Revealing Plasma Membrane Nano-Domains with Diffusion Analysis Methods

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Lipid Membrane Nano-Domains
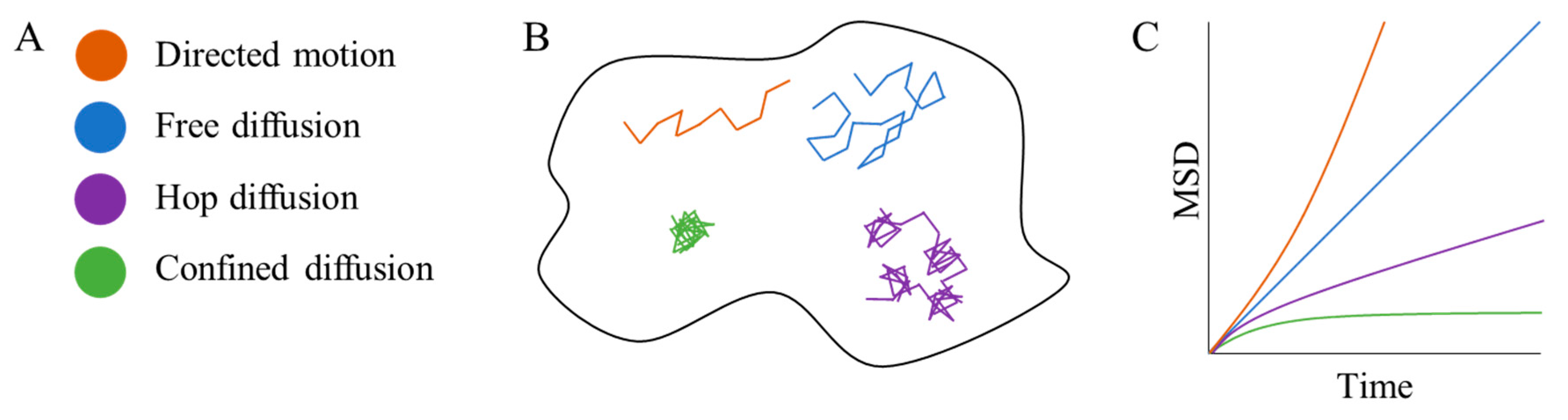
2. Single Particle Tracking
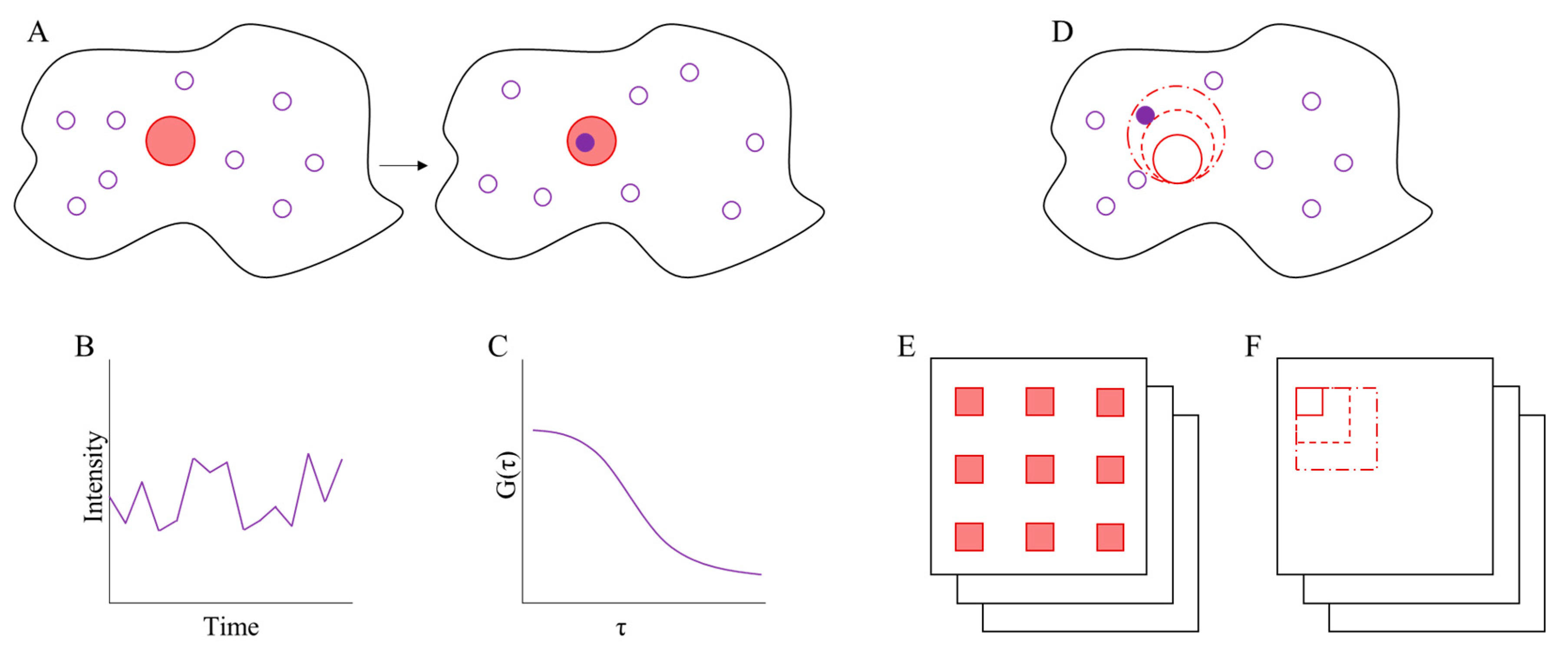
3. Fluorescence Correlation Spectroscopy
3.1. Imaging FCS
3.2. Spot Variation FCS
3.3. Binned-Imaging FCS
4. Image Correlation Spectroscopy
4.1. Spatio-Temporal Image Correlation Spectroscopy
4.2. Imaging Mean Square Displacement
4.3. PICS
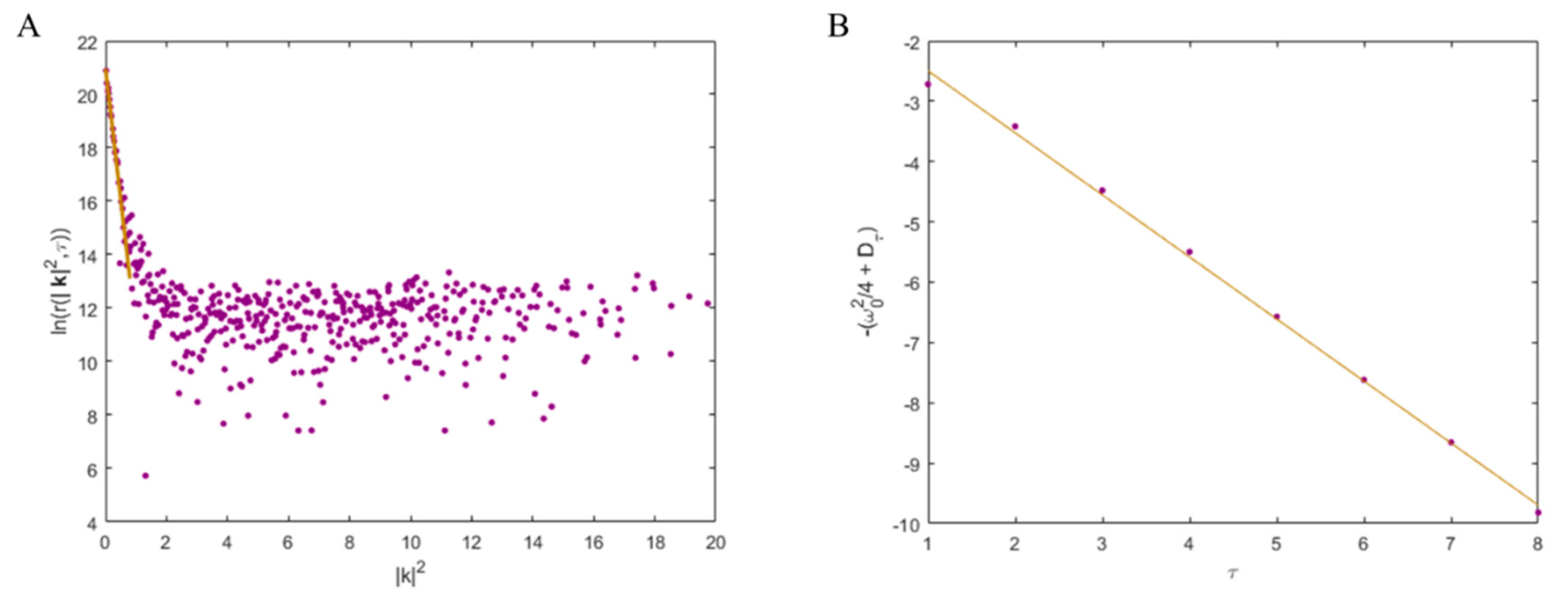
4.4. k-Space Image Correlation Spectroscopy
5. Fluorescence Recovery after Photobleaching
6. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singer, S.J.; Nicolson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Fischman, D.A.; Steck, T.L. Selective Solubilization of Proteins and Phospholipids from Red Blood Cell Membranes by Nonionic Detergents. J. Supramol. Cell. Biochem. 1973, 1, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Sada, R.; Kimura, H.; Fukata, Y.; Fukata, M.; Yamamoto, H.; Kikuchi, A. Dynamic Palmitoylation Controls the Microdomain Localization of the DKK1 Receptors CKAP4 and LRP6. Sci. Signal. 2019, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; Rose, J.K. Sorting of GPI-Anchored Proteins to Glycolipid-Enriched Membrane Subdomains during Transport to the Apical Cell Surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Sankaram, M.B.; Thompson, T.E. Cholesterol-Induced Fluid-Phase Immiscibility in Membranes. Proc. Natl. Acad. Sci. USA 1991, 88, 8686–8690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahya, N.; Scherfeld, D.; Bacia, K.; Poolman, B.; Schwille, P. Probing Lipid Mobility of Raft-Exhibiting Model Membranes by Fluorescence Correlation Spectroscopy. J. Biol. Chem. 2003, 278, 28109–28115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Ikonen, E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Pike, L.J. Rafts Defined: A Report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. [Google Scholar] [CrossRef] [Green Version]
- Simons, K.; Sampaio, J.L. Membrane Organization and Lipid Rafts. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–17. [Google Scholar] [CrossRef]
- Lingwood, D.; Simons, K. Lipid Rafts as a Membrane-Organizing Principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [Green Version]
- Goñi, F.M. “Rafts”: A Nickname for Putative Transient Nanodomains. Chem. Phys. Lipids 2019, 218, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Sheetz, M.P.; Elson, E.L. Single Particle Tracking. Analysis of Diffusion and Flow in Two-Dimensional Systems. Biophys. J. 1991, 60, 910–921. [Google Scholar] [CrossRef] [Green Version]
