oort, I.M.; Schreibelt, G.; Gorris, M.A.J.; Mehra, N.; Simons, M.; de Goede, A.L.; van Rossum, M.M.; Croockewit, A.J.; et al. Blood-derived dendritic cell vaccinations induce immune responses that correlate with clinical outcome in patients with chemo-naive castration-resistant prostate cancer. J. Immunother. Cancer 2019, 7, 302. [Google Scholar] [CrossRef]
- Tel, J.; Aarntzen, E.H.; Baba, T.; Schreibelt, G.; Schulte, B.M.; Benitez-Ribas, D.; Boerman, O.C.; Croockewit, S.; Oyen, W.J.; van Rossum, M.; et al. Natural human plasmacytoid dendritic cells induce antigen-specific T-cell responses in melanoma patients. Cancer Res. 2013, 73, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Prue, R.L.; Vari, F.; Radford, K.J.; Tong, H.; Hardy, M.Y.; D’Rozario, R.; Waterhouse, N.J.; Rossetti, T.; Coleman, R.; Tracey, C.; et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J. Immunother. 2015, 38, 71–76. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Teijeira, A.; Kandalaft, L.E.; Romero, P.; Coukos, G.; et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef]
- Breckpot, K.; Corthals, J.; Bonehill, A.; Michiels, A.; Tuyaerts, S.; Aerts, C.; Heirman, C.; Thielemans, K. Dendritic cells differentiated in the presence of IFN-{β} and IL-3 are potent inducers of an antigen-specific CD8+ T cell response. J. Leukoc. Biol. 2005, 78, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Calmeiro, J.; Carrascal, M.A.; Tavares, A.R.; Ferreira, D.A.; Gomes, C.; Falcao, A.; Cruz, M.T.; Neves, B.M. Dendritic cell vaccines for cancer immunotherapy: The role of human conventional type 1 dendritic cells. Pharmaceutics 2020, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Liu, X.; Sun, Y.; Zhou, P.; Wang, Y.; Zhang, Y. Dendritic cell targeted vaccines: Recent progresses and challenges. Hum. Vaccines Immunother. 2016, 12, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Beg, S.; Alharbi, K.S.; Alruwaili, N.K.; Alotaibi, N.H.; Almalki, W.H.; Alenezi, S.K.; Altowayan, W.M.; Alshammari, M.S.; Rahman, M. Nanotherapeutic systems for delivering cancer vaccines: Recent advances. Nanomedicine 2020, 15, 1527–1537. [Google Scholar] [CrossRef]
- Yang, G.; Chen, S.; Zhang, J. Bioinspired and biomimetic nanotherapies for the treatment of infectious diseases. Front. Pharmacol. 2019, 10, 751. [Google Scholar] [CrossRef]
- Soares, S.; Sousa, J.; Pais, A.; Vitorino, C. Nanomedicine: Principles, properties, and regulatory issues. Front. Chem. 2018, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Roche, K.C.; Tian, S.; Eblan, M.J.; McKinnon, K.P.; Caster, J.M.; Chai, S.; Herring, L.E.; Zhang, L.; Zhang, T.; et al. Antigen-capturing nanoparticles improve the abscopal effect and cancer immunotherapy. Nat. Nanotechnol. 2017, 12, 877–882. [Google Scholar] [CrossRef] [Green Version]
- Burris, H.A.; Patel, M.R.; Cho, D.C.; Clarke, J.M.; Gutierrez, M.; Zaks, T.Z.; Frederick, J.; Hopson, K.; Mody, K.; Binanti-Berube, A.; et al. A phase I multicenter study to assess the safety, tolerability, and immunogenicity of mRNA-4157 alone in patients with resected solid tumors and in combination with pembrolizumab in patients with unresectable solid tumors. J. Clin. Oncol. 2019, 37, 2523. [Google Scholar] [CrossRef]
- Xia, W.; Wang, J.; Xu, Y.; Jiang, F.; Xu, L. L-BLP25 as a peptide vaccine therapy in non-small cell lung cancer: A review. J. Thorac. Dis. 2014, 6, 1513–1520. [Google Scholar] [CrossRef]
