Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer



Abstract
:1. Introduction
2. Ca2+ Signaling
Calcium Ion Channels
3. CRAC Channels
3.1. STIM Proteins
STIM1 Structure
3.2. Orai Proteins
Orai Structure
3.3. Activation Mechanisms of the STIM1/Orai Signalling Machinery
3.4. STIM1 Activation
3.5. STIM1-Orai1 Coupling
3.5.1. STIM1 Domains Coupling to Orai1
3.5.2. Orai1 Domains Coupling to STIM1
3.6. Activation of the Orai1 Ion Channel
3.7. The Orai1 Pore
3.8. Unique Biophysical Properties of CRAC Channels
3.9. Pharmacology of CRAC Channels
4. Pathophysiological Functions of STIM1 and Orai1
4.1. STIM1 and Diseases
4.2. Orai1 and Diseases
4.3. CRAC Channels and Cancer
4.3.1. Breast Cancer
4.3.2. Prostate Cancer
4.3.3. Colon Cancer
4.3.4. Other Cancer Types
5. Ca2+-Activated K+ Channels
5.1. SK Channels
5.2. Activation Mechanism of a Human SK-Calmodulin Channel Complex
6. SK Channel Pharmacology
7. Ca2+-Activated K+ Channels in Diseases
7.1. SK Channel in Neurons and Neuronal Disease
7.2. SK Channels in Cancer
8. Individual and Collective Modulation of Ca2+ Sensitive Ion Channels in Lipid Rafts
8.1. CRAC Channel Regulation in Signalplexes in Lipid Rafts
8.1.1. Lipid Mediated STIM1-Orai1 Regulation
8.1.2. Proteins Modulating STIM1-Orai1 Function
8.1.3. CRAC Channel Components in Co-Regulation with Other Ion Channels
8.2. Ca2+-Activated K+ Channel Regulation in Microdomains
8.2.1. Modulation of KCa2+ Channels by Lipids
8.2.2. Accessory Proteins Modulating KCa2+ Channel Activity
8.2.3. KCa2+ Channel Activity in Co-Regulation with Other Ion Channels
9. SK3 and Orai1 Channel Interplay
9.1. SK3 and Orai1 Colocalization in Lipid Rafts in Cancer Cells
9.2. SK3/Orai1 Complex and Accessory Proteins
9.3. SK3/Orai1 Complex and cAMP-PKA Pathway
10. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1-EBIO | 1-Ethylbenzimidazolinone |
| 2-APB | 2-aminoethoxydiphenyl borate |
| 4-AP | 4-aminopyridine |
| Å | Angstrom (unit of length equal to 10−10 m) |
| aa | Amino acid |
| AC8 | Adenylyl cyclase 8 |
| Akt | Known as protein kinase B or PKB |
| ARC | Arachidonic acid (AA)-activated channels |
| ATP | Adenosine triphosphate |
| ANSGA | 4-Point mutation in hinge region aa position 261–265 |
| ATPase | Adenosine triphosphatase |
| BK | Large conductance, Ca2+-activated potassium channels |
| BTP2 | [N-{4-[3,5-bis(Trifluoromethyl)-1H-pyrazol-1-yl]phenyl}-4-methyl-1,2,3-thiadiazole-5-carboxamide] |
| Ca2+ | Calcium ion |
| CAD | Ca2+ release-activated Ca2+-activating domain |
| CaM | Calmodulin |
| cAMP | Cyclic adenosine monophosphate |
| CAR | Ca2+ accumulating region |
| CaV1 | Caveolin-1 |
| CC | Coiled-coil |
| Ccb9 | Coiled-coil domain containing region b9 |
| cEF | Canonical EF hand |
| CRAC | Ca2+ release-activated Ca2+ |
| CRACR2A | Calcium release activated channel regulator 2A |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats/Cas |
| Cs+ | Cesium ion |
| Cyppa | N-Cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-4 pyrimidinamine |
| ∆ | Represents deletion mutants |
| dOrai | Drosophila melanogaster Orai |
| DVF | Divalent-free |
| EDA | Ectodermal dysplasia |
| EGF | Epidermal growth factor |
| EGTA | Ethylene glycol tetraacetic acid |
| ER | Endoplasmic reticulum |
| ERα | Estrogen receptor-α |
| ERK1/2 | Extracellular-signal-regulated kinases 1 and 2 |
| ETON | Extended transmembrane Orai1 N-terminal |
| FCDI | Fast calcium-dependent inactivation |
| FRAP | Fluorescence recovery after photobleaching |
| FRET | Fluorescence resonance energy transfer |
| GBM | Glioblastoma multiforme cells |
| GoF | Gain of function |
| GSK | GlaxoSmithKline compounds |
| HEK | Human embryonic kidney |
| I/V | Current voltage relationship |
| ICa2+ | CRAC current |
| INa+ | Sodium current in sodium divalent free solution |
| ID | Inhibitory domain |
| IH | Inhibitory helix |
| IK | Intermediate Ca2+-activated K+ channels |
| IP3 | Inositol-triphosphate |
| IP3R | Inositol-triphosphate receptor |
| K+ | Potassium ion |
| KCa | Ca2+-activated K+ channels |
| Kir | Inward-rectifier potassium channels |
| L1-L3 | Loop 1–3 (of Orai channels) |
| L-type | Long-lasting calcium channel |
| LGC | Ligand gated calcium channels |
| LoF | Loss of function |
| MCU | Mitochondrial Ca2+ uniporter |
| MD simulations | Molecular dynamics simulations |
| Na+-DVF | Sodium divalent free |
| nEF | Non-canonical EF hand |
| NFAT | Nuclear factor of activated T cells |
| nAChR | Nicotinic acetylcholine receptors |
| NS309 | 3-Oxime-6,7-dichloro-1H-indole-2,3-dione |
| NS3893 | N-[(1R)-1,2,3,4-Tetrahydro-1-naphthalenyl]-1H-Benzimidazol-2-amine hydrochloride |
| NMR | Nuclear magnetic resonance |
| OASF | Orai-activating small fragment |
| Orai 1–3 | Orai proteins (also as O1–3) |
| P/S | Proline, serine |
| P-Akt | Phosphorylated known as protein kinase B or PKB |
| PI3K | Phosphoinositide 3-kinases |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PKA | Protein kinase A |
| PKC | Protein kinase C |
| PM | Plasma membrane |
| PMCA | Plasma membrane Ca2+ ATPase |
| Ref | References |
| S | Signal peptide |
| SAM | sterile α-motif |
| SARAF | Store-operated calcium entry associated regulatory factor |
| SCDI | Slow calcium-dependent inactivation |
| SCID | Severe combined immune deficiency |
| SigmaR1 | Sigma non-Opioid intracellular receptor 1 |
| SK channels | Small-conductance Ca(2+)-activated K(+) channels |
| SKA-111 | 5-Methylnaphtho[1¨C-d]thiazol-2-amine |
| SKA-121 | 5-Methylnaphth[2,1-d]oxazol-2-amine |
| SOAP | STIM-Orai association pocket |
| SOAR | STIM-Orai activating region |
| SOC | Store operated channel |
| SOCE | Store-operated calcium entry |
| SPCA2 | Secretory pathway Ca2+-ATPase |
| STIM | Stromal interaction molecule |
| STRMK | Stormorken syndrome |
| Synta66 | 4-Pyridinecarboxamide |
| TAM | Tubular Aggregate Myopathy |
| TM | Transmembrane helices |
| TRP | Transient receptor potential ion channel (C-canonical, M-melastatin, V-vallinoid) |
| VGCC | Voltage gated calcium channels |
References
- Hanna, M.G. Genetic neurological channelopathies. Nat. Clin. Pract. Neurol. 2006, 2, 252–263. [Google Scholar] [CrossRef]
- Spillane, J.; Kullmann, D.M.; Hanna, M.G. Genetic neurological channelopathies: Molecular genetics and linical phenotypes. J. Neurol. Neurosurg. Psychiatry 2016, 87, 37–48. [Google Scholar] [CrossRef]
- Moreau, A.; Chahine, M. A new cardiac channelopathy: From clinical phenotypes to molecular mechanisms associated with Nav1.5 gating pores. Front. Cardiovasc. Med. 2018, 5, 139. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.R. Neurological channelopathies. BMJ 1998, 316, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B. Channelopathies. Korean J. Pediatrics 2014, 57, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunzelmann, K. Ion channels and cancer. J. Membr. Biol. 2005, 205, 159–173. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, A.; Du, Q.; Liao, Q.; Shuai, Z.; Chen, C.; Yang, X.; Hu, Y.; Zhao, J.; Liu, S.; et al. Novel insights into ion channels in cancer stem cells (Review). Int. J. Oncol. 2018, 53, 1435–1441. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels in cancer: Are cancer hallmarks oncochannelopathies? Physiol. Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [Green Version]
- Ambudkar, I.S.; de Souza, L.B.; Ong, H.L. TRPC1, Orai1, and STIM1 in SOCE: Friends in tight spaces. Cell Calcium. 2017, 63, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Ong, H.L.; Ambudkar, I.S. The dynamic complexity of the TRPC1 channelosome. Channels 2011, 5, 424–431. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Fakler, B.; Kaczmarek, L.K.; Isom, L.L. More than a pore: Ion channel signaling complexes. J. Neurosci. 2014, 34, 15159–15169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacruz, R.S.; Feske, S. Diseases caused by mutations in ORAI1 and STIM. Ann. N. Y. Acad. Sci. 2015, 1356, 45–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzon, N.M.; Beam, K.G. Calcium channelopathies. Kidney Int. 2000, 57, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.M.; Simpson, R.U. Inhibition of cancer cell growth by calcium channel antagonists in the athymic mouse. Cancer Res. 1992, 52, 2413–2418. [Google Scholar] [PubMed]
- Pancrazio, J.J.; Viglione, M.P.; Tabbara, I.A.; Kim, Y.I. Voltage-dependent ion channels in small-cell lung cancer cells. Cancer Res. 1989, 49, 5901–5906. [Google Scholar] [PubMed]
- Pancrazio, J.J.; Tabbara, I.A.; Kim, Y.I. Voltage-activated K+ conductance and cell proliferation in small-cell lung cancer. Anticancer Res. 1993, 13, 1231–1234. [Google Scholar]
- Lee, S.C.; Deutsch, C.; Beck, W.T. Comparison of ion channels in multidrug-resistant and -sensitive human leukemic cells. Proc. Natl. Acad. Sci. USA 1988, 85, 2019–2023. [Google Scholar] [CrossRef] [Green Version]
- Batra, S.; Alenfall, J. Effect of diverse categories of drugs on human colon tumour cell proliferation. Anticancer Res. 1991, 11, 1221–1224. [Google Scholar]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: How it contributes to cancer hallmarks? Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell. Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell. Biol. 2003, 4, 517. [Google Scholar] [CrossRef] [Green Version]
- Berridge, M.J.; Bootman, M.D.; Lipp, P. Calcium—A life and death signal. Nature 1998, 395, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Rosado, J.A.; Sage, S.O. The actin cytoskeleton in store-mediated calcium entry. J. Physiol. 2000, 526 Pt 2, 221–229. [Google Scholar] [CrossRef]
- Jardin, I.; Diez-Bello, R.; Lopez, J.J.; Redondo, P.C.; Salido, G.M.; Smani, T.; Rosado, J.A. TRPC6 channels are required for proliferation, migration and invasion of breast cancer cell lines by modulation of Orai1 and Orai3 surface exposure. Cancers 2018, 10, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosado, J.A. Calcium entry pathways in non-excitable cells. Preface. Adv. Exp. Med. Biol. 2016, 898, vii–viii. [Google Scholar] [PubMed]
- Capiod, T.; Haiech, J.; Heizmann, C.W.; Krebs, J.; Mignen, O. Calcium and cell fate. Biochim. Biophys. Acta. 2016, 1863 (6 Pt B), 1335–1336. [Google Scholar] [CrossRef]
- Debant, M.; Hemon, P.; Brigaudeau, C.; Renaudineau, Y.; Mignen, O. Calcium signaling and cell fate: How can Ca2+ signals contribute to wrong decisions for chronic lymphocytic leukemic B lymphocyte outcome? Int. J. Dev. Biol. 2015, 59, 379–389. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.J. Calcium. Methods Mol. Biol. 2002, 172, 21–49. [Google Scholar]
- Vig, M.; Kinet, J.P. Calcium signaling in immune cells. Nat. Immunol. 2009, 10, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Parkash, J.; Asotra, K. Calcium wave signaling in cancer cells. Life Sci. 2010, 87, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vashisht, A.; Trebak, M.; Motiani, R.K. STIM and Orai proteins as novel targets for cancer therapy. A review in the theme: Cell and molecular processes in cancer metastasis. Am. J. Physiol. Cell Physiol. 2015, 309, C457–C469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tajada, S.; Villalobos, C. Calcium permeable channels in cancer hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef] [PubMed]
- Chantôme, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Guéguinou, M.; Pagès, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [Green Version]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion channels and the hallmarks of cancer. Trends Mol. Med. 2010, 16, 107–121. [Google Scholar] [CrossRef]
- Clarysse, L.; Guéguinou, M.; Potier-Cartereau, M.; Vandecasteele, G.; Bougnoux, P.; Chevalier, S.; Chantôme, A.; Vandier, C. cAMP-PKA inhibition of SK3 channel reduced both Ca2+ entry and cancer cell migration by regulation of SK3-Orai1 complex. Pflug. Arch. 2014, 466, 1921–1932. [Google Scholar] [CrossRef]
- Gilbert, S.M.; Oliphant, C.J.; Hassan, S.; Peille, A.L.; Bronsert, P.; Falzoni, S.; Di Virgilio, F.; McNulty, S.; Lara, R. ATP in the tumour microenvironment drives expression of nfP2X7, a key mediator of cancer cell survival. Oncogene 2019, 38, 194–208. [Google Scholar] [CrossRef] [Green Version]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Pinault, M.; Marionneau-Lambot, S.; Oullier, T.; Simon, G.; Couthon-Gourvès, H. New alkyl-lipid blockers of SK3 channels reduce cancer cell migration and occurrence of metastasis. Curr. Cancer Drug Targets 2011, 11, 1111–1125. [Google Scholar] [CrossRef]
- Jaffrès, P.A.; Gajate, C.; Bouchet, A.M.; Couthon-Gourvès, H.; Chantôme, A.; Potier-Cartereau, M.; Besson, P.; Bougnoux, P.; Mollinedo, F.; Vandier, C. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol. Ther. 2016, 165, 114–131. [Google Scholar] [CrossRef]
- Jardin, I.; Lopez, J.J.; Salido, G.M.; Rosado, J.A. Store-operated Ca2+ entry in breast cancer cells: Remodeling and functional role. Int. J. Mol. Sci. 2018, 19, 4053. [Google Scholar] [CrossRef] [Green Version]
- Mignen, O.; Constantin, B.; Potier-Cartereau, M.; Penna, A.; Gautier, M.; Guéguinou, M.; Renaudineau, Y.; Shoji, K.F.; Félix, R.; Bayet, E.; et al. Constitutive calcium entry and cancer: Updated views and insights. Eur. Biophys. J. 2017, 46, 395–413. [Google Scholar] [CrossRef] [PubMed]
- Parihar, A.S.; Coghlan, M.J.; Gopalakrishnan, M.; Shieh, C.C. Effects of intermediate-conductance Ca2+-activated K+ channel modulators on human prostate cancer cell proliferation. Eur. J. Pharmacol. 2003, 471, 157–164. [Google Scholar] [CrossRef]
- Steudel, F.A.; Mohr, C.J.; Stegen, B.; Nguyen, H.Y.; Barnert, A.; Steinle, M.; Beer-Hammer, S.; Koch, P.; Lo, W.Y.; Schroth, W.; et al. SK4 channels modulate Ca2+ signalling and cell cycle progression in murine breast cancer. Mol. Oncol. 2017, 11, 1172–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, K.; Zhong, X.G.; Xia, X.M.; Huang, J.H.; Fan, Y.F.; Yuan, R.X.; Xue, N.R.; Du, J.; Han, W.X.; Xu, A.M.; et al. Orai1 forms a signal complex with SK3 channel in gallbladder smooth muscle. Biochem. Biophys. Res. Commun. 2015, 466, 456–462. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Z.; Gu, M.; Feng, X.; Xu, H. Release and uptake mechanisms of vesicular Ca2+ stores. Protein Cell 2019, 10, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef]
- Pankratov, Y.; Lalo, U. Calcium permeability of ligand-gated Ca2+ channels. Eur. J. Pharmacol. 2014, 739, 60–73. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Venkatachalam, K.; Montell, C. TRP channels. Annu. Rev. Biochem. 2007, 76, 387–417. [Google Scholar] [CrossRef] [Green Version]
- Ranade, S.S.; Syeda, R.; Patapoutian, A. Mechanically activated ion channels. Neuron 2015, 87, 1162–1179. [Google Scholar] [CrossRef] [Green Version]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef] [Green Version]
- Lam, A.K.; Galione, A. The endoplasmic reticulum and junctional membrane communication during calcium signaling. Biochim. Biophys Acta 2013, 1833, 2542–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Martinoia, E.; Szabo, I. Organellar channels and transporters. Cell Calcium 2015, 58, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshima, H.; Venturi, E.; Sitsapesan, R. New and notable ion-channels in the sarcoplasmic/endoplasmic reticulum: Do they support the process of intracellular Ca2+ release? J. Physiol. 2015, 593, 3241–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.J.; Feske, S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J. Physiol. 2012, 590, 4157–4167. [Google Scholar] [CrossRef]
- Zhang, S.L.; Yu, Y.; Roos, J.; Kozak, J.A.; Deerinck, T.J.; Ellisman, M.H.; Stauderman, K.A.; Cahalan, M.D. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 2005, 437, 902–905. [Google Scholar] [CrossRef]
- Stanisz, H.; Vultur, A.; Herlyn, M.; Roesch, A.; Bogeski, I. The role of Orai-STIM calcium channels in melanocytes and melanoma. J. Physiol. 2016, 594, 2825–2835. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Dragoni, S.; Turin, I.; Laforenza, U.; Potenza, D.M.; Bottino, C.; Glasnov, T.N.; Prestia, M.; Ferulli, F.; Saitta, A.; Mosca, A.; et al. Store-operated Ca2+ entry does not control proliferation in primary cultures of human metastatic renal cellular carcinoma. Biomed. Res. Int. 2014, 2014, 739494. [Google Scholar] [CrossRef] [Green Version]