- Kusumi, A.; Sako, Y. Cell Surface Organization by the Membrane Skeleton. Curr. Opin. Cell Biol. 1996, 8, 566–574. [Google Scholar] [CrossRef]
- Albrecht, D.; Winterflood, C.M.; Sadeghi, M.; Tschager, T.; Noé, F.; Ewers, H. Nanoscopic Compartmentalization of Membrane Protein Motion at the Axon Initial Segment. J. Cell Biol. 2016, 215, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumi, A.; Suzuki, K.G.N.; Kasai, R.S.; Ritchie, K.; Fujiwara, T.K. Hierarchical Mesoscale Domain Organization of the Plasma Membrane. Trends Biochem. Sci. 2011, 36, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Adebiyi, A.; Soni, H.; John, T.A.; Yang, F. Lipid Rafts Are Required for Signal Transduction by Angiotensin II Receptor Type 1 in Neonatal Glomerular Mesangial Cells. Exp. Cell Res. 2014, 324, 92–104. [Google Scholar] [CrossRef]
- Varshney, P.; Yadav, V.; Saini, N. Lipid Rafts in Immune Signalling: Current Progress and Future Perspective. Immunology 2016, 149, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Kalappurakkal, J.M.; Anilkumar, A.A.; Patra, C.; van Zanten, T.S.; Sheetz, M.P.; Mayor, S. Integrin Mechano-Chemical Signaling Generates Plasma Membrane Nanodomains That Promote Cell Spreading. Cell 2019, 177, 1738–1756. [Google Scholar] [CrossRef] [Green Version]
- Helms, J.B.; Zurzolo, C. Lipids as Targeting Signals: Lipid Rafts and Intracellular Trafficking. Traffic 2004, 5, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Raghu, H.; Sodadasu, P.K.; Malla, R.R.; Gondi, C.S.; Estes, N.; Rao, J.S. Localization of UPAR and MMP-9 in Lipid Rafts Is Critical for Migration, Invasion and Angiogenesis in Human Breast Cancer Cells. BMC Cancer 2010, 10, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rios, F.J.O.; Ferracini, M.; Pecenin, M.; Koga, M.M.; Wang, Y.; Ketelhuth, D.F.J.; Jancar, S. Uptake of OxLDL and IL-10 Production by Macrophages Requires PAFR and CD36 Recruitment into the Same Lipid Rafts. PLoS ONE 2013, 8, e76893. [Google Scholar] [CrossRef] [PubMed]
- Sezgin, E.; Levental, I.; Mayor, S.; Eggeling, C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafzadeh, P.; Parmryd, I. Methods Applicable to Membrane Nanodomain Studies? Essays Biochem. 2015, 57, 57–68. [Google Scholar] [PubMed]