- Butts, C.; Socinski, M.A.; Mitchell, P.L.; Thatcher, N.; Havel, L.; Krzakowski, M.; Nawrocki, S.; Ciuleanu, T.E.; Bosquee, L.; Trigo, J.M.; et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): A randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Singer, C.F.; Pfeiler, G.; Hubalek, M.; Bartsch, R.; Stoger, H.; Pichler, A.; Petru, E.; Bjelic-Radisic, V.; Greil, R.; Rudas, M.; et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: Results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur. J. Cancer 2020, 132, 43–52. [Google Scholar] [CrossRef]
- Le Gall, C.M.; Weiden, J.; Eggermont, L.J.; Figdor, C.G. Dendritic cells in cancer immunotherapy. Nat. Mater. 2018, 17, 474–475. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Patel, J.M.; Vartabedian, V.F.; Bozeman, E.N.; Caoyonan, B.E.; Srivatsan, S.; Pack, C.D.; Dey, P.; D’Souza, M.J.; Yang, L.; Selvaraj, P. Plasma membrane vesicles decorated with glycolipid-anchored antigens and adjuvants via protein transfer as an antigen delivery platform for inhibition of tumor growth. Biomaterials 2016, 74, 231–244. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Cancer Vaccine | Strategy | Vaccine Type | Advantages | Disadvantages |
|---|---|---|---|---|
| Preventive | Viral antigens-based vaccines | HBV HPV (Gardasil, Cervarix) | Highly efficacious Excellent safety profile Highly immunogenic | Restricted to cancers with known etiopathogenic agents |
| Retired antigens-based vaccines | AMHR2-ED α-lactalbumin | Specific for adult onset non- viral associated cancers Highly specific Immunogenic | Only applicable to cancer types with known retired antigens | |
| Embryonic material-based vaccines | Intact ES cells Intact IPSCs ES cell exosomes | Comprehensive immune responses against multiple antigens; Broad spectrum (off-the shelf) | Complex and costly manufacture procedure | |
| Therapeutic | Cell-based vaccines | Gvax Sipuleucel-T Algenpantucel-L STINGVAX | High antigenic immunogenic potency; Control of antigen presentation | Risk for vaccine-triggered adversary effects; Complex and costly manufacture procedure |
| Viral vector- or bacterial vector-based vaccines | PROSTVAC ALVAC | High antigenic immunogenic potency; Broad spectrum (off-the-shelf); Suitable for large-scale manufacture | Host-induced immune responses to vectors; Safety concerns for accidental infection; Risk for vaccine-triggered adversary effects | |
| Peptide-based vaccines | CTAG1B MAGE-A3 BIRC5 WT1 Peptide-based mutant neo-epitopes (personalized vaccines) | Low risk for vaccine-triggered adversary effects; Suitable for large-scale manufacture | Modest antigenic immunogenic potency; Restriction in HLA haplotype subtype | |
| DNA- or RNA-based vaccines | RNA-based neo-epitopes (personalized vaccines) RNA-based TAAs (NY-EXO-1, MAGE-A3, Tyrosinase) | Flexible to deliver multiple antigens; No restriction in HLA haplotype subtype; Comprehensive T cell and B cell responses; Suitable for large-scale manufacture | Modest antigenic immunogenic potency; Stringent temperature requirements for storage and transport of RNA-based vaccines |
| Number | Phase | Cancer Type | Vaccine | Outcome | Reference |
|---|---|---|---|---|---|
| NCT00089856 | III | Metastatic Prostate Cancer | Allogeneic prostate cancer cells overexpressing GM-CSF (GVAX) | Terminated (<30% chance of meeting primary endpoint) | [32] |
| NCT01836432 | III | Metastatic Pancreatic Cancer | Pancreatic ductal adenocarcinoma cells expressing aGT (Algenpantucel-L) | No improvement in overall survival | [35] |
| NCT00676507 | III | Advanced NSCLC | Allogeneic NSCLC cells with reduced TGFβ2 expression (belagenpumatucel-L) | Terminated without meeting the survival endpoint | [55] |