- Stanisz, H.; Saul, S.; Müller, C.S.; Kappl, R.; Niemeyer, B.A.; Vogt, T.; Hoth, M.; Roesch, A.; Bogeski, I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014, 27, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Flourakis, M.; Lehen’Kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Van Denabeele, F.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosado, J.A. Introduction: Overview of the pathophysiological implications of store-operated calcium entry in mammalian cells. Adv. Exp. Med. Biol. 2017, 993, 391–395. [Google Scholar] [PubMed]
- Gueguinou, M.; Gambade, A.; Felix, R.; Chantome, A.; Fourbon, Y.; Bougnoux, P.; Weber, G.; Potier-Cartereau, M.; Vandier, C. Lipid rafts, KCa/ClCa/Ca2+ channel complexes and EGFR signaling: Novel targets to reduce tumor development by lipids? Biochim. Biophys. Acta. 2015, 1848 (10 Pt B), 2603–2620. [Google Scholar] [CrossRef] [Green Version]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef]
- Butorac, C.; Krizova, A.; Derler, I. Review: Structure and activation mechanisms of CRAC channels. Adv. Exp. Med. Biol. 2020, 1131, 547–604. [Google Scholar]
- Liou, J.; Kim, M.L.; Heo, W.D.; Jones, J.T.; Myers, J.W.; Ferrell, J.E.; Meyer, T., Jr. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 2005, 15, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.J.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Prakriya, M.; Feske, S.; Gwack, Y.; Srikanth, S.; Rao, A.; Hogan, P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature 2006, 443, 230–233. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal structure of the calcium release-activated calcium channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef] [Green Version]
- Lis, A.; Peinelt, C.; Beck, A.; Parvez, S.; Monteilh-Zoller, M.; Fleig, A.; Penner, R. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 2007, 17, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S.M.; Ashmole, I.; Smallwood, D.T.; Leyland, M.L.; Bradding, P. Orai/CRACM1 and KCa3.1 ion channels interact in the human lung mast cell plasma membrane. Cell Commun. Signal. 2015, 13, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, Y.; Hayashi, K.; Fujii, Y.; Mizushima, A.; Watarai, H.; Wakamori, M.; Numaga, T.; Mori, Y.; Iino, M.; Hikida, M.; et al. Coupling of STIM1 to store-operated Ca2+ entry through its constitutive and inducible movement in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2006, 103, 16704–16709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Lu, J.; Xu, P.; Xie, X.; Chen, L.; Xu, T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J. Biol. Chem. 2007, 282, 29448–29456. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Zheng, L.; Li, G.Y.; Plevin, M.J.; Ikura, M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell 2008, 135, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.M.; Buchanan, J.; Luik, R.M. Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 2006, 174, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Barr, V.A.; Bernot, K.M.; Srikanth, S.; Gwack, Y.; Balagopalan, L.; Regan, C.K.; Helman, D.J.; Sommers, C.L.; Oh-Hora, M.; Rao, A.; et al. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: Puncta and distal caps. Mol. Biol. Cell. 2008, 19, 2802–2817. [Google Scholar] [CrossRef]
- Muik, M.; Frischauf, I.; Derler, I.; Fahrner, M.; Bergsmann, J.; Eder, P.; Schindl, R.; Hesch, C.; Polzinger, B.; Fritsch, R.; et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J. Biol. Chem. 2008, 283, 8014–8022. [Google Scholar] [CrossRef] [Green Version]
- Muik, M.; Fahrner, M.; Schindl, R.; Stathopulos, P.; Frischauf, I.; Derler, I.; Plenk, P.; Lackner, B.; Groschner, K.; Ikura, M.; et al. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 2011, 30, 1678–1689. [Google Scholar] [CrossRef]
- Yuan, J.P.; Zeng, W.; Dorwart, M.R.; Choi, Y.J.; Worley, P.F.; Muallem, S. SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nat. Cell Biol. 2009, 11, 337–343. [Google Scholar] [CrossRef]
- Berna-Erro, A.; Braun, A.; Kraft, R.; Kleinschnitz, C.; Schuhmann, M.K.; Stegner, D.; Wultsch, T.; Eilers, J.; Meuth, S.G.; Stoll, G.; et al. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Sci. Signal. 2009, 2, ra67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.T.; Manji, S.S.; Parker, N.J.; Hancock, M.S.; Van Stekelenburg, L.; Eid, J.P.; Senior, P.V.; Kazenwadel, J.S.; Shandala, T.; Saint, R. Identification and characterization of the STIM (stromal interaction molecule) gene family: Coding for a novel class of transmembrane proteins. Biochem. J. 2001, 357 Pt 3, 673–685. [Google Scholar] [CrossRef]
- Rosado, J.A.; Diez, R.; Smani, T.; Jardin, I. STIM and Orai1 variants in store-operated calcium entry. Front. Pharmacol. 2015, 6, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Yen, M.; Sadaghiani, A.M.; Malmersjö, S.; Park, C.Y.; E Dolmetsch, R.; Lewis, R.S. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. J. Cell Biol. 2015, 209, 653–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miederer, A.-M.; AlAnsary, D.; Schwär, G.; Lee, P.-H.; Jung, M.; Helms, V.; Niemeyer, B.A. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat. Commun. 2015, 6, 6899. [Google Scholar] [CrossRef] [Green Version]
- Brandman, O.; Liou, J.; Park, W.S.; Meyer, T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 2007, 13, 1327–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wissenbach, U.; Philipp, S.E.; Gross, S.A.; Cavalie, A.; Flockerzi, V. Primary structure, chromosomal localization and expression in immune cells of the murine ORAI and STIM genes. Cell Calcium 2007, 42, 439–446. [Google Scholar] [CrossRef]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-Hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef] [Green Version]
- Feske, S. ORAI1 and STIM1 deficiency in human and mice: Roles of store-operated Ca2+ entry in the immune system and beyond. Immunol. Rev. 2009, 231, 189–209. [Google Scholar] [CrossRef]
- Vig, M.; I DeHaven, W.; Bird, G.S.; Billingsley, J.M.; Wang, H.; E Rao, P.; Hutchings, A.B.; Jouvin, M.-H.; Putney, J.W.; Kinet, J.-P. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat. Immunol. 2008, 9, 89–96. [Google Scholar] [CrossRef] [Green Version]
- McCarl, C.-A.; Picard, C.; Khalil, S.; Kawasaki, T.; Röther, J.; Papolos, A.; Kutok, J.; Hivroz, C.; LeDeist, F.; Plogmann, K.; et al. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clin. Immunol. 2009, 124, 1311–1318e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziadek, M.A.; Johnstone, L.S. Biochemical properties and cellular localisation of STIM proteins. Cell Calcium 2007, 42, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Grosse, J.; Braun, A.; Varga-Szabo, D.; Beyersdorf, N.; Schneider, B.; Zeitlmann, L.; Hanke, P.; Schropp, P.; Mühlstedt, S.; Zorn, C.; et al. An EF hand mutation in Stim1 causes premature platelet activation and bleeding in mice. J. Clin. Investig. 2007, 117, 3540–3550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Stiber, J.; Hawkins, A.F.; Zhang, Z.-S.; Wang, S.; Burch, J.; Graham, V.; Ward, C.C.; Seth, M.; Finch, E.; Malouf, N.N.; et al. STIM1 signalling controls store-operated calcium entry required for development and contractile function in skeletal muscle. Nat. Cell Biol. 2008, 10, 688–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, P.; Yang, S. The store-operated calcium channels in cancer metastasis: From cell migration, invasion to metastatic colonization. Front. Biosci. 2018, 23, 1241–1256. [Google Scholar]
- Hewavitharana, T.; Deng, X.; Soboloff, J.; Gill, D.L. Role of STIM and Orai proteins in the store-operated calcium signaling pathway. Cell Calcium. 2007, 42, 173–182. [Google Scholar] [CrossRef]
- Nilsson, I.; Lara, P.; Hessa, T.; Johnson, A.E.; von Heijne, G.; Karamyshev, A.L. The code for directing proteins for translocation across ER membrane: SRP cotranslationally recognizes specific features of a signal sequence. J. Mol. Biol. 2015, 427, 1191–1201. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Li, G.Y.; Plevin, M.J.; Ames, J.B.; Ikura, M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J. Biol. Chem. 2006, 281, 35855–35862. [Google Scholar] [CrossRef] [Green Version]
- Novello, M.J.; Zhu, J.; Feng, Q.; Ikura, M.; Stathopulos, P.B. Structural elements of stromal interaction molecule function. Cell Calcium 2018, 73, 88–94. [Google Scholar] [CrossRef]
- Zheng, L.; Stathopulos, P.B.; Schindl, R.; Li, G.Y.; Romanin, C.; Ikura, M. Auto-inhibitory role of the EF-SAM domain of STIM proteins in store-operated calcium entry. Proc. Natl. Acad. Sci. USA 2011, 108, 1337–1342. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Wei, M.; He, L.; Liu, C.; Wu, B.; Zhang, S.L.; Jing, J.; Liang, X.; Senes, A.; Tan, P.; et al. Inside-out Ca2+ signalling prompted by STIM1 conformational switch. Nat. Commun. 2015, 6, 7826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullins, F.M.; Lewis, R.S. The inactivation domain of STIM1 is functionally coupled with the Orai1 pore to enable Ca2+-dependent inactivation. J. Gen. Physiol. 2016, 147, 153–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Fahrner, M.; Muik, M.; Lackner, B.; Schindl, R.; Groschner, K.; Romanin, C. A Ca2+release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J. Biol. Chem. 2009, 284, 24933–24938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, P.G.; Lewis, R.S.; Rao, A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010, 28, 491–533. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Momburg, F.; Engel, U.; Temmerman, K.; Nickel, W.; Seedorf, M. A conserved, lipid-mediated sorting mechanism of yeast Ist2 and mammalian STIM proteins to the peripheral ER. Traffic 2009, 10, 1802–1818. [Google Scholar] [CrossRef]
- Muik, M.; Fahrner, M.; Derler, I.; Schindl, R.; Bergsmann, J.; Frischauf, I.; Groschner, K.; Romanin, C. A Cytosolic homomerization and a modulatory domain within STIM1 C terminus determine coupling to ORAI1 channels. J. Biol. Chem. 2009, 284, 8421–8426. [Google Scholar] [CrossRef] [Green Version]
- Park, C.Y.; Hoover, P.J.; Mullins, F.M.; Bachhawat, P.; Covington, E.D.; Raunser, S.; Walz, T.; Garcia, K.C.; Dolmetsch, R.E.; Lewis, R.S. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai. Cell 2009, 136, 876–890. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Lange, I.; Feske, S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem. Biophys. Res. Commun. 2009, 385, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Yang, X.; Li, S.; Lin, Z.; Wang, Z.; Dong, C.; Shen, Y. The inhibitory helix controls the intramolecular conformational switching of the C-terminus of STIM. PLoS ONE 2013, 8, e74735. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Jin, H.; Cai, X.; Li, S.; Shen, Y. Structural and mechanistic insights into the activation of Stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 5657–5662. [Google Scholar] [CrossRef] [Green Version]
- Stathopulos, P.B.; Schindl, R.; Fahrner, M.; Zheng, L.; Gasmi-Seabrook, G.M.; Muik, M.; Romanin, C.; Ikura, M. STIM1/Orai1 coiled-coil interplay in the regulation of store-operated calcium entry. Nat. Commun. 2013, 4, 2963. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Wang, X.; Loktionova, N.A.; Cai, X.; Nwokonko, R.M.; Vrana, E.; Wang, Y.; Rothberg, B.S.; Gill, D.L. STIM1 dimers undergo unimolecular coupling to activate Orai1 channels. Nat. Commun. 2015, 6, 8395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Cai, X.; Nwokonko, R.M.; Loktionova, N.A.; Wang, Y.; Gill, D.L. The STIM-Orai coupling interface and gating of the Orai1 channel. Cell Calcium 2017, 63, 8–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butorac, C.; Muik, M.; Derler, I.; Stadlbauer, M.; Lunz, V.; Krizova, A.; Lindinger, S.; Schober, R.; Frischauf, I.; Bhardwaj, R.; et al. A novel STIM1-Orai1 gating interface essential for CRAC channel activation. Cell Calcium 2019, 79, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, H.; Zhang, M.; Deng, Y.; Wang, H.; Lu, J.; Xu, T.; Xu, P. An aromatic amino acid in the coiled-coil 1 domain plays a crucial role in the auto-inhibitory mechanism of STIM. Biochem. J. 2013, 454, 401–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.; Sun, L.; Hubrack, S.; Selvaraj, S.; Machaca, K. Intramolecular shielding maintains the ER Ca2+ sensor STIM1 in an inactive conformation. J. Cell Sci. 2013, 126 Pt 11, 2401–2410. [Google Scholar] [CrossRef] [Green Version]
- Korzeniowski, M.K.; Manjarres, I.M.; Varnai, P.; Balla, T. Activation of STIM1-Orai1 involves an intramolecular switching mechanism. Sci. Signal. 2010, 3, ra82. [Google Scholar] [CrossRef] [Green Version]
- Fahrner, M.; Muik, M.; Schindl, R.; Butorac, C.; Stathopulos, P.; Zheng, L.; Jardin, I.; Ikura, M.; Romanin, C. A coiled-coil clamp controls both conformation and clustering of stromal interaction molecule 1 (STIM1). J. Biol. Chem. 2014, 289, 33231–33244. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, G.; Yu, Y.; Chen, X.; Ji, R.; Lu, J.; Li, X.; Zhang, X.; Yang, X.; Shen, Y. Molecular understanding of calcium permeation through the open Orai channel. PLoS Biol. 2019, 17, e3000096. [Google Scholar] [CrossRef]
- Hou, X.; Burstein, S.R.; Long, S. Structures reveal opening of the store-operated calcium channel Orai. bioRxiv 2018. bioRxiv:elife.36758. [Google Scholar]
- Hou, X.; Outhwaite, I.R.; Pedi, L.; Long, S.B. Cryo-EM structure of the calcium release-activated calcium channel Orai in an open conformation. eLife 2020, 9, e62772. [Google Scholar] [CrossRef] [PubMed]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, M.; Pandey, S.K.; Muik, M.; Traxler, L.; Butorac, C.; Stadlbauer, M.; Zayats, V.; Krizova, A.; Plenk, P.; Frischauf, I.; et al. Communication between N terminus and loop2 tunes Orai activation. J. Biol. Chem. 2018, 293, 1271–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.J.; Feske, S. Physiological and pathophysiological functions of SOCE in the immune system. Front. Biosci. 2012, 4, 2253–2268. [Google Scholar] [CrossRef]
- Shaw, P.J.; Qu, B.; Hoth, M.; Feske, S. Molecular regulation of CRAC channels and their role in lymphocyte function. Cell. Mol. Life Sci. 2013, 70, 2637–2656. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Hoebe, K.; Sauer, K. New therapeutic targets in immune disorders: ItpkB, Orai1 and UNC93B. Expert Opin. Ther. Targets 2008, 12, 391–413. [Google Scholar] [CrossRef]
- Gross, S.A.; Wissenbach, U.; Philipp, S.E.; Freichel, M.; Cavalié, A.; Flockerzi, V. Murine ORAI2 Splice Variants Form Functional Ca2+Release-activated Ca2+(CRAC) Channels. J. Biol. Chem. 2007, 282, 19375–19384. [Google Scholar] [CrossRef] [Green Version]
- Hoth, M.; Niemeyer, B.A. The neglected CRAC proteins: Orai2, Orai3, and STIM. Curr. Top Membr. 2013, 71, 237–71. [Google Scholar]
- Motiani, R.K.; A Stolwijk, J.; Newton, R.L.; Zhang, X.; Trebak, M. Emerging roles of Orai3 in pathophysiology. Channels 2013, 7, 392–401. [Google Scholar] [CrossRef] [Green Version]
- McNally, B.A.; Somasundaram, A.; Yamashita, M.; Prakriya, M. Gated regulation of CRAC channel ion selectivity by STIM. Nat. Cell Biol. 2012, 482, 241–245. [Google Scholar] [CrossRef]
- Frischauf, I.; Litviňuková, M.; Schober, R.; Zayats, V.; Svobodová, B.; Bonhenry, D.; Lunz, V.; Cappello, S.; Tociu, L.; Reha, D.; et al. Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription. Sci. Signal. 2017, 10, eaao0358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Yeung, P.S.-W.; Ing, C.E.; McNally, B.A.; Pomès, R.; Prakriya, M. STIM1 activates CRAC channels through rotation of the pore helix to open a hydrophobic gate. Nat. Commun. 2017, 8, 14512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Stanley, C.; Isacoff, E.Y. Critical role for Orai1 C-terminal domain and TM4 in CRAC channel gating. Cell Res. 2015, 25, 963–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Cai, X.; Loktionova, N.A.; Wang, X.; Nwokonko, R.M.; Wang, X.; Wang, Y.; Rothberg, B.S.; Trebak, M.; Gill, D.L. The STIM1-binding site nexus remotely controls Orai1 channel gating. Nat. Commun. 2016, 7, 13725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, J.; Bulla, M.; Urquhart, J.E.; Malfatti, E.; Williams, S.G.; O’Sullivan, J.; Szlauer, A.; Koch, C.; Baranello, G.; Mora, M.; et al. ORAI1 Mutations with distinct channel gating defects in tubular aggregate myopathy. Hum. Mutat. 2017, 38, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Noguchi, S.; Hara, Y.; Hayashi, Y.K.; Motomura, K.; Miyatake, S.; Murakami, N.; Tanaka, S.; Yamashita, S.; Kizu, R.; et al. Dominant mutations in ORAI1 cause tubular aggregate myopathy with hypocalcemia via constitutive activation of store-operated Ca2+ channels. Hum. Mol. Genet. 2015, 24, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Yeung, P.S.-W.; Yamashita, M.; E Ing, C.; Pomès, R.; Freymann, D.M.; Prakriya, M. Mapping the functional anatomy of Orai1 transmembrane domains for CRAC channel gating. Proc. Natl. Acad. Sci. USA 2018, 115, E5193–E5202. [Google Scholar] [CrossRef] [Green Version]