- Geerts, H.; De Brabander, M.; Nuydens, R.; Geuens, S.; Moeremans, M.; De Mey, J.; Hollenbeck, P. Nanovid Tracking: A New Automatic Method for the Study of Mobility in Living Cells Based on Colloidal Gold and Video Microscopy. Biophys. J. 1987, 52, 775–782. [Google Scholar] [CrossRef] [Green Version]
- Schindler, H.; Feher, G. Imaging of Single Molecule Diffusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2926–2929. [Google Scholar]
- Schütz, G.J.; Kada, G.; Pastushenko, V.P.; Schindler, H. Properties of Lipid Microdomains in a Muscle Cell Membrane Visualized by Single Molecule Microscopy. EMBO J. 2000, 19, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [Green Version]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-Density Mapping of Single-Molecule Trajectories with Photoactivated Localization Microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef] [Green Version]
- Clausen, M.P.; Arnspang, E.C.; Ballou, B.; Bear, J.E.; Lagerholm, B.C. Simultaneous Multi-Species Tracking in Live Cells with Quantum Dot Conjugates. PLoS ONE 2014, 9, e97671. [Google Scholar] [CrossRef]
- Kusumi, A.; Sako, Y.; Yamamoto, M. Confined Lateral Diffusion of Membrane Receptors. Biophys. J. 1993, 65, 2021–2040. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, T.; Ritchie, K.; Murakoshi, H.; Jacobson, K.; Kusumi, A. Phospholipids Undergo Hop Diffusion in Compartmentalized Cell Membrane. J. Cell Biol. 2002, 157, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Jaqaman, K.; Kuwata, H.; Touret, N.; Collins, R.; Trimble, W.S.; Danuser, G.; Grinstein, S. Cytoskeletal Control of CD36 Diffusion Promotes Its Receptor and Signaling Function. Cell 2011, 146, 593–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, K.G.N.; Fujiwara, T.K.; Edidin, M.; Kusumi, A. Dynamic Recruitment of Phospholipase Cγ at Transiently Immobilized GPI-Anchored Receptor Clusters Induces IP3-Ca2+ Signaling: Single-Molecule Tracking Study 2. J. Cell Biol. 2007, 177, 731–742. [Google Scholar] [CrossRef]
- Liu, P.; Weinreb, V.; Ridilla, M.; Betts, L.; Patel, P.; De Silva, A.M.; Thompson, N.L.; Jacobson, K. Rapid, Directed Transport of DC-SIGN Clusters in the Plasma Membrane. Sci. Adv. 2017, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, K.I.; Churchman, L.S.; Spudich, J.A.; Flyvbjerg, H. Optimized Localization Analysis for Single-Molecule Tracking and Super-Resolution Microscopy. Nat. Methods 2010, 7, 377–381. [Google Scholar] [CrossRef] [Green Version]