| NCT00796445 | III | Melanoma | Recombinant MAGE-A3 and AS15 immunostimulant | Terminated early for the lack of efficacy | [71] |
| NCT00409188 | III | Stage III NSCLC | Lipopeptide with MUC1 peptide sequence (Tecemotide) | Clinically relevant prolonged overall survival | [82] |
| NCT00683670 | I | Advanced melanoma | Autologous dendritic cells loaded with patient-specific neoantigens | Diverse neoantigen-specific T cell receptor repertoire | [104] |
| NCT01970358 | I | Advanced melanoma | Twenty predicted personal tumor neoantigens | Induction of CD4+ and CD8+ T cells targeting neoantigens | [105] |
| NCT02035956 | I | Advanced melanoma | Poly-neoepitopic coding RNA of an individual patient | T cell responses against multiple vaccine neoepitopes | [106] |
| NCT02149225 | I | Newly diagnosed glioblastoma | Unmutated antigen library and personalized neoepitopes | Sustained CD8+ and CD4+ T cell responses | 110 |
| NCT01896869 | II | Metastatic Pancreatic Cancer | GM-CSF-secreting allogeneic pancreatic tumor cells and ipilimumab | No improvement in overall survival | [113] |
| NCT01832870 | I | metastatic prostate cancer | Autologous dendritic cells loaded with PA2024 (Sipuleucel-T) and ipilimumab | Increase in tumor-specific antibodies | [114] |
| NCT01322490 | III | Metastatic prostate cancer | Poxviruses expressing PSA and costimulatory molecules (PROSTVAC) | No effect on alive without events and overall survival | [118] |
| NCT00113984 | I | Metastatic prostate cancer | PROSTVAC and ipilimumab | Enhancement in co-stimulation of the immune system | [119] |
| NCT01302496 | II | Advanced melanoma | Autologous dendritic cells electroporated with synthetic mRNA and ipilimumab | Highly durable tumor responses | [120] |
| NCT00094653 | III | Metastatic melanoma | Glycoprotein 100 peptide and ipilimumab | No effect in overall survival | [122] |
| NCT00084656 | II | Advanced melanoma | Multipeptides (tyrosinase/gp100/MART-1) and ipilimumab | No improvement in the clinical outcomes | [124] |
| NCT01176461 | I | Advanced melanoma | Multipeptides (MART-1/NY-ESO-1/gp100) and Nivolumab | No immunological responses obtained | [125] |
| NCT02410733 | I | Advanced melanoma | Liposomal RNA of 4 non-mutated TAAs (NY-ESO-1/MAGE-A3/tyrosinase/TPTE) and anti-PD-1 antibody | Strong CD4+ and CD8+ T cell immunity against antigens | [127] |
| NCT01976585 | I | Low-Grade Lymphoma | Flt3L, radiotherapy, and TLR3 agonist | Increase in the durable remission rates | [189] |
| NCT03313778 | I | Melanoma | Lipid-encapsulated mRNA | Safe and well-tolerated | [224] |
| NCT00409188 | III | NSCLC | MUC1 liposomal-based vaccine | No difference in overall sutvival | [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donninger, H.; Li, C.; Eaton, J.W.; Yaddanapudi, K. Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines 2021, 9, 668. https://doi.org/10.3390/vaccines9060668
Donninger H, Li C, Eaton JW, Yaddanapudi K. Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines. 2021; 9(6):668. https://doi.org/10.3390/vaccines9060668
Chicago/Turabian StyleDonninger, Howard, Chi Li, John W. Eaton, and Kavitha Yaddanapudi. 2021. "Cancer Vaccines: Promising Therapeutics or an Unattainable Dream" Vaccines 9, no. 6: 668. https://doi.org/10.3390/vaccines9060668
APA StyleDonninger, H., Li, C., Eaton, J. W., & Yaddanapudi, K. (2021). Cancer Vaccines: Promising Therapeutics or an Unattainable Dream. Vaccines, 9(6), 668. https://doi.org/10.3390/vaccines9060668