- Nesin, V.; Wiley, G.; Kousi, M.; Ong, E.-C.; Lehmann, T.; Nicholl, D.J.; Suri, M.; Shahrizaila, N.; Katsanis, N.; Gaffney, P.M.; et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc. Natl. Acad. Sci. USA 2014, 111, 4197–4202. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Plenk, P.; Fahrner, M.; Muik, M.; Jardin, I.; Schindl, R.; Gruber, H.J.; Groschner, K.; Romanin, C. The Extended Transmembrane Orai1 N-terminal (ETON) region combines binding interface and gate for Orai1 activation by STIM. J. Biol. Chem. 2013, 288, 29025–29034. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Nwokonko, R.M.; Baraniak, J.H., Jr.; Trebak, M.; Lee, K.P.K.; Gill, D.L. The remote allosteric control of Orai channel gating. PLoS Biol. 2019, 17, e3000413. [Google Scholar] [CrossRef] [Green Version]
- Covington, E.D.; Wu, M.M.; Lewis, R.S. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM. Mol. Biol. Cell 2010, 21, 1897–1907. [Google Scholar] [CrossRef] [Green Version]
- Grigoriev, I.; Gouveia, S.M.; Van Der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.O.; Putney, J.W.; Hoogenraad, C.C.; et al. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 2008, 18, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.M.; Covington, E.D.; Lewis, R.S. Single-molecule analysis of diffusion and trapping of STIM1 and Orai1 at endoplasmic reticulum–plasma membrane junctions. Mol. Biol. Cell 2014, 25, 3672–3685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, A.; Park, S.; Shin, D.M.; Muallem, S. Orai1 and STIM1 in ER/PM junctions: Roles in pancreatic cell function and dysfunction. Am. J. Physiol. Physiol. 2016, 310, C414–C422. [Google Scholar] [CrossRef] [PubMed]
- Subedi, K.P.; Ong, H.L.; Ambudkar, I.S. Assembly of ER-PM junctions: A critical determinant in the regulation of SOCE and TRPC. Atherosclerosis 2017, 981, 253–276. [Google Scholar] [CrossRef]
- Chang, C.-L.; Chen, Y.-J.; Liou, J. ER-plasma membrane junctions: Why and how do we study them? Biochim. Biophys. Acta 2017, 1864, 1494–1506. [Google Scholar] [CrossRef]
- Hogan, P.G. The STIM1-ORAI1 microdomain. Cell Calcium 2015, 58, 357–67. [Google Scholar] [CrossRef] [Green Version]
- Schober, R.; Waldherr, L.; Schmidt, T.; Graziani, A.; Stilianu, C.; Legat, L.; Groschner, K.; Schindl, R. STIM1 and Orai1 regulate Ca2+ microdomains for activation of transcription. Biochim. Biophys. Acta Bioenerg. 2019, 1866, 1079–1091. [Google Scholar] [CrossRef]
- Prakriya, M.; Lewis, R.S. Regulation of CRAC channel activity by recruitment of silent channels to a high open-probability gating mode. J. Gen. Physiol. 2006, 128, 373–386. [Google Scholar] [CrossRef]
- Samanta, K.; Kar, P.; Mirams, G.R.; Parekh, A.B. Ca2+ channel re-localization to plasma-membrane microdomains strengthens activation of Ca2+-dependent nuclear gene expression. Cell Rep. 2015, 12, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Rosato, A.S.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signaling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Kar, P.; Nelson, C.; Parekh, A.B. CRAC Channels drive digital activation and provide analog control and synergy to Ca2+-dependent gene regulation. Curr. Biol. 2012, 22, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Northrop, J.P.; Ho, S.N.; Chen, L.; Thomas, D.J.; Timmerman, L.A.; Nolan, G.P.; Admon, A.; Crabtree, G.R. NF-AT components define a family of transcription factors targeted in T-cell activation. Nat. Cell Biol. 1994, 369, 497–502. [Google Scholar] [CrossRef]
- Srikanth, S.; Gwack, Y. Orai1-NFAT signalling pathway triggered by T cell receptor stimulation. Mol. Cells 2013, 35, 182–194. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.T.; Liu, X.; Ong, H.L.; Swaim, W.; Ambudkar, I.S. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLoS Biol. 2011, 9, e1001025. [Google Scholar] [CrossRef] [Green Version]
- Lemonnier, L.; Prevarskaya, N.; Shuba, Y.; Abeele, F.V.; Nilius, B.; Mazurier, J.; Skryma, R. Ca2+ modulation of volume-regulated anion channels: Evidence for colocalization with store-operated channels. FASEB J. 2001, 16, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ng, S.W.; Di Capite, J.; Singaravelu, K.; Parekh, A.B. Sustained activation of the tyrosine kinase Syk by antigen in mast cells requires local Ca2+Influx through Ca2+Release-activated Ca2+Channels. J. Biol. Chem. 2008, 283, 31348–31355. [Google Scholar] [CrossRef] [Green Version]
- Gwack, Y.; Feske, S.; Srikanth, S.; Hogan, P.G.; Rao, A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007, 42, 145–156. [Google Scholar] [CrossRef]
- Srikanth, S.; Ribalet, B.; Gwack, Y. Regulation of CRAC channels by protein interactions and post-translational modification. Channels 2013, 7, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Vaeth, M.; Kahlfuss, S.; Feske, S. CRAC channels and calcium signaling in T cell-mediated immunity. Trends Immunol. 2020, 41, 878–901. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Zheng, L.; Ikura, M. Stromal Interaction Molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J. Biol. Chem. 2008, 284, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Hirve, N.; Rajanikanth, V.; Hogan, P.G.; Gudlur, A. Coiled-coil formation conveys a STIM1 signal from ER lumen to cytoplasm. Cell Rep. 2018, 22, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Frischauf, I.; Muik, M.; Derler, I.; Bergsmann, J.; Fahrner, M.; Schindl, R.; Groschner, K.; Romanin, C. Molecular determinants of the coupling between STIM1 and Orai channels: Differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 2009, 284, 21696–21706. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, Y.; Zhou, Y.; Hendron, E.; Mancarella, S.; Andrake, M.D.; Rothberg, B.S.; Soboloff, J.; Gill, D.L. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Yen, M.; Lewis, R.S. Physiological CRAC channel activation and pore properties require STIM1 binding to all six Orai1 subunits. J. Gen. Physiol. 2018, 150, 1373–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirado-Lee, L.; Yamashita, M.; Prakriya, M. Conformational changes in the Orai1 C-Terminus evoked by STIM1 binding. PLoS ONE 2015, 10, e0128622. [Google Scholar] [CrossRef] [PubMed]
- Krizova, A.; Maltan, L.; Derler, I. Critical parameters maintaining authentic CRAC channel hallmarks. Eur. Biophys. J. 2019, 48, 425–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I.; Butorac, C.; Krizova, A.; Stadlbauer, M.; Muik, M.; Fahrner, M.; Frischauf, I.; Romanin, C. Authentic CRAC channel activity requires STIM1 and the conserved portion of the Orai N terminus. J. Biol. Chem. 2017, 293, 1259–1270. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Isacoff, E.Y. Cooperative Binding of Stromal Interaction Molecule 1 (STIM1) to the N and C termini of calcium release-activated calcium modulator 1 (Orai1). J. Biol. Chem. 2015, 291, 334–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, Y.; Song, R.; Xu, J.; Yuan, Y.; Liu, J.; Li, J.; Zheng, S.; Liu, T.; Lu, B.; et al. Toward a model for activation of Orai channel. iScience 2019, 16, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, P.S.; Yamashita, M.; Prakriya, M. Molecular basis of allosteric Orai1 channel activation by STIM. J. Physiol. 2020, 598, 1707–1723. [Google Scholar] [CrossRef] [PubMed]
- Yeung, P.S.-W.; Prakriya, M. The exquisitely cooperative nature of Orai1 channel activation. J. Gen. Physiol. 2018, 150, 1352–1355. [Google Scholar] [CrossRef] [PubMed]
- Bulla, M.; Gyimesi, G.; Kim, J.; Bhardwaj, R.; Hediger, M.A.; Frieden, M.; Demaurex, N. ORAI1 channel gating and selectivity is differentially altered by natural mutations in the first or third transmembrane domain. J. Physiol. 2019, 597, 561–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiffner, A.; Schober, R.; Höglinger, C.; Bonhenry, D.; Pandey, S.; Lunz, V.; Sallinger, M.; Frischauf, I.; Fahrner, M.; Lindinger, S.; et al. A series of Orai1 gating checkpoints in transmembrane and cytosolic regions requires clearance for CRAC channel opening. bioRxiv 2020. bioRxiv:207183. [Google Scholar]
- Frischauf, I.; Zayats, V.; Deix, M.; Hochreiter, A.; Jardin, I.; Muik, M.; Lackner, B.; Svobodová, B.; Pammer, T.; Litviňuková, M.; et al. A calcium-accumulating region, CAR, in the channel Orai1 enhances Ca2+ permeation and SOCE-induced gene transcription. Sci. Signal. 2015, 8, ra131. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; Navarro-Borelly, L.; McNally, B.A.; Prakriya, M. Orai1 mutations alter ion permeation and Ca2+-dependent fast inactivation of CRAC channels: Evidence for coupling of permeation and gating. J. Gen. Physiol. 2007, 130, 525–540. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Nwokonko, R.M.; Loktionova, N.A.; Abdulqadir, R.; Baraniak, J.H.; Wang, Y.; Trebak, M.; Zhou, Y.; Gill, D.L. Pore properties of Orai1 calcium channel dimers and their activation by the STIM1 ER calcium sensor. J. Biol. Chem. 2018, 293, 12962–12974. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ramachandran, S.; Oh-Hora, M.; Rao, A.; Hogan, P.G. Pore architecture of the ORAI1 store-operated calcium channel. Proc. Natl. Acad. Sci. USA 2010, 107, 4896–4901. [Google Scholar] [CrossRef] [Green Version]
- McNally, B.A.; Yamashita, M.; Engh, A.; Prakriya, M. Structural determinants of ion permeation in CRAC channels. Proc. Natl. Acad. Sci. USA 2009, 106, 22516–22521. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, M.; E Ing, C.; Yeung, P.S.-W.; Maneshi, M.M.; Pomès, R.; Prakriya, M. The basic residues in the Orai1 channel inner pore promote opening of the outer hydrophobic gate. J. Gen. Physiol. 2020, 152. [Google Scholar] [CrossRef] [PubMed]
- Hoth, M.; Penner, R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nat. Cell Biol. 1992, 355, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Prakriya, M. The molecular physiology of CRAC channels. Immunol. Rev. 2009, 231, 88–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoth, M.; Penner, R. Calcium release-activated calcium current in rat mast cells. J. Physiol. 1993, 465, 359–386. [Google Scholar] [CrossRef]
- Bakowski, D.; Parekh, A.B. Permeation through store-operated CRAC channels in divalent-free solution: Potential problems and implications for putative CRAC channel genes. Cell Calcium 2002, 32, 379–391. [Google Scholar] [CrossRef]
- Lepple-Wienhues, A.; Cahalan, M. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys. J. 1996, 71, 787–794. [Google Scholar] [CrossRef] [Green Version]
- McCleskey, E.W.; Almers, W. The Ca channel in skeletal muscle is a large pore. Proc. Natl. Acad. Sci. USA 1985, 82, 7149–7153. [Google Scholar] [CrossRef] [Green Version]
- Mullins, F.M.; Yen, M.; Lewis, R.S. Correction: Orai1 pore residues control CRAC channel inactivation independently of calmodulin. J. Gen. Physiol. 2016, 147, 289. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J. Gen. Physiol. 2002, 119, 487–507. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Shoemaker, R.L.; Marchase, R.B.; Blalock, J.E. Ca2+ modulation of Ca2+ release-activated Ca2+ channels is responsible for the inactivation of its monovalent cation current. Biophys. J. 2004, 86, 805–814. [Google Scholar] [CrossRef] [Green Version]
- Yeung, P.S.-W.; Yamashita, M.; Prakriya, M. Pore opening mechanism of CRAC channels. Cell Calcium 2017, 63, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derler, I. Gating and permeation of Orai channels. Front. Biosci. 2012, 17, 1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweifach, A.; Lewis, R.S. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci. USA 1993, 90, 6295–6299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malasics, A.; Gillespie, D.; Nonner, W.; Henderson, D.; Eisenberg, B.; Boda, D. Protein structure and ionic selectivity in calcium channels: Selectivity filter size, not shape, matters. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2471–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boda, D.; Valiskó, M.; Eisenberg, B.; Nonner, W.; Henderson, D.; Gillespie, D. Combined effect of pore radius and protein dielectric coefficient on the selectivity of a calcium channel. Phys. Rev. Lett. 2007, 98, 168102. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. CRAC channels: Activation, permeation, and the search for a molecular identity. Cell Calcium 2003, 33, 311–321. [Google Scholar] [CrossRef]
- Zweifach, A.; Lewis, R.S. Slow Calcium-dependent inactivation of depletion-activated calcium current. store-dependent and -independent mechanisms. J. Biol. Chem. 1995, 270, 14445–14451. [Google Scholar] [CrossRef] [Green Version]
- Zweifach, A.; Lewis, R.S. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J. Gen. Physiol. 1995, 105, 209–226. [Google Scholar] [CrossRef] [Green Version]
- Stauderman, K.A. CRAC channels as targets for drug discovery and development. Cell Calcium 2018, 74, 147–159. [Google Scholar] [CrossRef]
- Tian, C.; Du, L.; Zhou, Y.; Li, M. Store-operated CRAC channel inhibitors: Opportunities and challenges. Futur. Med. Chem. 2016, 8, 817–832. [Google Scholar] [CrossRef] [Green Version]
- McNally, B.A.; Prakriya, M. Permeation, selectivity and gating in store-operated CRAC channels. J. Physiol. 2012, 590, 4179–4191. [Google Scholar] [CrossRef] [PubMed]
- Yeromin, A.V.; Zhang, S.L.; Jiang, W.; Yu, Y.; Safrina, O.; Cahalan, M.D. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 2006, 443, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Franzius, D.; Hoth, M.; Penner, R. Non-specific effects of calcium entry antagonists in mast cells. Pflug. Arch. 1994, 428, 433–438. [Google Scholar] [CrossRef]
- Goto, J.-I.; Suzuki, A.Z.; Ozaki, S.; Matsumoto, N.; Nakamura, T.; Ebisui, E.; Fleig, A.; Penner, R.; Mikoshiba, K. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca2+ entry via STIM proteins. Cell Calcium 2010, 47, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.-T.; Venkatachalam, K.; Parys, J.B.; Gill, D.L. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-Aminoethoxydiphenyl borate in DT40 lymphocytes. J. Biol. Chem. 2002, 277, 6915–6922. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lewis, R.S. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J. Physiol. 2001, 536 Pt 1, 3–19. [Google Scholar] [CrossRef]
- Amcheslavsky, A.; Safrina, O.; Cahalan, M.D. Orai3 TM3 point mutation G158C alters kinetics of 2-APB–induced gating by disulfide bridge formation with TM2 C101. J. Gen. Physiol. 2013, 142, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Schindl, R.; Bergsmann, J.; Frischauf, I.; Derler, I.; Fahrner, M.; Muik, M.; Fritsch, R.; Groschner, K.; Romanin, C. 2-Aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J. Biol. Chem. 2008, 283, 20261–20267. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Borelly, L.; Somasundaram, A.; Yamashita, M.; Ren, D.; Miller, R.J.; Prakriya, M. STIM1-Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J. Physiol. 2008, 586, 5383–5401. [Google Scholar] [CrossRef]
- Peinelt, C.; Lis, A.; Beck, A.; Fleig, A.; Penner, R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J. Physiol. 2008, 586, 3061–3073. [Google Scholar] [CrossRef]
- DeHaven, W.I.; Smyth, J.T.; Boyles, R.R.; Bird, G.S.; Putney, J.W., Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 2008, 283, 19265–19273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, R.B.; Rychkov, G.; Barritt, G.J. Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem. J. 2001, 354 Pt 2, 285–290. [Google Scholar] [CrossRef]
- Iwasaki, H.; Mori, Y.; Hara, Y.; Uchida, K.; Zhou, H.; Mikoshiba, K. 2-Aminoethoxydiphenyl borate (2-APB) inhibits capacitative calcium entry independently of the function of inositol 1,4,5-trisphosphate receptors. Recept. Channels 2001, 7, 429–439. [Google Scholar] [PubMed]