- Arnspang, E.C.; Sengupta, P.; Mortensen, K.I.; Jensen, H.H.; Hahn, U.; Jensen, E.B.V.; Lippincott-Schwartz, J.; Nejsum, L.N. Regulation of Plasma Membrane Nanodomains of the Water Channel Aquaporin-3 Revealed by Fixed and Live Photoactivated Localization Microscopy. Nano Lett. 2019, 19, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Clausen, M.P.; Lagerholm, B.C. Visualization of Plasma Membrane Compartmentalization by High-Speed Quantum Dot Tracking. Nano Lett. 2013, 13, 2332–2337. [Google Scholar] [CrossRef]
- Vestergaard, C.L.; Blainey, P.C.; Flyvbjerg, H. Optimal Estimation of Diffusion Coefficients from Single-Particle Trajectories. Phys. Rev. E 2014, 89, 022726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestergaard, C.L.; Pedersen, J.N.; Mortensen, K.I.; Flyvbjerg, H. Estimation of Motility Parameters from Trajectory Data: A Condensate of Our Recent Results. Eur. Phys. J. Spec. Top. 2015, 224, 1151–1168. [Google Scholar] [CrossRef] [Green Version]
- Adler, J.; Shevchuk, A.I.; Novak, P.; Korchev, Y.E.; Parmryd, I. Plasma Membrane Topography and Interpretation of Single-Particle Tracks. Nat. Methods 2010, 7, 170–171. [Google Scholar] [CrossRef]
- Ram, S.; Prabhat, P.; Chao, J.; Sally Ward, E.; Ober, R.J. High Accuracy 3D Quantum Dot Tracking with Multifocal Plane Microscopy for the Study of Fast Intracellular Dynamics in Live Cells. Biophys. J. 2008, 95, 6025–6043. [Google Scholar] [CrossRef] [Green Version]
- Magde, D.; Elson, E.; Webb, W.W. Thermodynamic Fluctuations in a Reacting System—Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett. 1972, 29, 705–708. [Google Scholar] [CrossRef]
- Petersen, N.O. Scanning Fluorescence Correlation Spectroscopy. I. Theory and Simulation of Aggregation Measurements. Biophys. J. 1986, 49, 809–815. [Google Scholar] [CrossRef] [Green Version]
- Rigler, R.; Mets, Ü.; Widengren, J.; Kask, P. Fluorescence Correlation Spectroscopy with High Count Rate and Low Background: Analysis of Translational Diffusion. Eur. Biophys. J. 1993, 22, 169–175. [Google Scholar] [CrossRef]
- Winkler, P.M.; Regmi, R.; Flauraud, V.; Brugger, J.; Rigneault, H.; Wenger, J.; García-Parajo, M.F. Optical Antenna-Based Fluorescence Correlation Spectroscopy to Probe the Nanoscale Dynamics of Biological Membranes. J. Phys. Chem. Lett. 2018, 9, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regmi, R.; Winkler, P.M.; Flauraud, V.; Borgman, K.J.E.; Manzo, C.; Brugger, J.; Rigneault, H.; Wenger, J.; García-Parajo, M.F. Planar Optical Nanoantennas Resolve Cholesterol-Dependent Nanoscale Heterogeneities in the Plasma Membrane of Living Cells. Nano Lett. 2017, 17, 6295–6302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannan, B.; Guo, L.; Sudhaharan, T.; Ahmed, S.; Maruyama, I.; Wohland, T. Spatially Resolved Total Internal Reflection Fluorescence Correlation Microscopy Using an Electron Multiplying Charge-Coupled Device Camera. Anal. Chem. 2007, 79, 4463–4470. [Google Scholar] [CrossRef]
- Krieger, J.W.; Singh, A.P.; Bag, N.; Garbe, C.S.; Saunders, T.E.; Langowski, J.; Wohland, T. Imaging Fluorescence (Cross-) Correlation Spectroscopy in Live Cells and Organisms. Nat. Protoc. 2015, 10, 1948–1974. [Google Scholar] [CrossRef]
- Bag, N.; Holowka, D.A.; Baird, B.A. Imaging FCS Delineates Subtle Heterogeneity in Plasma Membranes of Resting Mast Cells. Mol. Biol. Cell 2020, 31, 709–723. [Google Scholar] [CrossRef]
- Wawrezinieck, L.; Rigneault, H.; Marguet, D.; Lenne, P.F. Fluorescence Correlation Spectroscopy Diffusion Laws to Probe the Submicron Cell Membrane Organization. Biophys. J. 2005, 89, 4029–4042. [Google Scholar] [CrossRef] [Green Version]
- Lenne, P.F.; Wawrezinieck, L.; Conchonaud, F.; Wurtz, O.; Boned, A.; Guo, X.J.; Rigneault, H.; He, H.T.; Marguet, D. Dynamic Molecular Confinement in the Plasma Membrane by Microdomains and the Cytoskeleton Meshwork. EMBO J. 2006, 25, 3245–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.-T.; Marguet, D. Detecting Nanodomains in Living Cell Membrane by Fluorescence Correlation Spectroscopy. Annu. Rev. Phys. Chem. 2011, 62, 417–436. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the Diffraction Resolution Limit by Stimulated Emission: Stimulated-Emission-Depletion Fluorescence Microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef]
- Eggeling, C.; Ringemann, C.; Medda, R.; Schwarzmann, G.; Sandhoff, K.; Polyakova, S.; Belov, V.N.; Hein, B.; Von Middendorff, C.; Schönle, A.; et al. Direct Observation of the Nanoscale Dynamics of Membrane Lipids in a Living Cell. Nature 2009, 457, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Pralle, A. Continuous Monitoring of Membrane Protein Micro- Domain Association during Cell Signaling. arXiv 2011, arXiv:1101.5087v1. [Google Scholar]
- Huang, H.; Simsek, M.F.; Jin, W.; Pralle, A. Effect of Receptor Dimerization on Membrane Lipid Raft Structure Continuously Quantified on Single Cells by Camera Based Fluorescence Correlation Spectroscopy. PLoS ONE 2015, 10, e0121777. [Google Scholar] [CrossRef]
- Jin, W.; Simsek, M.F.; Pralle, A. Quantifying Spatial and Temporal Variations of the Cell Membrane Ultra-Structure by BimFCS. Methods 2018, 140–141, 151–160. [Google Scholar] [CrossRef]