- Zhang, X.; Xin, P.; Yoast, R.E.; Emrich, S.M.; Johnson, M.T.; Pathak, T.; Benson, J.C.; Azimi, I.; Gill, D.L.; Monteith, G.R.; et al. Distinct pharmacological profiles of ORAI1, ORAI2, and ORAI3 channels. Cell Calcium 2020, 91, 102281. [Google Scholar] [CrossRef]
- Hendron, E.; Wang, X.; Zhou, Y.; Cai, X.; Goto, J.-I.; Mikoshiba, K.; Baba, Y.; Kurosaki, T.; Wang, Y.; Gill, D.L. Potent functional uncoupling between STIM1 and Orai1 by dimeric 2-aminodiphenyl borinate analogs. Cell Calcium 2014, 56, 482–492. [Google Scholar] [CrossRef] [Green Version]
- Parvez, S.; Beck, A.; Peinelt, C.; Soboloff, J.; Lis, A.; Monteilh-Zoller, M.; Gill, D.L.; Fleig, A.; Penner, R. STIM2 protein mediates distinct store-dependent and store-independent modes of CRAC channel activation. FASEB J. 2008, 22, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Zitt, C.; Strauss, B.; Schwarz, E.C.; Spaeth, N.; Rast, G.; Hatzelmann, A.; Hoth, M. Potent inhibition of Ca2+Release-activated Ca2+Channels and T-lymphocyte activation by the pyrazole derivative BTP. J. Biol. Chem. 2004, 279, 12427–12437. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, J.; Ohga, K.; Yoshino, T.; Takezawa, R.; Ichikawa, A.; Kubota, H.; Yamada, T. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J. Immunol. 2003, 170, 4441–4449. [Google Scholar] [CrossRef] [Green Version]
- Schleifer, H.; Doleschal, B.; Lichtenegger, M.; Oppenrieder, R.; Derler, I.; Frischauf, I.; Glasnov, T.N.; O Kappe, C.; Romanin, C.; Groschner, K. Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol. 2012, 167, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- Takezawa, R.; Cheng, H.; Beck, A.; Ishikawa, J.; Launay, P.; Kubota, H.; Kinet, J.-P.; Fleig, A.; Yamada, T.; Penner, R. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol. Pharmacol. 2006, 69, 1413–1420. [Google Scholar] [CrossRef]
- He, L.-P.; Hewavitharana, T.; Soboloff, J.; Spassova, M.A.; Gill, D.L. A functional link between store-operated and TRPC channels revealed by the 3,5-Bis(trifluoromethyl)pyrazole DERIVATIVE, BTP. J. Biol. Chem. 2005, 280, 10997–11006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashmole, I.; Duffy, S.M.; Leyland, M.L.; Morrison, V.S.; Begg, M.; Bradding, P. CRACM/Orai ion channel expression and function in human lung mast cells. J. Allergy Clin. Immunol. 2012, 129, 1628–1635.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, L.V.; Bax, H.J.; Russell, L.J.; Barrett, V.J.; Walton, S.E.; Deakin, A.M.; Thomson, S.A.; Lucas, F.; Solari, R.; House, D.; et al. Characterization of selective calcium-release activated calcium channel blockers in mast cells and T-cells from human, rat, mouse and guinea-pig preparations. Eur. J. Pharmacol. 2013, 704, 49–57. [Google Scholar] [CrossRef]
- Derler, I.; Schindl, R.; Fritsch, R.; Heftberger, P.; Riedl, M.C.; Begg, M.; House, D.; Romanin, C. The action of selective CRAC channel blockers is affected by the Orai pore geometry. Cell Calcium 2013, 53, 139–151. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Rovedatti, L.; Kaur, R.; Spencer, J.P.; Brown, J.T.; Morisset, V.D.; Biancheri, P.; Leakey, N.A.B.; Wilde, J.I.; Scott, L.; et al. Targeting gut T cell Ca2+ Release-activated Ca2+ channels inhibits T cell cytokine production and T-Box transcription factor T-Bet in inflammatory bowel disease. J. Immunol. 2009, 183, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Waldherr, L.; Tiffner, A.; Mishra, D.; Sallinger, M.; Schober, R.; Frischauf, I.; Schmidt, T.; Handl, V.; Sagmeister, P.; Köckinger, M.; et al. Blockage of store-operated Ca2+ influx by Synta66 is mediated by direct inhibition of the Ca2+ selective Orai1 pore. Cancers 2020, 12, 2876. [Google Scholar] [CrossRef]
- Smyth, J.T.; DeHaven, W.I.; Bird, G.S.; Putney, J.W., Jr. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J. Cell Sci. 2008, 121, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Ohana, L.; Newell, E.W.; Stanley, E.F.; Schlichter, L.C. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels 2009, 3, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Zakharov, S.I.; Smani, T.; Dobrydneva, Y.; Monje, F.; Fichandler, C.; Blackmore, P.F.; Bolotina, V.M. Diethylstilbestrol is a potent inhibitor of store-operated channels and capacitative Ca2+ influx. Mol. Pharmacol. 2004, 66, 702–707. [Google Scholar]
- Holowka, N.B.; Korzeniowski, M.K.; Bryant, K.L.; Baird, B. Polyunsaturated fatty acids inhibit stimulated coupling between the ER Ca2+ sensor STIM1 and the Ca2+ channel protein Orai1 in a process that correlates with inhibition of stimulated STIM1 oligomerization. Biochim. Biophys. Acta 2014, 1841, 1210–1216. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Panicker, S.; Lau, K.-Y.; Apparsundaram, S.; Patel, V.A.; Chen, S.-L.; Soto, R.; Jung, J.K.; Ravindran, P.; Okuhara, D.; et al. Characterization of a novel CRAC inhibitor that potently blocks human T cell activation and effector functions. Mol. Immunol. 2013, 54, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Bruen, C.; Schnaus, M.; Zhang, J.; Ali, S.; Lind, A.; Stoecker, Z.; Stauderman, K.; Hebbar, S. Auxora versus standard of care for the treatment of severe or critical COVID-19 pneumonia: Results from a randomized controlled trial. Crit. Care 2020, 24, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Derler, I.; Fritsch, R.; Schindl, R.; Romanin, C. CRAC inhibitors: Identification and potential. Expert Opin. Drug Discov. 2008, 3, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Djuric, S.W.; Bamaung, N.Y.; Basha, A.; Liu, H.; Luly, J.R.; Madar, D.J.; Sciotti, R.J.; Tu, N.P.; Wagenaar, F.L.; Wiedeman, P.E.; et al. 3,5-Bis(trifluoromethyl)pyrazoles: A novel class of NFAT transcription factor regulator. J. Med. Chem. 2000, 43, 2975–2981. [Google Scholar] [CrossRef]
- Merritt, J.E.; Armstrong, W.P.; Benham, C.D.; Hallam, T.J.; Jacob, R.; Jaxa-Chamiec, A.; Leigh, B.K.; McCarthy, S.A.; Moores, K.E.; Rink, T.J. SK&F 96365, a novel inhibitor of receptor-mediated calcium entry. Biochem. J. 1990, 271, 515–522. [Google Scholar]
- Van Kruchten, R.; Braun, A.P.; Feijge, M.A.H.; Kuijpers, M.J.E.; Rivera-Galdos, R.; Kraft, P.; Stoll, G.; Kleinschnitz, C.; Bevers, E.M.; Nieswandt, B.; et al. Antithrombotic potential of blockers of store-operated calcium channels in platelets. Arter. Thromb. Vasc. Biol. 2012, 32, 1717–1723. [Google Scholar] [CrossRef] [Green Version]
- Morin, G.; Bruechle, N.O.; Singh, A.R.; Knopp, C.; Jedraszak, G.; Elbracht, M.; Brémond-Gignac, D.; Hartmann, K.; Sevestre, H.; Deutz, P.; et al. Gain-of-function mutation in STIM1 (P.R304W) is associated with Stormorken syndrome. Hum. Mutat. 2014, 35, 1221–1232. [Google Scholar] [CrossRef]
- Boehm, J.; Chevessier, F.; De Paula, A.M.; Koch, C.; Attarian, S.; Feger, C.; Hantaï, D.; Laforêt, P.; Ghorab, K.; Vallat, J.-M.; et al. Constitutive activation of the calcium sensor STIM1 causes tubular-aggregate myopathy. Am. J. Hum. Genet. 2013, 92, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Böhm, J.; Chevessier, F.; Koch, C.; Peche, G.A.; Mora, M.; Morandi, L.; Pasanisi, B.; Moroni, I.; Tasca, G.; Fattori, F.; et al. Clinical, histological and genetic characterisation of patients with tubular aggregate myopathy caused by mutations in STIMJ. Med. Genet. 2014, 51, 824–833. [Google Scholar] [CrossRef]
- Hedberg, C.; Niceta, M.; Fattori, F.; Lindvall, B.; Ciolfi, A.; D’Amico, A.; Tasca, G.; Petrini, S.; Tulinius, M.; Tartaglia, M.; et al. Childhood onset tubular aggregate myopathy associated with de novo STIM1 mutations. J. Neurol. 2014, 261, 870–876. [Google Scholar] [CrossRef]
- Morin, G.; Biancalana, V.; Echaniz-Laguna, A.; Noury, J.; Lornage, X.; Moggio, M.; Ripolone, M.; Violano, R.; Marcorelles, P.; Maréchal, D.; et al. Tubular aggregate myopathy and Stormorken syndrome: Mutation spectrum and genotype/phenotype correlation. Hum. Mutat. 2020, 41, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Misceo, D.; Holmgren, A.; Louch, W.E.; Holme, P.A.; Mizobuchi, M.; Morales, R.J.; De Paula, A.M.; Stray-Pedersen, A.; Lyle, R.; Dalhus, B.; et al. A Dominant STIM1 mutation causes Stormorken syndrome. Hum. Mutat. 2014, 35, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Stadlbauer, M.; Muik, M.; Rathner, P.; Stathopulos, P.; Ikura, M.; Müller, N.; Romanin, C. A dual mechanism promotes switching of the Stormorken STIM1 R304W mutant into the activated state. Nat. Commun. 2018, 9, 825. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.C.; Rossius, M.; Zitzelsberger, M.; Vorgerd, M.; Müller-Felber, W.; Ertl-Wagner, B.; Zhang, Y.; Brinkmeier, H.; Senderek, J.; Schoser, B. 50 years to diagnosis: Autosomal dominant tubular aggregate myopathy caused by a novel STIM1 mutation. Neuromuscul. Disord. 2015, 25, 577–584. [Google Scholar] [CrossRef]
- Fuchs, S.; Rensing-Ehl, A.; Speckmann, C.; Bengsch, B.; Schmitt-Graeff, A.; Bondzio, I.; Maul-Pavicic, A.; Bass, T.; Vraetz, T.; Strahm, B.; et al. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J. Immunol. 2012, 188, 1523–1533. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Choi, M.; Richardson, A.; Reid, B.; Seymen, F.; Yildirim, M.; Tuna, E.; Gençay, K.; Simmer, J.; Hu, J. STIM1 and SLC24A4 are critical for enamel maturation. J. Dent. Res. 2014, 93, 94S–100S. [Google Scholar] [CrossRef] [Green Version]
- Picard, C.; McCarl, C.A.; Papolos, A.; Khalil, S.; Lüthy, K.; Hivroz, C.; LeDeist, F.; Rieux-Laucat, F.; Rechavi, G.; Rao, A.; et al. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009, 360, 1971–1980. [Google Scholar] [CrossRef] [Green Version]
- Garibaldi, M.; Fattori, F.; Riva, B.; Labasse, C.; Brochier, G.; Ottaviani, P.; Sacconi, S.; Vizzaccaro, E.; Laschena, F.; Romero, N.; et al. A novel gain-of-function mutation inORAI1causes late-onset tubular aggregate myopathy and congenital miosis. Clin. Genet. 2017, 91, 780–786. [Google Scholar] [CrossRef]
- Lian, J.; Cuk, M.; Kahlfuss, S.; Kozhaya, L.; Vaeth, M.; Rieux-Laucat, F.; Picard, C.; Benson, M.J.; Jakovcevic, A.; Bilic, K.; et al. ORAI1 mutations abolishing store-operated Ca2+ entry cause anhidrotic ectodermal dysplasia with immunodeficiency. J. Allergy Clin. Immunol. 2018, 142, 1297–1310.e11. [Google Scholar] [CrossRef]
- Schober, R.; Bonhenry, D.; Lunz, V.; Zhu, J.; Tiffner, A.; Frischauf, I.; Fahrner, M.; Zhang, M.; Waldherr, L.; Schmidt, T.; et al. Sequential activation of STIM1 links Ca2+ with luminal domain unfolding. Sci. Signal. 2019, 12, eaax3194. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lkhagvadorj, S.; Lee, M.-R.; Hwang, K.-H.; Chung, H.C.; Jung, J.H.; Cha, S.-K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Z.-Y.; Zhong, L.-X.; Feng, M.; Wang, J.-F.; Liu, D.-B.; Xiong, J.-P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar] [PubMed]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantonero, C.; Sanchez-Collado, J.; Nuñez, M.A.G.; Salido, G.; Lopez, J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca2+ entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Abeele, F.V.; Lehen’Kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Haglund, F.; Ma, R.; Huss, M.; Sulaiman, L.; Lu, M.; Nilsson, I.-L.; Höög, A.; Juhlin, C.C.; Hartman, J.; Larsson, C. Evidence of a functional estrogen receptor in parathyroid adenomas. J. Clin. Endocrinol. Metab. 2012, 97, 4631–4639. [Google Scholar] [CrossRef] [Green Version]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell. Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Holzmann, C.; Kilch, T.; Kappel, S.; Armbrüster, A.; Jung, V.; Stöckle, M.; Bogeski, I.; Schwarz, E.C.; Peinelt, C. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget 2013, 4, 2096–2107. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, X.; Feng, B.; Liu, N.; Wu, Q.; Han, Y.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene 2015, 34, 4808–4820. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-Y.; Sun, J.; Huang, M.-Y.; Wang, Y.-S.; Hou, M.-F.; Sun, Y.; He, H.; Krishna, N.; Chiu, S.-J.; Lin, S.; et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene 2015, 34, 4358–4367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W.-J. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-T.; Chiu, W.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-F.; Liu, Y.-L.; Chang, H.-C.; Chen, Y.-T.; Chiu, W.; Liu, K.-Y.; Chang, J.-Y.; Shen, M.-R. Microtubule-associated histone deacetylase 6 supports the calcium store sensor STIM1 in mediating malignant cell behaviors. Cancer Res. 2013, 73, 4500–4509. [Google Scholar] [CrossRef] [Green Version]
- Ruano, Y.; Mollejo, M.; Ribalta, T.; Fiaño, C.; I Camacho, F.; Gomez, E.; De Lope, A.R.; Hernández-Moneo, J.-L.; Martinez, P.; Meléndez, B. Identification of novel candidate target genes in amplicons of Glioblastoma multiforme tumors detected by expression and CGH microarray profiling. Mol. Cancer 2006, 5, 39. [Google Scholar] [CrossRef] [Green Version]
- Aytes, A.; Molleví, D.G.; Martínez-Iniesta, M.; Nadal, M.; Vidal, A.; Morales, A.; Salazar, R.; Capellá, G.; Villanueva, A. Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype. Mol. Carcinog. 2011, 51, 746–753. [Google Scholar] [CrossRef]
- Ashida, S.; Orloff, M.S.; Bebek, G.; Zhang, L.; Zheng, P.; Peehl, D.M.; Eng, C. Integrated analysis reveals critical genomic regions in prostate tumor microenvironment associated with clinicopathologic phenotypes. Clin. Cancer Res. 2012, 18, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Waroux, O.; Massotte, L.; Alleva, L.; Graulich, A.; Thomas, E.; Liégeois, J.F.; Scuvée-Moreau, J.; Seutin, V. SK channels control the firing pattern of midbrain dopaminergic neurons in vivo. Eur. J. Neurosci. 2005, 22, 3111–3121. [Google Scholar] [CrossRef]
- Chen, M.X.; Gorman, S.A.; Benson, B.; Singh, K.; Hieble, J.P.; Michel, M.C.; Tate, S.N.; Trezise, D.J. Small and intermediate conductance Ca 2+ -activated K + channels confer distinctive patterns of distribution in human tissues and differential cellular localisation in the colon and corpus cavernosum. Naunyn Schmiedeberg’s Arch. Pharmacol. 2004, 369, 602–615. [Google Scholar] [CrossRef]
- Ji, H.; Shepard, P.D. SK Ca2+-activated K+ channel ligands alter the firing pattern of dopamine-containing neurons in vivo. Neuroscience 2006, 140, 623–633. [Google Scholar] [CrossRef]
- Gueguinou, M.; Crottès, D.; Chantôme, A.; Rapetti-Mauss, R.; Potier-Cartereau, M.; Clarysse, L.; Girault, A.; Fourbon, Y.; Jézéquel, P.; Guérin-Charbonnel, C.; et al. The SigmaR1 chaperone drives breast and colorectal cancer cell migration by tuning SK3-dependent Ca2+ homeostasis. Oncogene 2017, 36, 3640–3647. [Google Scholar] [CrossRef] [PubMed]
- Guéguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantôme, A.; Haelters, J.P.; Jaffrès, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohr, C.J.; Steudel, F.A.; Gross, D.; Ruth, P.; Lo, W.-Y.; Hoppe, R.; Schroth, W.; Brauch, H.; Huber, S.M.; Lukowski, R. Cancer-associated intermediate conductance Ca2+-Activated K+ channel KCa3. Cancers 2019, 11, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Yang, X.; Yin, Q.; Yi, J.; Shen, W.; Zhao, L.; Zhu, Z.; Liu, J. Inhibition of SK4 potassium channels suppresses cell proliferation, migration and the epithelial-mesenchymal transition in triple-negative breast cancer cells. PLoS ONE 2016, 11, e0154471. [Google Scholar] [CrossRef] [PubMed]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological roles and therapeutic potential of Ca2+ activated potassium channels in the nervous system. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Liu, X.; Chang, Y.; Reinhart, P.H.; Sontheimer, H. Cloning and characterization of glioma BK, a novel BK channel isoform highly expressed in human glioma cells. J. Neurosci. 2002, 22, 1840–1849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basrai, D.; Kraft, R.; Bollensdorff, C.; Liebmann, L.; Benndorf, K.; Patt, S. BK channel blockers inhibit potassium-induced proliferation of human astrocytoma cells. NeuroReport 2002, 13, 403–407. [Google Scholar] [CrossRef]
- Ransom, C.B.; Sontheimer, H. BK Channels in human glioma cells. J. Neurophysiol. 2001, 85, 790–803. [Google Scholar] [CrossRef] [Green Version]
- NCI’s Genomic Data Commons (GDC). PanCanAtlas Publications: TCGA PanCancerAtlas; NCI’s Genomic Data Commons: Bethesda, MD, USA. Available online: https://portal.gdc.cancer.gov/ (accessed on 14 December 2020).
- Jardin, I.; Rosado, J.A. STIM and calcium channel complexes in cancer. Biochim. Biophys. Acta 2016, 1863, 1418–1426. [Google Scholar] [CrossRef]
- Pla, A.F.; Kondratska, K.; Prevarskaya, N. STIM and ORAI proteins: Crucial roles in hallmarks of cancer. Am. J. Physiol. Physiol. 2016, 310, C509–C519. [Google Scholar] [CrossRef]
- Hoth, M. CRAC channels, calcium, and cancer in light of the driver and passenger concept. Biochim. Biophys. Acta 2016, 1863, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Moccia, F.; Zuccolo, E.; Poletto, V.; Turin, I.; Guerra, G.; Pedrazzoli, P.; Rosti, V.; Porta, C.; Montagna, D. Targeting stim and Orai proteins as an alternative approach in anticancer therapy. Curr. Med. Chem. 2016, 23, 1. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, J.J.; Huang, X.-Y. Orai1 and STIM1 Are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef] [Green Version]
- Kappel, S.; Marques, I.J.; Zoni, E.; Stokłosa, P.; Peinelt, C.; Mercader, N.; Julio, M.K.-D.; Borgström, A. Store-operated Ca2+ entry as a prostate cancer biomarker—A riddle with perspectives. Curr. Mol. Biol. Rep. 2017, 3, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobradillo, D.; Hernández-Morales, M.; Ubierna, D.; Moyer, M.P.; Núñez, L.; Villalobos, C. A Reciprocal Shift in Transient Receptor Potential Channel 1 (TRPC1) and Stromal Interaction Molecule 2 (STIM2) contributes to Ca2+Remodeling and cancer hallmarks in colorectal carcinoma cells. J. Biol. Chem. 2014, 289, 28765–28782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccolo, E.; Laforenza, U.; Ferulli, F.; Pellavio, G.; Scarpellino, G.; Tanzi, M.; Turin, I.; Faris, P.; Lucariello, A.; Maestri, M.; et al. Stim and Orai mediate constitutive Ca2+ entry and control endoplasmic reticulum Ca2+ refilling in primary cultures of colorectal carcinoma cells. Oncotarget 2018, 9, 31098–31119. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.-H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Zhang, Z.; Wang, R.; Ma, W.; Yang, Y.; Wei, J.; Wei, Y. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J. Exp. Clin. Cancer Res. 2013, 32, 20. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Chen, L.; Zhao, P.; Zhou, H.; Zhang, C.; Yu, S.; Lin, Y.; Yang, X. Store-operated Ca2+ entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J. Exp. Clin. Cancer Res. 2014, 33, 98. [Google Scholar] [CrossRef] [Green Version]
- Umemura, M.; Baljinnyam, E.; Feske, S.; De Lorenzo, M.S.; Xie, L.-H.; Feng, X.; Oda, K.; Makino, A.; Fujita, T.; Yokoyama, U.; et al. Store-operated Ca2+ Entry (SOCE) regulates melanoma proliferation and cell migration. PLoS ONE 2014, 9, e89292. [Google Scholar] [CrossRef] [PubMed]
- Ay, A.S.; Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 constitutes a native store-operated calcium entry that regulates non-small cell lung adenocarcinoma cell proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, M.; Xu, L.; Lin, D.; Cai, S.; Zou, F. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM. Exp. Toxicol. Pathol. 2013, 65, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Trombetta-Lima, M.; Krabbendam, I.E.; Dolga, A.M. Calcium-activated potassium channels: Implications for aging and age-related neurodegeneration. Int. J. Biochem. Cell Biol. 2020, 123, 105748. [Google Scholar] [CrossRef] [PubMed]
- Lallet-Daher, H.; Roudbaraki, M.; Bavencoffe, A.; Mariot, P.; Gackiere, F.; Bidaux, G.; Urbain, R.; Gosset, P.; Delcourt, P.; Fleurisse, L.; et al. Intermediate-conductance Ca2+-activated K+ channels (IKCa1) regulate human prostate cancer cell proliferation through a close control of calcium entry. Oncogene 2009, 28, 1792–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chantome, A.; Girault, A.; Potier-Cartereau, M.; Collin, C.; Vaudin, P.; Pagès, J.-C.; Vandier, C.; Joulin, V. KCa2.3 channel-dependent hyperpolarization increases melanoma cell motility. Exp. Cell Res. 2009, 315, 3620–3630. [Google Scholar] [CrossRef]
- Potier, M.-C.; Tran, T.A.; Chantome, A.; Girault, A.; Joulin, V.; Bougnoux, P.; Vandier, C.; Pierre, F. Altered SK3/KCa2.3-mediated migration in adenomatous polyposis coli (Apc) mutated mouse colon epithelial cells. Biochem. Biophys. Res. Commun. 2010, 397, 42–47. [Google Scholar] [CrossRef]
- Gackière, F.; Warnier, M.; Katsogiannou, M.; Derouiche, S.; Delcourt, P.; Dewailly, E.; Slomianny, C.; Humez, S.; Prevarskaya, N.; Roudbaraki, M.; et al. Functional coupling between large-conductance potassium channels and Cav3.2 voltage-dependent calcium channels participates in prostate cancer cell growth. Biol. Open 2013, 2, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-Activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef] [Green Version]
- Berkefeld, H.; Sailer, C.A.; Bildl, W.; Rohde, V.; Thumfart, J.-O.; Eble, S.; Klugbauer, N.; Reisinger, E.; Bischofberger, J.; Oliver, D.; et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-Activated K+ signaling. Science 2006, 314, 615–620. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Ngo-Anh, T.J.; Bruening-Wright, A.; Maylie, J.; Adelman, J.P. Small conductance Ca2+-activated K+ channels and calmodulin—Cell surface expression and gating. J. Biol. Chem. 2003, 278, 25940–25946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.K.; Bomben, V.C.; Sontheimer, H. Expression and function of calcium-activated potassium channels in human glioma cells. Glia 2006, 54, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wulff, H.; MacKinnon, R. Activation mechanism of a human SK-calmodulin channel complex elucidated by cryo-EM structures. Science 2018, 360, 508–513. [Google Scholar] [CrossRef]
- Guéguinou, M.; Chantôme, A.; Fromont, G.; Bougnoux, P.; Vandier, C.; Potier-Cartereau, M. KCa and Ca2+ channels: The complex thought. Biochim. Biophys. Acta 2014, 1843, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Maylie, J.; Bond, C.T.; Herson, P.S.; Lee, C.W.-S.; Adelman, J.P. Small conductance Ca2+-activated K+Channels and calmodulin. J. Physiol. 2004, 554, 255–261. [Google Scholar] [CrossRef]
- Xia, X.-M.; Fakler, B.; Rivard, A.L.; A Wayman, G.; Johnsonpais, T.L.; E Keen, J.; Ishii, T.; Hirschberg, B.; Bond, C.T.; Lutsenko, S.V.; et al. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nat. Cell Biol. 1998, 395, 503–507. [Google Scholar] [CrossRef]
- A Schumacher, M.; Rivard, A.F.; Bächinger, H.P.; Adelman, J.P. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nat. Cell Biol. 2001, 410, 1120–1124. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Crum, M.; Miller, M.C. Crystal structures of apocalmodulin and an apocalmodulin/SK potassium channel gating domain complex. Structure 2004, 12, 849–860. [Google Scholar] [CrossRef]
- Tidow, H.; Nissen, P. Structural diversity of calmodulin binding to its target sites. FEBS J. 2013, 280, 5551–5565. [Google Scholar] [CrossRef]
- Adelman, J.P. SK channels and calmodulin. Channels 2015, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Messier, C.; Mourre, C.; Bontempi, B.; Sif, J.; Lazdunski, M.; Destrade, C. Effect of apamin, a toxin that inhibits Ca2+-dependent K+ channels, on learning and memory processes. Brain Res. 1991, 551, 322–326. [Google Scholar] [CrossRef]
- Grunnet, M.; Jespersen, T.; Angelo, K.; Frøkjaer-Jensen, C.; A Klaerke, D.; Olesen, S.P.; Jensen, B.S. Pharmacological modulation of SK3 channels. Neuropharmacology 2001, 40, 879–887. [Google Scholar] [CrossRef]
- Hougaard, C.; Eriksen, B.L.; Jørgensen, S.; Johansen, T.H.; Dyhring, T.; Madsen, L.S.; Strøbaek, D.; Christophersen, P. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br. J. Pharmacol. 2007, 151, 655–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skibsbye, L.; Poulet, C.; Diness, J.G.; Bentzen, B.H.; Yuan, L.; Kappert, U.; Matschke, K.; Wettwer, E.; Ravens, U.; Grunnet, M.; et al. Small-conductance calcium-activated potassium (SK) channels contribute to action potential repolarization in human atria. Cardiovasc. Res. 2014, 103, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strøbæk, R.; Teuber, L.; Jørgensen, T.D.; Ahring, P.K.; Kjær, K.; Hansen, R.S.; Olesen, M.S.; Christophersen, P.; Skaaning-Jensen, B.; Ahring, P.K. Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 2004, 1665, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Potier-Cartereau, M.; Chantome, A.; Joulin, V.; Girault, A.; Roger, S.; Besson, P.; Jourdan, M.-L.; LeGuennec, J.-Y.; Bougnoux, P.; Vandier, C. The SK3/KCa2.3 potassium channel is a new cellular target for edelfosine. Br. J. Pharmacol. 2010, 162, 464–479. [Google Scholar] [CrossRef] [Green Version]
- Christophersen, P.; Wulff, H. Pharmacological gating modulation of small- and intermediate-conductance Ca2+-activated K+channels (KCa2.x and KCa3.1). Channels 2015, 9, 336–343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Pascal, J.M.; Schumann, M.; Armen, R.S.; Zhang, J.-F. Identification of the functional binding pocket for compounds targeting small-conductance Ca2+-activated potassium channels. Nat. Commun. 2012, 3, 1021. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of small- and intermediate-conductance calcium-activated potassium channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 219–240. [Google Scholar] [CrossRef] [Green Version]
- Kouba, S.; Braire, J.; Félix, R.; Chantôme, A.; Jaffrès, P.-A.; Lebreton, J.; Dubreuil, D.; Pipelier, M.; Zhang, X.; Trebak, M.; et al. Lipidic synthetic alkaloids as SK3 channel modulators. Synthesis and biological evaluation of 2-substituted tetrahydropyridine derivatives with potential anti-metastatic activity. Eur. J. Med. Chem. 2020, 186, 111854. [Google Scholar] [CrossRef]
- Herrera, F.E.; Sevrain, C.M.; Jaffrès, P.-A.; Couthon, H.; Grélard, A.; Dufourc, E.J.; Chantôme, A.; Potier-Cartereau, M.; Vandier, C.; Bouchet, A.M. Singular interaction between an antimetastatic agent and the lipid bilayer: The Ohmline case. ACS Omega 2017, 2, 6361–6370. [Google Scholar] [CrossRef]