- Petersen, N.O.; Höddelius, P.L.; Wiseman, P.W.; Seger, O.; Magnusson, K.E. Quantitation of Membrane Receptor Distributions by Image Correlation Spectroscopy: Concept and Application. Biophys. J. 1993, 65, 1135–1146. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, P.W.; Petersen, N.O. Image Correlation Spectroscopy. II. Optimization for Ultrasensitive Detection of Preexisting Platelet-Derived Growth Factor-β Receptor Oligomers on Intact Cells. Biophys. J. 1999, 76, 963–977. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, P.W. Image Correlation Spectroscopy. Compr. Biophys. 2012, 2, 246–259. [Google Scholar]
- Srivastava, M.; Petersen, N.O. Image Cross-Correlation Spectroscopy: A New Experimental Biophysical Approach to Measurement of Slow Diffusion of Fluorescent Molecules. Methods Cell Sci. 1996, 18, 47–54. [Google Scholar] [CrossRef]
- Wiseman, P.W.; Squier, J.A.; Ellisman, M.H.; Wilson, K.R. Two-Photo Image Correlation Spectroscopy and Image Cross-Correlation Spectroscopy. J. Microsc. 2000, 200, 14–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, B.; Costantino, S.; Wiseman, P.W. Spatiotemporal Image Correlation Spectroscopy (STICS) Theory, Verification, and Application to Protein Velocity Mapping in Living CHO Cells. Biophys. J. 2005, 88, 3601–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolin, D.L.; Ronis, D.; Wiseman, P.W. K-Space Image Correlation Spectroscopy: A Method for Accurate Transport Measurements Independent of Fluorophore Photophysics. Biophys. J. 2006, 91, 3061–3075. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Sengupta, P.; Wiseman, P.W.; Brown, C.M.; Horwitz, A.R.; Gratton, E. Fluctuation Correlation Spectroscopy with a Laser-Scanning Microscope: Exploiting the Hidden Time Structure. Biophys. J. 2005, 88, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Digman, M.A.; Brown, C.M.; Sengupta, P.; Wiseman, P.W.; Horwitz, A.R.; Gratton, E. Measuring Fast Dynamics in Solutions and Cells with a Laser Scanning Microscope. Biophys. J. 2005, 89, 1317–1327. [Google Scholar] [CrossRef] [Green Version]
- Semrau, S.; Schmidt, T. Particle Image Correlation Spectroscopy (PICS): Retrieving Nanometer-Scale Correlations from High-Density Single-Molecule Position Data. Biophys. J. 2007, 92, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Ciccotosto, G.D.; Kozer, N.; Chow, T.T.Y.; Chon, J.W.M.; Clayton, A.H.A. Aggregation Distributions on Cells Determined by Photobleaching Image Correlation Spectroscopy. Biophys. J. 2013, 104, 1056–1064. [Google Scholar] [CrossRef] [Green Version]