- Adeagbo, A.S. 1-Ethyl-2-benzimidazolinone stimulates endothelial K(Ca) channels and nitric oxide formation in rat mesenteric vessels. Eur. J. Pharmacol. 1999, 379, 151–159. [Google Scholar] [CrossRef]
- Dimitriadi, M.; Kye, M.J.; Kalloo, G.; Yersak, J.M.; Sahin, M.; Hart, A.C. The neuroprotective drug riluzole acts via small conductance Ca2+-activated K+ channels to ameliorate defects in spinal muscular atrophy models. J. Neurosci. 2013, 33, 6557–6562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarra, D.V.; Cornwell, T.; Solodushko, V.; Brown, A.; Taylor, M.S. CyPPA, a positive modulator of small-conductance Ca2+-activated K+ channels, inhibits phasic uterine contractions and delays preterm birth in mice. Am. J. Physiol. Physiol. 2011, 301, C1027–C1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, N.; Brown, B.M.; Oliván-Viguera, A.; Singh, V.; Olmstead, M.M.; Valero, M.S.; Köhler, R.; Wulff, H. New positive Ca2+-activated K+ channel gating modulators with selectivity for KCa3. Mol. Pharmacol. 2014, 86, 342–57. [Google Scholar] [CrossRef] [Green Version]
- Strøbaek, D.; Hougaard, C.; Johansen, T.H.; Sørensen, U.S.; Nielsen, E.Ø.; Nielsen, K.S.; Taylor, R.D.T.; Pedarzani, P.; Christophersen, P. Inhibitory gating modulation of small conductance Ca2+-Activated K+ Channels by the synthetic compound (R)-N-(Benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces after hyperpolarizing current in hippocampal CA1 neurons. Mol. Pharmacol. 2006, 70, 1771–1782. [Google Scholar] [CrossRef] [Green Version]
- Burnham, M.P.; Bychkov, R.; Félétou, M.; Richards, G.R.; Vanhoutte, P.M.; Weston, A.H.; Edwards, G. Characterization of an apamin-sensitive small-conductance Ca2+-activated K+ channel in porcine coronary artery endothelium: Relevance to EDHF. Br. J. Pharmacol. 2002, 135, 1133–1143. [Google Scholar] [CrossRef] [Green Version]
- Wulff, H.; Miller, M.J.; Hänsel, W.; Grissmer, S.; Cahalan, M.D.; Chandy, K.G. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: A potential immunosuppressant. Proc. Natl. Acad. Sci. USA 2000, 97, 8151–8156. [Google Scholar] [CrossRef] [Green Version]
- Oliván-Viguera, A.; Valero, M.S.; Murillo, M.D.; Wulff, H.; García-Otín, Á.-L.; Arbonés-Mainar, J.-M.; Köhler, R.N. Novel phenolic inhibitors of small/intermediate-conductance Ca2+-Activated K+ Channels, KCa3.1 and KCa2. PLoS ONE 2013, 8, e58614. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.J.; Rauer, H.; Tomita, H.; Gargus, J.J.; Gutman, G.A.; Cahalan, M.D.; Chandy, K.G. Nuclear localization and dominant-negative suppression by a mutant SKCa3 N-terminal channel fragment identified in a patient with schizophrenia. J. Biol. Chem. 2001, 276, 27753–27756. [Google Scholar] [CrossRef] [Green Version]
- Bowen, T.; Williams, N.; Norton, N.; Spurlock, G.; Wittekindt, O.H.; Dj, M.-R.; Brzustowicz, L.; Hoogendoorn, B.; Zammit, S.; Jones, G.; et al. Mutation screening of the KCNN3 gene reveals a rare frameshift mutation. Mol. Psychiatry 2001, 6, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, M.S.; Skinner, F.; Arshadmansab, M.F.; Garcia, I.; Mello, C.F.; Knaus, H.-G.; Ermolinsky, B.S.; Otalora, L.F.P.; Garrido-Sanabria, E.R. Altered expression and function of small-conductance (SK) Ca2+-activated K+ channels in pilocarpine-treated epileptic rats. Brain Res. 2010, 1348, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Dolga, A.M.; De Andrade, A.; Meissner, L.; Knaus, H.-G.; Hollerhage, M.; Christophersen, P.; Zischka, H.; Plesnila, N.; Hoglinger, G.U.; Culmsee, C. Subcellular expression and neuroprotective effects of SK channels in human dopaminergic neurons. Cell Death Dis. 2014, 5, e999. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Fischer, D.; Noelker, C.; Vulinović, F.; Grünewald, A.; Chevarin, C.; Klein, C.; Oertel, W.H.; Hirsch, E.C.; Michel, P.P.; Hartmann, A. Bee venom and its component apamin as neuroprotective agents in a parkinson disease mouse model. PLoS ONE 2013, 8, e61700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.I.; Yang, E.J.; Lee, M.S.; Kim, Y.S.; Huh, Y.; Cho, I.H.; Kang, S.; Koh, H.K. Bee venom reduces neuroinflammation in the MPTP-induced model of Parkinson’s disease. Int. J. Neurosci. 2011, 121, 209–217. [Google Scholar] [CrossRef]
- Doo, A.-R.; Kim, S.-T.; Moon, W.; Yin, C.S.; Chae, Y.; Park, H.-K.; Lee, H.; Park, H.-J. Neuroprotective effects of bee venom pharmaceutical acupuncture in acute 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced mouse model of Parkinson’s disease. Neurol. Res. 2010, 32, 88–91. [Google Scholar] [CrossRef]
- Bauer, C.K.; Schneeberger, P.E.; Kortüm, F.; Altmüller, J.; Santos-Simarro, F.; Baker, L.; Keller-Ramey, J.; White, S.M.; Campeau, P.M.; Gripp, K.W.; et al. Gain-of-function mutations in KCNN3 Encoding the small-conductance Ca2+-Activated K+ Channel SK3 cause Zimmermann-Laband syndrome. Am. J. Hum. Genet. 2019, 104, 1139–1157. [Google Scholar] [CrossRef] [Green Version]
- Carignani, C.; Roncarati, R.; Rimini, R.; Terstappen, G.C. Pharmacological and molecular characterisation of SK3 channels in the TE671 human medulloblastoma cell line. Brain Res. 2002, 939, 11–18. [Google Scholar] [CrossRef]
- Potier, M.; Joulin, V.; Roger, S.; Besson, P.; Jourdan, M.-L.; LeGuennec, J.-Y.; Bougnoux, P.; Vandier, C. Identification of SK3 channel as a new mediator of breast cancer cell migration. Mol. Cancer Ther. 2006, 5, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Tajima, N.; Schönherr, K.; Niedling, S.; Kaatz, M.; Kanno, H.; Schönherr, R.; Heinemann, S.H. Ca2+-activated K+ channels in human melanoma cells are up-regulated by hypoxia involving hypoxia-inducible factor-1α and the von Hippel-Lindau protein. J. Physiol. 2006, 571, 349–359. [Google Scholar] [CrossRef]
- Meyer, R.; Schönherr, R.; Gavrilova-Ruch, O.; Wohlrab, W.; Heinemann, S.H. Identification of ether à go-go and calcium-activated potassium channels in human melanoma cells. J. Membr. Biol. 1999, 171, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Girault, A.; Haelters, J.-P.; Potier-Cartereau, M.; Chantôme, A.; Jaffres, P.-A.; Bougnoux, P.; Joulin, V.; Vandier, C. Targeting SKCa channels in cancer: Potential new therapeutic approaches. Curr. Med. Chem. 2012, 19, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar] [CrossRef] [PubMed]
- Cuddapah, V.A.; Sontheimer, H. Ion channels and tranporters in cancer. Ion channels and the control of cancer cell migration. Am. J. Physiol. Physiol. 2011, 301, C541–C549. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of ion channels and transporters in cell migration. Physiol. Rev. 2012, 92, 1865–1913. [Google Scholar] [CrossRef]
- Pardo, L.A.; Stühmer, W. The roles of K+ channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Villalonga, N.; Ferreres, J.C.; Argilés, J.M.; Condom, E.; Felipe, A. Potassium channels are a new target field in anticancer drug design. Recent Pat. Anticancer Drug Discov. 2007, 2, 212–223. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumor metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Monteith, G.; Davis, F.M.; Roberts-Thomson, S.J. Calcium channels and pumps in cancer: Changes and consequences. J. Biol. Chem. 2012, 287, 31666–31673. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Chen, Y.-T.; Chiu, W.; Shen, M.-R. Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 2013, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Contreras, G.F.; Castillo, K.; Enrique, N.; Carrasquel-Ursulaez, W.; Castillo, J.P.; Milesi, V.; Neely, A.; Alvarez, O.; Ferreira, G.; Gonzalez, C.; et al. A BK (Slo1) channel journey from molecule to physiology. Channels 2013, 7, 442–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, M.; Gasparoli, L.; Arcangeli, A. Potassium channels: Novel emerging biomarkers and targets for therapy in cancer. Recent Pat. Anticancer Drug Discov. 2013, 8, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Weaver, A.K.; Liu, X.; Sontheimer, H. Role for calcium-activated potassium channels (BK) in growth control of human malignant glioma cells. J. Neurosci. Res. 2004, 78, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontheimer, H. Ion channels and amino acid transporters support the growth and invasion of primary brain tumors. Mol. Neurobiol. 2004, 29, 61–72. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Chen, L.; Zhang, X.; See, L.-H.; Koch, C.M.; Yen, C.; Tong, J.J.; Spiegel, L.; Nguyen, K.C.; Servoss, A.; et al. Genomic amplification and oncogenic properties of the KCNK9 potassium channel gene. Cancer Cell 2003, 3, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Oeggerli, M.; Tian, Y.; Ruiz, C.; Wijker, B.; Sauter, G.; Obermann, E.; Güth, U.; Zlobec, I.; Sausbier, M.; Kunzelmann, K.; et al. Role of KCNMA1 in breast cancer. PLoS ONE 2012, 7, e41664. [Google Scholar] [CrossRef]
- Bloch, M.; Ousingsawat, J.; Simon, R.; Schraml, P.; Gasser, T.C.; Mihatsch, M.J.; Kunzelmann, K.; Bubendorf, L. KCNMA1 gene amplification promotes tumor cell proliferation in human prostate cancer. Oncogene 2006, 26, 2525–2534. [Google Scholar] [CrossRef] [Green Version]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabò, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef]
- Thurber, A.E.; Nelson, M.; Frost, C.L.; Levin, M.; Brackenbury, W.J.; Kaplan, D.L. IK channel activation increases tumor growth and induces differential behavioral responses in two breast epithelial cell lines. Oncotarget 2017, 8, 42382–42397. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as signaling hubs in cancer cell survival/death and invasion: Implications in tumor progression and therapy. J. Lipid Res. 2020, 61, 611–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pathak, T.; Yoast, R.; Emrich, S.; Xin, P.; Nwokonko, R.M.; Johnson, M.; Wu, S.; Delierneux, C.; Gueguinou, M.; et al. A calcium/cAMP signaling loop at the ORAI1 mouth drives channel inactivation to shape NFAT induction. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gamper, N.; Rohacs, T. Phosphoinositide sensitivity of ion channels, a functional perspective. Subcell. Biochem. 2012, 59, 289–333. [Google Scholar] [CrossRef] [PubMed]