- Kolin, D.L.; Wiseman, P.W. Advances in Image Correlation Spectroscopy: Measuring Number Densities, Aggregation States, and Dynamics of Fluorescently Labeled Macromolecules in Cells. Cell Biochem. Biophys. 2007, 49, 141–164. [Google Scholar] [CrossRef]
- Meddens, M.B.M.; Pandzic, E.; Slotman, J.A.; Guillet, D.; Joosten, B.; Mennens, S.; Paardekooper, L.M.; Houtsmuller, A.B.; Van Den Dries, K.; Wiseman, P.W.; et al. Actomyosin-Dependent Dynamic Spatial Patterns of Cytoskeletal Components Drive Mesoscale Podosome Organization. Nat. Commun. 2016, 7, 1–17. [Google Scholar] [CrossRef]
- Pandžić, E.; Abu-Arish, A.; Whan, R.M.; Hanrahan, J.W.; Wiseman, P.W. Velocity Landscape Correlation Resolves Multiple Flowing Protein Populations from Fluorescence Image Time Series. Methods 2018, 140–141, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Pandžić, E.; Rossy, J.; Gaus, K. Tracking Molecular Dynamics without Tracking: Image Correlation of Photo-Activation Microscopy. Methods Appl. Fluoresc. 2015, 3, 014006. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, C.; Piazza, V.; Gratton, E.; Beltram, F.; Cardarelli, F. Probing Short-Range Protein Brownian Motion in the Cytoplasm of Living Cells. Nat. Commun. 2014, 5, 5891. [Google Scholar] [CrossRef] [PubMed]
- Gröner, N.; Capoulade, J.; Cremer, C.; Wachsmuth, M. Measuring and Imaging Diffusion with Multiple Scan Speed Image Correlation Spectroscopy. Opt. Express 2010, 18, 21225. [Google Scholar] [CrossRef] [Green Version]
- Digiacomo, L.; Digman, M.A.; Gratton, E.; Caracciolo, G. Development of an Image Mean Square Displacement (IMSD)-Based Method as a Novel Approach to Study the Intracellular Trafficking of Nanoparticles. Acta Biomater. 2016, 42, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Bohórquez-Hernández, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol Modulates the Cellular Localization of Orai1 Channels and Its Disposition among Membrane Domains. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1481–1490. [Google Scholar] [CrossRef]
- Arnspang, E.C.; Schwartzentruber, J.; Clausen, M.P.; Wiseman, P.W.; Lagerholm, B.C. Bridging the Gap between Single Molecule and Ensemble Methods for Measuring Lateral Dynamics in the Plasma Membrane. PLoS ONE 2013, 8, e78096. [Google Scholar] [CrossRef]
- Arnspang, E.C.; Login, F.H.; Koffman, J.S.; Sengupta, P.; Nejsum, L.N. AQP2 Plasma Membrane Diffusion Is Altered by the Degree of AQP2-S256 Phosphorylation. Int. J. Mol. Sci. 2016, 17, 1804. [Google Scholar] [CrossRef] [Green Version]