- Hille, B.; Dickson, E.J.; Kruse, M.; Vivas, O.; Suh, B.-C. Phosphoinositides regulate ion channels. Biochim. Biophys. Acta 2015, 1851, 844–856. [Google Scholar] [CrossRef]
- Levitan, I.; Fang, Y.; Rosenhouse-Dantsker, A.; Romanenko, V. Cholesterol and ion channels. Subcell. Biochem. 2010, 51, 509–549. [Google Scholar] [CrossRef] [Green Version]
- Morales-Lázaro, S.L.; Rosenbaum, T. Multiple mechanisms of regulation of transient receptor potential ion channels by cholesterol. Curr. Top. Membr. 2017, 80, 139–161. [Google Scholar] [CrossRef]
- Liou, J.; Fivaz, M.; Inoue, T.; Meyer, T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc. Natl. Acad. Sci. USA 2007, 104, 9301–9306. [Google Scholar] [CrossRef] [Green Version]
- Chvanov, M.; Walsh, C.M.; Haynes, L.P.; Voronina, S.G.; Lur, G.; Gerasimenko, J.V.; Barraclough, R.; Rudland, P.S.; Petersen, O.H.; Burgoyne, R.D.; et al. ATP depletion induces translocation of STIM1 to puncta and formation of STIM1–ORAI1 clusters: Translocation and re-translocation of STIM1 does not require ATP. Pflug. Arch. 2008, 457, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Chvanov, M.; Haynes, L.P.; Petersen, O.H.; Tepikin, A.V.; Burgoyne, R.D. Role of phosphoinositides in STIM1 dynamics and store-operated calcium entry. Biochem. J. 2009, 425, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Korzeniowski, M.K.; Popovic, M.A.; Szentpetery, Z.; Varnai, P.; Stojilkovic, S.S.; Balla, T. Dependence of STIM1/Orai1-mediated calcium entry on plasma membrane phosphoinositides. J. Biol. Chem. 2009, 284, 21027–21035. [Google Scholar] [CrossRef] [Green Version]
- Katz, Z.B.; Zhang, C.; Quintana, A.; Lillemeier, Z.B.K.L.N.A.B.B.F.; Hogan, P.G. Septins organize endoplasmic reticulum-plasma membrane junctions for STIM1-ORAI1 calcium signalling. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Choi, S.; Maléth, J.J.; Park, S.; Ahuja, M.; Muallem, S. The ER/PM microdomain, PI(4,5)P2 and the regulation of STIM1–Orai1 channel function. Cell Calcium 2015, 58, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, E.J. RASSF4: Regulator of plasma membrane PI(4,5)P. J. Cell Biol. 2017, 216, 1879–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-J.; Chang, C.-L.; Lee, W.-R.; Liou, J. RASSF4 controls SOCE and ER–PM junctions through regulation of PI(4,5)P. J. Cell Biol. 2017, 216, 2011–2025. [Google Scholar] [CrossRef]
- Deb, B.K.; Pathak, T.; Hasan, G. Store-independent modulation of Ca2+ entry through Orai by Septin. Nat. Commun. 2016, 7, 11751. [Google Scholar] [CrossRef] [Green Version]
- Maléth, J.; Choi, S.; Muallem, S.; Ahuja, M. Translocation between PI(4,5)P2-poor and PI(4,5)P2-rich microdomains during store depletion determines STIM1 conformation and Orai1 gating. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Schroeder, C. Cholesterol-binding viral proteins in virus entry and morphogenesis. Cholest. Bind. Cholest. Transp. Proteins 2010, 51, 77–108. [Google Scholar]
- Levitan, I.; Singh, D.K.; Rosenhouse-Dantsker, A. Cholesterol binding to ion channels. Front. Physiol. 2014, 5, 65. [Google Scholar] [CrossRef] [Green Version]
- Derler, I.; Jardin, I.; Stathopulos, P.B.; Muik, M.; Fahrner, M.; Zayats, V.; Pandey, S.K.; Poteser, M.; Lackner, B.; Absolonova, M.; et al. Cholesterol modulates Orai1 channel function. Sci. Signal. 2016, 9, ra10. [Google Scholar] [CrossRef] [Green Version]
- Bóhorquez-Hernández, A.; Gratton, E.; Pacheco, J.; Asanov, A.; Vaca, L. Cholesterol modulates the cellular localization of Orai1 channels and its disposition among membrane domains. Biochim. Biophys. Acta 2017, 1862, 1481–1490. [Google Scholar] [CrossRef]
- Pacheco, J.; Dominguez, L.; Bohórquez-Hernández, A.; Asanov, A.; Vaca, L. A cholesterol-binding domain in STIM1 modulates STIM1-Orai1 physical and functional interactions. Sci. Rep. 2016, 6, 29634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, R.L. Faculty Opinions recommendation of Cholesterol deficiency in a mouse model of Smith-Lemli-Opitz syndrome reveals increased mast cell responsiveness. Fac. Opin. 2006, 203, 1161–1171. [Google Scholar] [CrossRef]
- Lopez, J.J.; Albarran, L.; Gómez, L.J.; Smani, T.; Salido, G.M.; Rosado, J.A. Molecular modulators of store-operated calcium entry. Biochim. Biophys. Acta 2016, 1863, 2037–2043. [Google Scholar] [CrossRef] [PubMed]
- Howie, D.; Nolan, K.F.; Daley, S.R.; Butterfield, E.; Adams, E.; Garcia-Rueda, H.; Thompson, C.; Saunders, N.J.; Cobbold, S.P.; Tone, Y.; et al. MS4A4B Is a GITR-associated membrane adapter, expressed by regulatory T cells, which modulates T cell activation. J. Immunol. 2009, 183, 4197–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.R.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nat. Cell Biol. 2013, 499, 238–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivats, S.; Balasuriya, D.; Pasche, M.; Vistal, G.; Edwardson, J.M.; Taylor, C.W.; Murrell-Lagnado, R.D. Sigma1 receptors inhibit store-operated Ca2+ entry by attenuating coupling of STIM1 to Orai. J. Cell Biol. 2016, 213, 65–79. [Google Scholar] [CrossRef]
- Jardin, I.; Salido, G.M.; Rosado, J.A. Role of lipid rafts in the interaction between hTRPC1, Orai1 and STIM. Channels 2008, 2, 401–403. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Plummer, N.W.; George, M.D.; Abramowitz, J.; Zhu, M.X.; Birnbaumer, L. A role for Orai in TRPC-mediated Ca2+ entry suggests that a TRPC:Orai complex may mediate store and receptor operated Ca2+ entry. Proc. Natl. Acad. Sci. USA 2009, 106, 3202–3206. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.C.L.; Willoughby, D.; Ciruela, A.; Ayling, L.-J.; Pagano, M.; Wachten, S.; Tengholm, A.; Cooper, D.M.F. Capacitative Ca2+Entry via Orai1 and Stromal Interacting Molecule 1 (STIM1) regulates adenylyl cyclase type. Mol. Pharmacology 2009, 75, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacol. 2008, 55, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, S.; Jung, H.-J.; Kim, K.-D.; Souda, P.; Whitelegge, J.; Gwack, Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF Inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galán, C.; Woodard, G.E.; Dionisio, N.; Salido, G.M.; Rosado, J.A. Lipid rafts modulate the activation but not the maintenance of store-operated Ca2+ entry. Biochim. Biophys. 2010, 1803, 1083–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krapivinsky, G.; Stotz, S.C.; Manasian, Y.; Clapham, D.E.; Krapivinsky, L. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 19234–19239. [Google Scholar] [CrossRef] [Green Version]
- Albarran, L.; Lopez, J.J.; Ben Amor, N.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Albarran, L.; Regodón, S.; Salido, G.M.; López, J.J.; Rosado, J.A. Role of STIM1 in the surface expression of SARAF. Channels 2016, 11, 84–88. [Google Scholar] [CrossRef]
- Jha, A.; Ahuja, M.; Maléth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER–PM junctions identifies STIMATE as a regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef] [Green Version]
- Quintana, A.; Rajanikanth, V.; Farber-Katz, S.; Gudlur, A.; Zhang, C.; Jing, J.; Zhou, Y.; Rao, A.; Hogan, P.G. TMEM110 regulates the maintenance and remodeling of mammalian ER–plasma membrane junctions competent for STIM–ORAI signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E7083–E7092. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, S.; Jew, M.; Kim, K.-D.; Yee, M.-K.; Abramson, J.; Gwack, Y. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, G.; Yang, Y.; Fu, S.; Liu, X.; Kang, H.; Yang, X.; Su, X.-C.; Shen, Y. Calmodulin dissociates the STIM1-Orai1 complex and STIM1 oligomers. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.Y.; Wang, B.; Wang, X.; Cheng, A.; Kolb, S.; Stanford, J.L.; Wright, J.L. Vigorous physical activity is associated with lower risk of metastatic–Lethal progression in prostate cancer and hypomethylation in the CRACR2A Gene. Cancer Epidemiol. Biomark. Prev. 2019, 28, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone-Corsi, P. The Cyclic AMP Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011148. [Google Scholar] [CrossRef]
- Kawasaki, T.; Ueyama, T.; Lange, I.; Feske, S.; Saito, N. Protein Kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+Level via the store-operated Ca2+Channel. J. Biol. Chem. 2010, 285, 25720–25730. [Google Scholar] [CrossRef] [Green Version]
- Yazbeck, P.; Tauseef, M.; Kruse, K.; Amin, M.-R.; Sheikh, R.; Feske, S.; Komarova, Y.; Mehta, D. STIM1 phosphorylation at Y361 recruits Orai1 to STIM1 puncta and induces Ca2+ entry. Sci. Rep. 2017, 7, srep42758. [Google Scholar] [CrossRef] [Green Version]
- Lopez, E.; Frischauf, I.; Jardin, I.; Derler, I.; Muik, M.; Cantonero, C.; Salido, G.M.; Smani, T.; Rosado, J.A.; Redondo, P.C. STIM1 phosphorylation at Y(316) modulates its interaction with SARAF and the activation of SOCE and I CRAC. J. Cell Sci. 2019, 132, jcs226019. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Sun, L.; Machaca, K. Orai1 internalization and STIM1 clustering inhibition modulate SOCE inactivation during meiosis. Proc. Natl. Acad. Sci. USA 2009, 106, 17401–17406. [Google Scholar] [CrossRef] [Green Version]
- Manji, S.S.; Parker, N.J.; Williams, R.T.; Van Stekelenburg, L.; Pearson, R.B.; Dziadek, M.; Smith, P.J. STIM1: A novel phosphoprotein located at the cell surface. Biochim. Biophys. Acta 2000, 1481, 147–155. [Google Scholar] [CrossRef]
- Cambier, J.; Yarkoni, Y. Faculty Opinions recommendation of phosphorylation of STIM1 underlies suppression of store-operated calcium entry during mitosis. Fac. Opin. 2009, 11, 1465–1472. [Google Scholar] [CrossRef]