- Abu-Arish, A.; Pandzic, E.; Goepp, J.; Matthes, E.; Hanrahan, J.W.; Wiseman, P.W. Cholesterol Modulates CFTR Confinement in the Plasma Membrane of Primary Epithelial Cells. Biophys. J. 2015, 109, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Peters, R.; Peters, J.; Tews, K.H.; Bähr, W. A Microfluorimetric Study of Translational Diffusion in Erythrocyte Membranes. Biochim. Biophys. Acta Biomembr. 1974, 367, 282–294. [Google Scholar] [CrossRef]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility Measurement by Analysis of Fluorescence Photobleaching Recovery Kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef] [Green Version]
- Braeckmans, K.; Remaut, K.; Vandenbroucke, R.E.; Lucas, B.; De Smedt, S.C.; Demeester, J. Line FRAP with the Confocal Laser Scanning Microscope for Diffusion Measurements in Small Regions of 3-D Samples. Biophys. J. 2007, 92, 2172–2183. [Google Scholar] [CrossRef] [Green Version]
- Oyola-Cintrón, J.; Caballero-Rivera, D.; Ballester, L.; Baéz-Pagán, C.A.; Martínez, H.L.; Vélez-Arroyo, K.P.; Quesada, O.; Lasalde-Dominicci, J.A. Lateral Diffusion, Function, and Expression of the Slow Channel Congenital Myasthenia Syndrome AC418W Nicotinic Receptor Mutation with Changes in Lipid Raft Components. J. Biol. Chem. 2015, 290, 26790–26800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zidovetzki, R.; Levitan, I. Use of Cyclodextrins to Manipulate Plasma Membrane Cholesterol Content: Evidence, Misconceptions and Control Strategies. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1311–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kure, J.L.; Andersen, C.B.; Mortensen, K.I.; Wiseman, P.W.; Arnspang, E.C. Revealing Plasma Membrane Nano-Domains with Diffusion Analysis Methods. Membranes 2020, 10, 314. https://doi.org/10.3390/membranes10110314
Kure JL, Andersen CB, Mortensen KI, Wiseman PW, Arnspang EC. Revealing Plasma Membrane Nano-Domains with Diffusion Analysis Methods. Membranes. 2020; 10(11):314. https://doi.org/10.3390/membranes10110314
Chicago/Turabian StyleKure, Jakob L., Camilla B. Andersen, Kim I. Mortensen, Paul W. Wiseman, and Eva C. Arnspang. 2020. "Revealing Plasma Membrane Nano-Domains with Diffusion Analysis Methods" Membranes 10, no. 11: 314. https://doi.org/10.3390/membranes10110314
APA StyleKure, J. L., Andersen, C. B., Mortensen, K. I., Wiseman, P. W., & Arnspang, E. C. (2020). Revealing Plasma Membrane Nano-Domains with Diffusion Analysis Methods. Membranes, 10(11), 314. https://doi.org/10.3390/membranes10110314