- Pozo-Guisado, E.; Campbell, D.G.; Deak, M.; Alvarez-Barrientos, A.; Morrice, N.A.; Alvarez, I.S.; Alessi, D.R.; Martín-Romero, F.J. Phosphorylation of STIM1 at ERK1/2 target sites modulates store-operated calcium entry. J. Cell Sci. 2010, 123 Pt 18, 3084–3093. [Google Scholar] [CrossRef] [Green Version]
- Dionisio, N.; Galán, C.; Jardin, I.; Salido, G.M.; Rosado, J.A. Lipid rafts are essential for the regulation of SOCE by plasma membrane resident STIM1 in human platelets. Biochim. Biophys. Acta 2011, 1813, 431–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brownlow, S.L.; Harper, A.G.; Harper, M.T.; Sage, S.O. A role for hTRPC1 and lipid raft domains in store-mediated calcium entry in human platelets. Cell Calcium 2004, 35, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, S.L.; Sage, S.O. Transient receptor potential protein subunit assembly and membrane distribution in human platelets. Thromb. Haemost. 2005, 94, 839–845. [Google Scholar] [PubMed]
- Ávila-Medina, J.; Calderón-Sánchez, E.; González-Rodríguez, P.; Monje-Quiroga, F.; Rosado, J.A.; Castellano, A.; Ordóñez, A.; Smani, T. Orai1 and TRPC1 proteins co-localize with CaV1.2 Channels to form a signal complex in vascular smooth muscle cells. J. Biol. Chem. 2016, 291, 21148–21159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderón-Sánchez, E.M.; Ávila-Medina, J.; Callejo-García, P.; Ordóñez, A.; Ordóñez, A.; Smani, T. Role of Orai1 and L-type CaV1.2 channels in Endothelin-1 mediated coronary contraction under ischemia and reperfusion. Cell Calcium 2020, 86, 102157. [Google Scholar] [CrossRef] [PubMed]
- Dang, D.; Prasad, H.; Rao, R. Secretory pathway Ca2+-ATPases promote in vitro microcalcifications in breast cancer cells. Mol. Carcinog. 2017, 56, 2474–2485. [Google Scholar] [CrossRef]
- Zhang, M.; Meng, X.Y.; Cui, M.; Pascal, J.M.; Logothetis, D.E.; Zhang, J.F. Selective phosphorylation modulates the PIP2 sensitivity of the CaM-SK channel complex. Nat. Chem. Biol. 2014, 10, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Zhang, Q.; Timofeyev, V.; Zhang, Z.; Young, J.N.; Shin, H.-S.; Knowlton, A.A.; Chiamvimonvat, N. Molecular coupling of a Ca 2+ -Activated K + Channel to L-Type Ca2+ Channels via α-Actinin. Circ. Res. 2007, 100, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Timofeyev, V.; Tuteja, D.; Xu, D.; Lu, L.; Zhang, Q.; Zhang, Z.; Singapuri, A.; Albert, T.R.; Rajagopal, A.V.; et al. Ablation of a Ca2+-activated K+ channel (SK2 channel) results in action potential prolongation in atrial myocytes and atrial fibrillation. J. Physiol. 2009, 587, 1087–1100. [Google Scholar] [CrossRef]
- Gonzalez-Perez, V.; Lingle, C.J. Regulation of BK channels by beta and gamma subunits. Annu. Rev. Physiol. 2019, 81, 113–137. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, V.; Xia XM, Lingle CJ. Functional regulation of BK potassium channels by gamma1 auxiliary subunits. Proc. Natl. Acad. Sci. USA 2014, 111, 4868–4873. [CrossRef] [PubMed] [Green Version]
- Issa, N.P.; Hudspeth, A.J. Clustering of Ca2+ channels and Ca2+-activated K+ channels at fluorescently labeled presynaptic active zones of hair cells. Proc. Natl. Acad. Sci. USA 1994, 91, 7578–7582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaranayake, H.; Saunders, J.C.; Greene, M.I.; Navaratnam, D.S. Ca2+ and K+ (BK) channels in chick hair cells are clustered and colocalized with apical–basal and tonotopic gradients. J. Physiol. 2004, 560, 13–20. [Google Scholar] [CrossRef]
- Sun, X.; Gu, X.Q.; Haddad, G.G. Calcium influx via L- and N-Type calcium channels activates a transient large-conductance Ca2+-Activated K+Current in mouse neocortical pyramidal neurons. J. Neurosci. 2003, 23, 3639–3648. [Google Scholar] [CrossRef] [Green Version]
- Prakriya, M.; Lingle, C.J. BK channel activation by brief depolarizations requires Ca2+ influx through L- and Q-type Ca2+ channels in rat chromaffin cells. J. Neurophysiol. 1999, 81, 2267–2278. [Google Scholar] [CrossRef] [Green Version]
- Wolfart, J.; Roeper, J. Selective coupling of T-Type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic Midbrain neurons. J. Neurosci. 2002, 22, 3404–3413. [Google Scholar] [CrossRef] [Green Version]
- Grunnet, M.; Kaufmann, W.A. Coassembly of big conductance Ca2+-activated K+Channels and L-type Voltage-gated Ca2+Channels in rat brain. J. Biol. Chem. 2004, 279, 36445–36453. [Google Scholar] [CrossRef] [Green Version]
- Marrion, N.V.; Tavalin, S.J. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nat. Cell Biol. 1998, 395, 900–905. [Google Scholar] [CrossRef]
- Rehak, R.; Bartoletti, T.M.; Engbers, J.D.T.; Berecki, G.; Turner, R.W.; Zamponi, G.W. Low Voltage Activation of KCa1.1 Current by Cav3-KCa1.1 Complexes. PLoS ONE 2013, 8, e61844. [Google Scholar] [CrossRef] [Green Version]
- Engbers, J.D.; Zamponi, G.W.; Turner, R.W. Modeling interactions between voltage-gated Ca2+ channels and KCa1.1 channels. Channels 2013, 7, 524–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Yamamura, H.; Ohya, S.; Imaizumi, Y. Caveolin-1 facilitates the direct coupling between large conductance Ca2+-activated K+(BKCa) and Cav1.2 Ca2+Channels and their clustering to regulate membrane excitability in vascular myocytes. J. Biol. Chem. 2013, 288, 36750–36761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, A.K.; Olsen, M.L.; McFerrin, M.B.; Sontheimer, H. BK channels are linked to inositol 1,4,5-triphosphate receptors via lipid rafts: A novel mechanism for coupling [Ca2+](i) to ion channel activation. J. Biol. Chem. 2007, 282, 31558–31568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.; Schlichter, L.C. Selective Activation of KCa3.1 and CRAC Channels by P2Y2 receptors promotes Ca2+ signaling, store refilling and migration of rat microglial cells. PLoS ONE 2013, 8, e62345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammadi, M.; Chopin, V.; Matifat, F.; Dhennin-Duthille, I.; Chasseraud, M.; Sevestre, H.; Ouadid-Ahidouch, H. Human ether à-gogo K+ channel 1 (hEag1) regulates MDA-MB-231 breast cancer cell migration through Orai1-dependent calcium entry. J. Cell. Physiol. 2012, 227, 3837–3846. [Google Scholar] [CrossRef]
- Chen, M.; Li, J.; Jiang, F.; Fu, J.; Xia, X.; Du, J.; Hu, M.; Huang, J.; Shen, B. Orai1 forms a signal complex with BKCa channel in mesenteric artery smooth muscle cells. Physiol. Rep. 2016, 4, e12682. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Yin, N.; Chen, X.; Huang, H.; Liao, Y. Functional coupling between BKCa and SOC channels. Tissue Cell 2020, 66, 101394. [Google Scholar] [CrossRef]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and glioblastoma: In vitro studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef]
- Ambudkar, I.S. Cellular domains that contribute to Ca2+ entry events. Sci. Signal. 2004, 2004, pe32. [Google Scholar] [CrossRef]
- Gudlur, A.; Quintana, A.; Zhou, Y.; Hirve, N.; Mahapatra, S.; Hogan, P.G. STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAI1 channel. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kourrich, S.; Hayashi, T.; Chuang, J.-Y.; Tsai, S.-Y.; Su, T.-P.; Bonci, A. Dynamic interaction between Sigma-1 Receptor and Kv1.2 shapes neuronal and behavioral responses to cocaine. Cell 2013, 152, 236–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crottès, D.; Martial, S.; Rapetti-Mauss, R.; Pisani, D.F.; Loriol, C.; Pellissier, B.; Martin, P.; Chevet, E.; Borgese, F.; Soriani, O. Sig1R protein regulates hERG channel expression through a post-translational mechanism in leukemic cells. J. Biol. Chem. 2011, 286, 27947–27958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crottès, D.; Guizouarn, H.; Martin, P.; Borgese, F.; Soriani, O. The sigma-1 receptor: A regulator of cancer cell electrical plasticity? Front. Physiol. 2013, 4, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crottès, D.; Rapetti-Mauss, R.; Alcaraz-Perez, F.; Tichet, M.; Gariano, G.; Martial, S.; Guizouarn, H.; Pellissier, B.; Loubat, A.; Popa, A.; et al. SIGMAR1 regulates membrane electrical activity in response to extracellular matrix stimulation to drive cancer cell invasiveness. Cancer Res. 2015, 76, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soriani, O.; Rapetti-Mauss, R. Sigma 1 receptor and ion channel dynamics in cancer. Atherosclerosis 2017, 964, 63–77. [Google Scholar] [CrossRef]
- Cross, B.M.; Hack, A.; Reinhardt, T.A.; Rao, R. SPCA2 Regulates Orai1 trafficking and store independent Ca2+ entry in a model of lactation. PLoS ONE 2013, 8, e67348. [Google Scholar] [CrossRef]
- Smaardijk, S.; Chen, J.; Wuytack, F.; Vangheluwe, P. SPCA2 couples Ca2+ influx via Orai1 to Ca2+ uptake into the Golgi/secretory pathway. Tissue Cell 2017, 49, 141–149. [Google Scholar] [CrossRef]
- O’Connor, K.L.; Shaw, L.M.; Mercurio, A.M. Release of cAMP gating by the alpha6beta4 integrin stimulates lamellae formation and the chemotactic migration of invasive carcinoma cells. J. Cell Biol. 1998, 143, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, K.L.; Nguyen, B.K.; Mercurio, A.M. RhoA function in lamellae formation and migration is regulated by the alpha6beta4 integrin and cAMP metabolism. J. Cell Biol. 2000, 148, 253–258. [Google Scholar] [CrossRef]
- O’Connor, K.L.; Mercurio, A.M. Protein kinase A regulates Rac and is required for the growth factor-stimulated migration of carcinoma cells. J. Biol. Chem. 2001, 276, 47895–47900. [Google Scholar] [CrossRef] [Green Version]











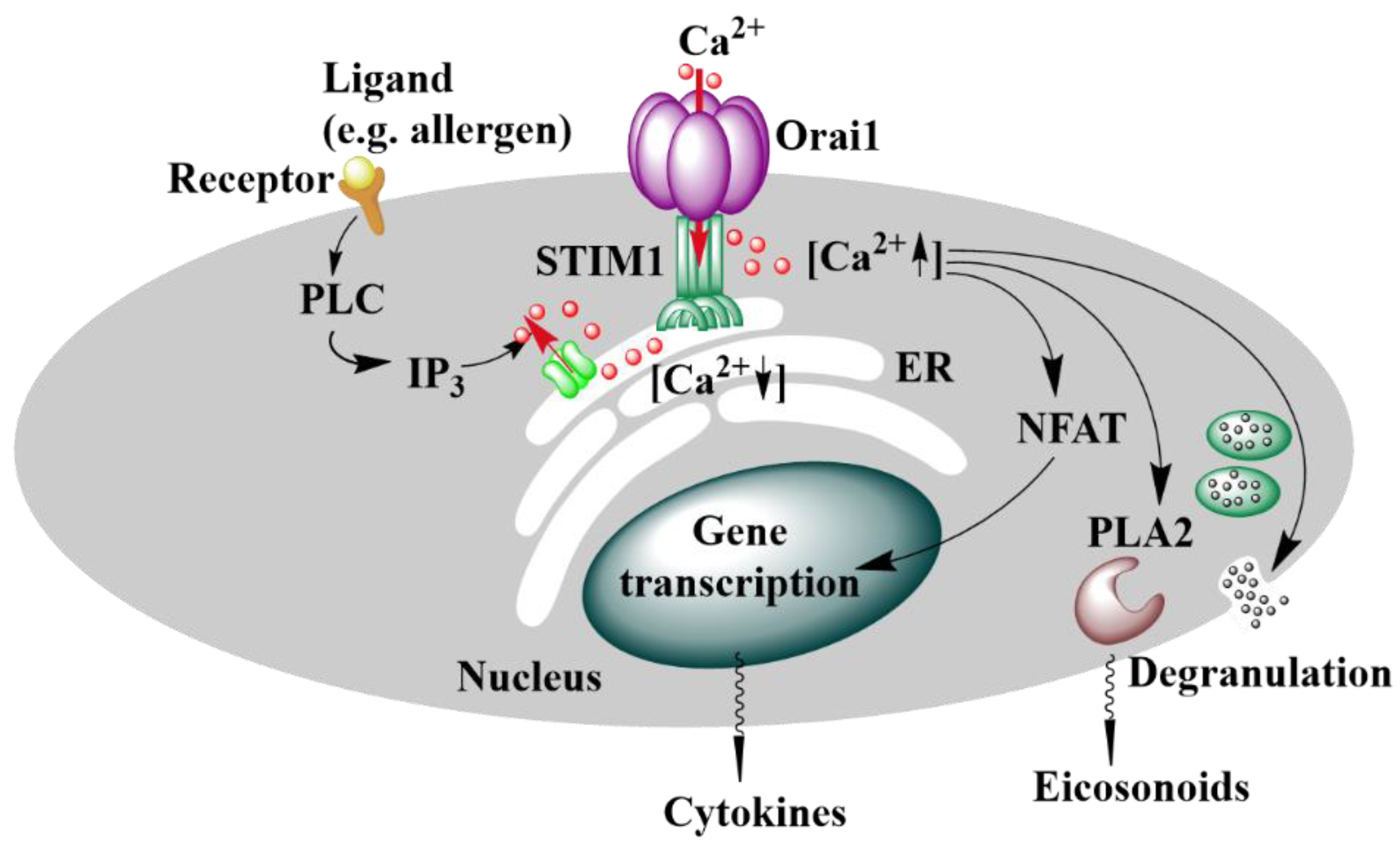{kind=link}
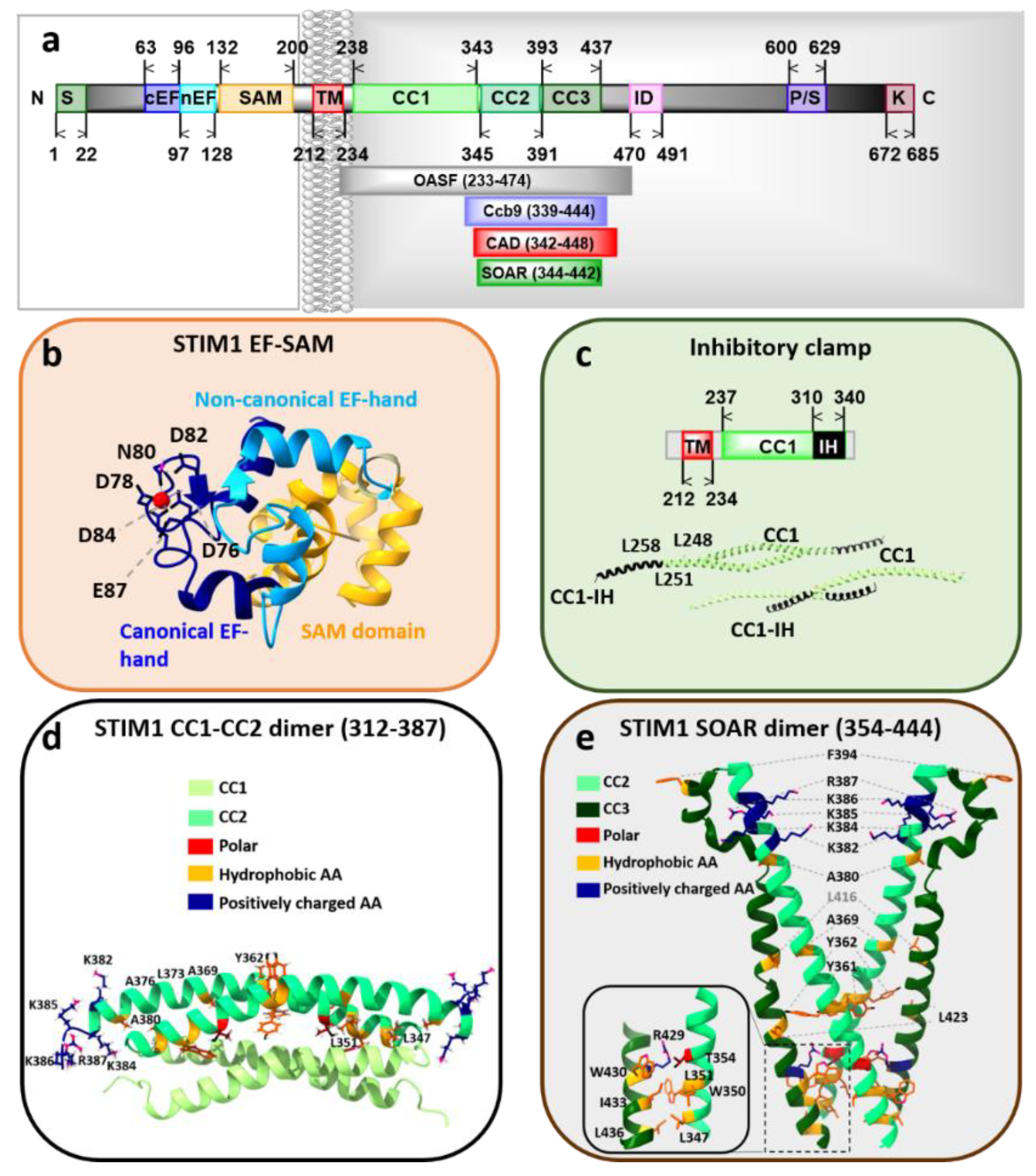{kind=link}
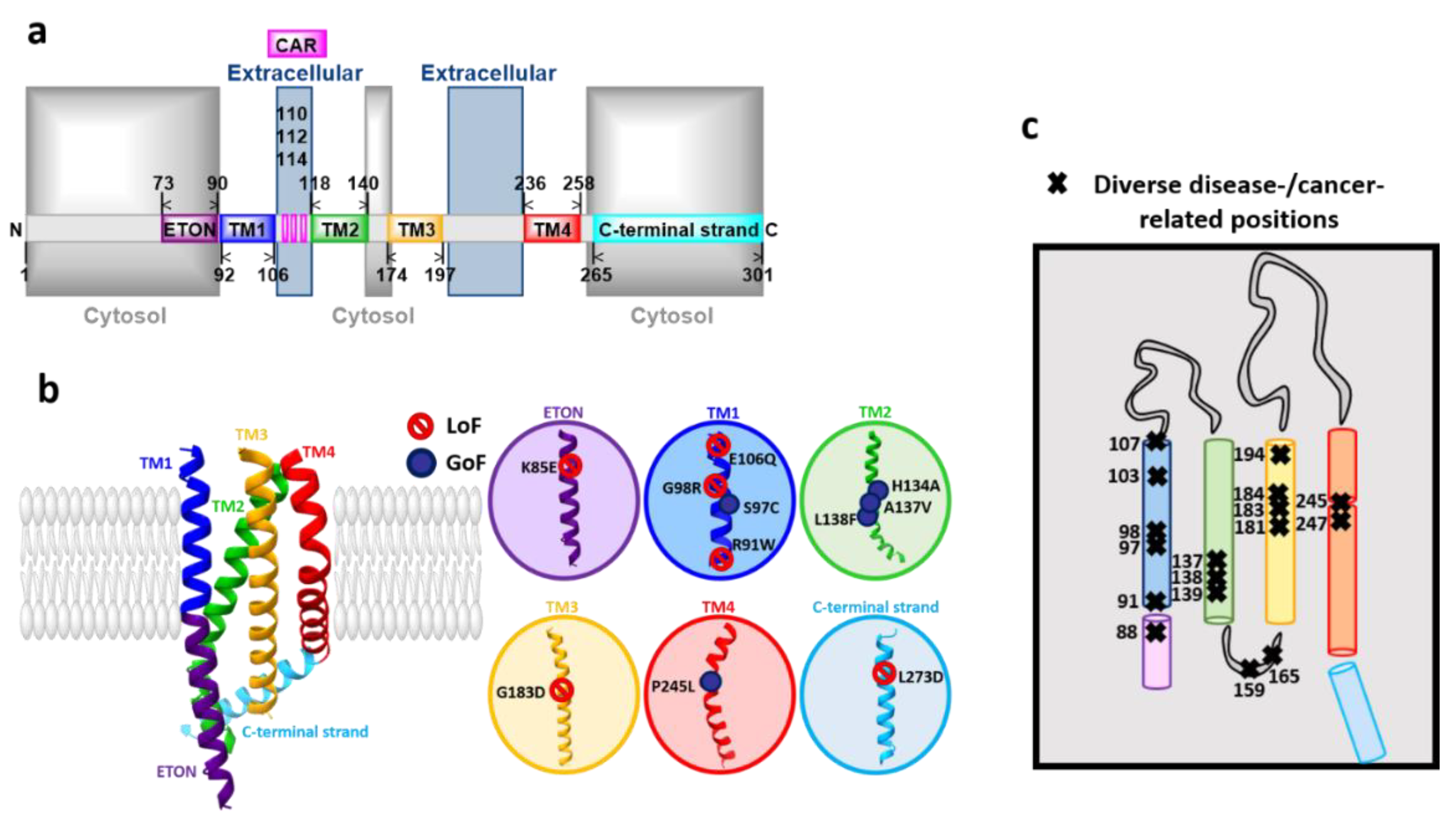{kind=link}
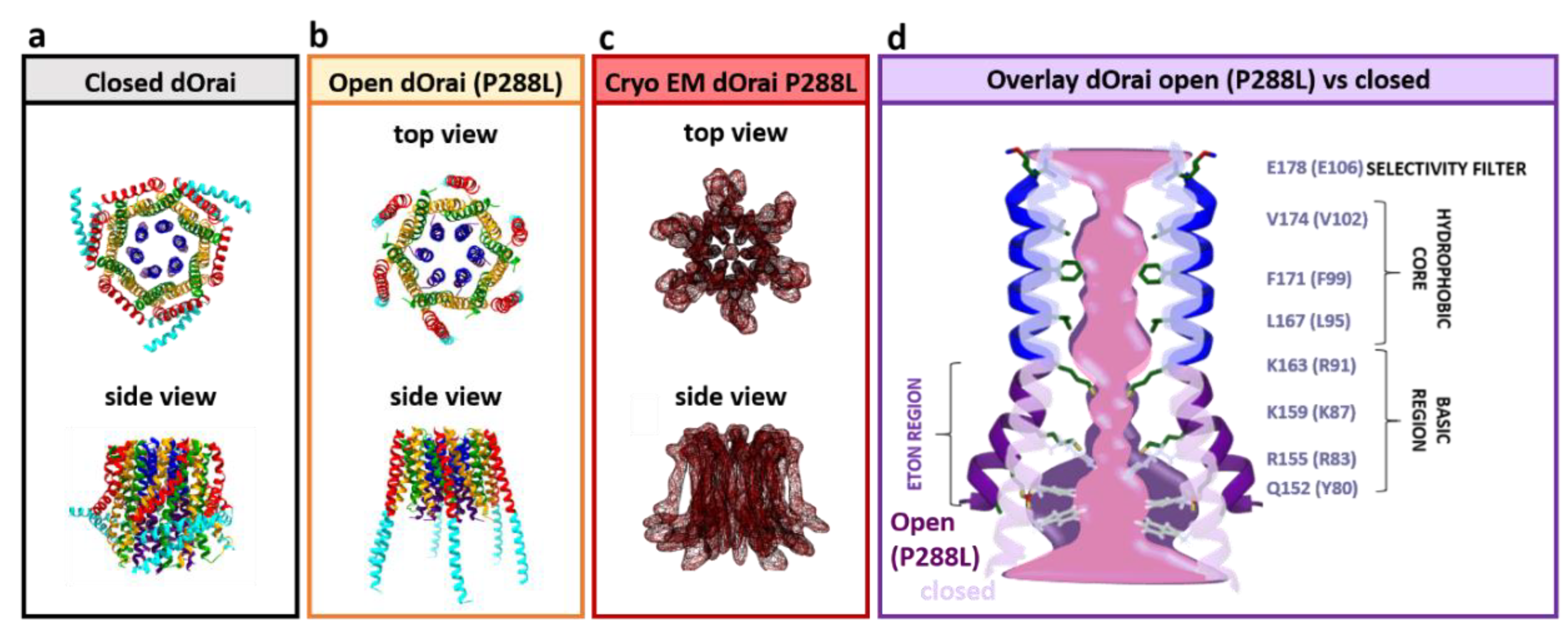{kind=link}
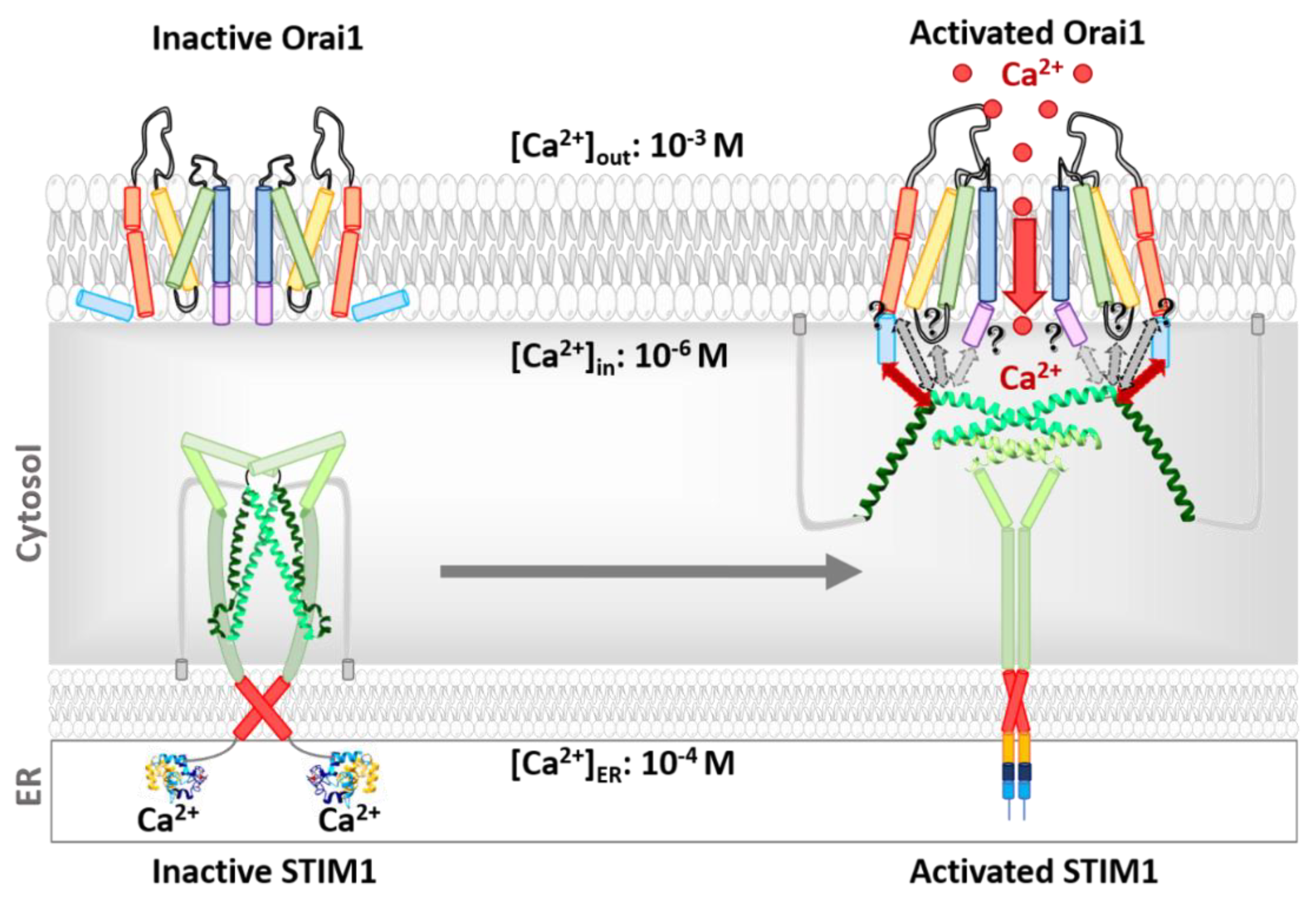{kind=link}
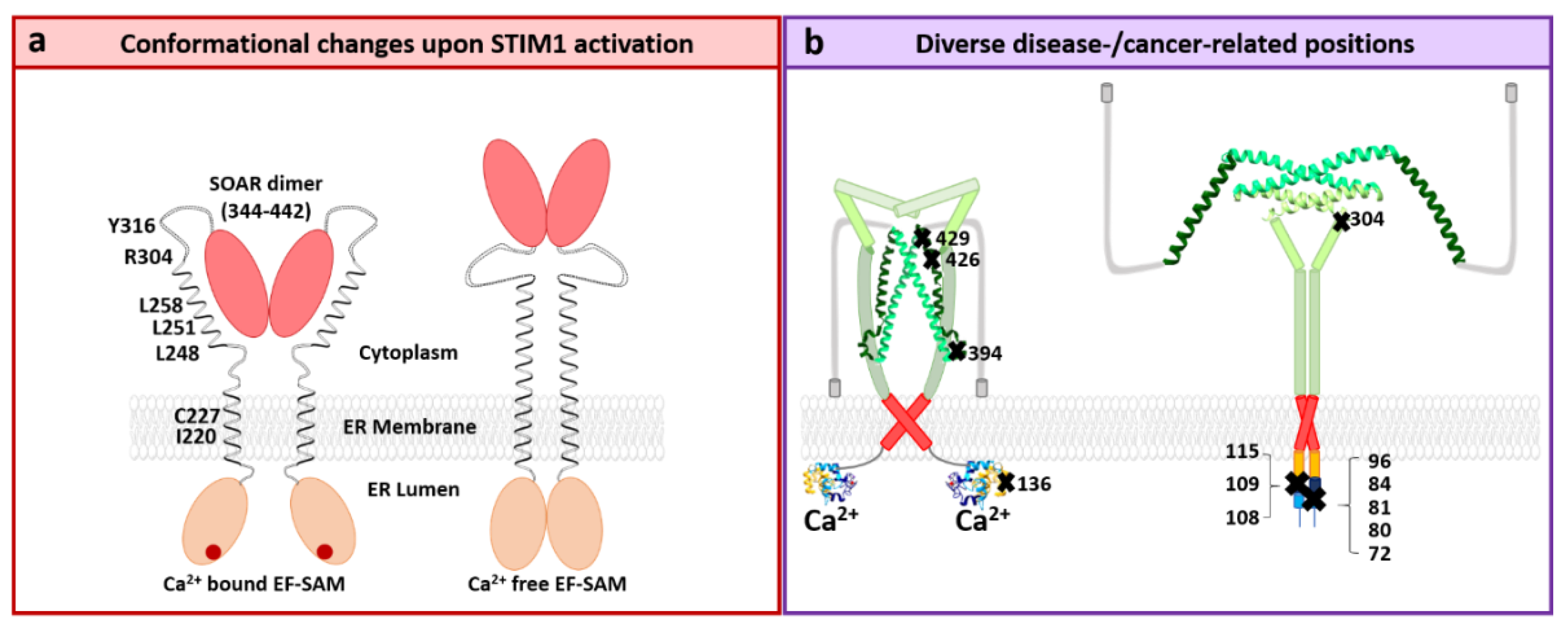{kind=link}
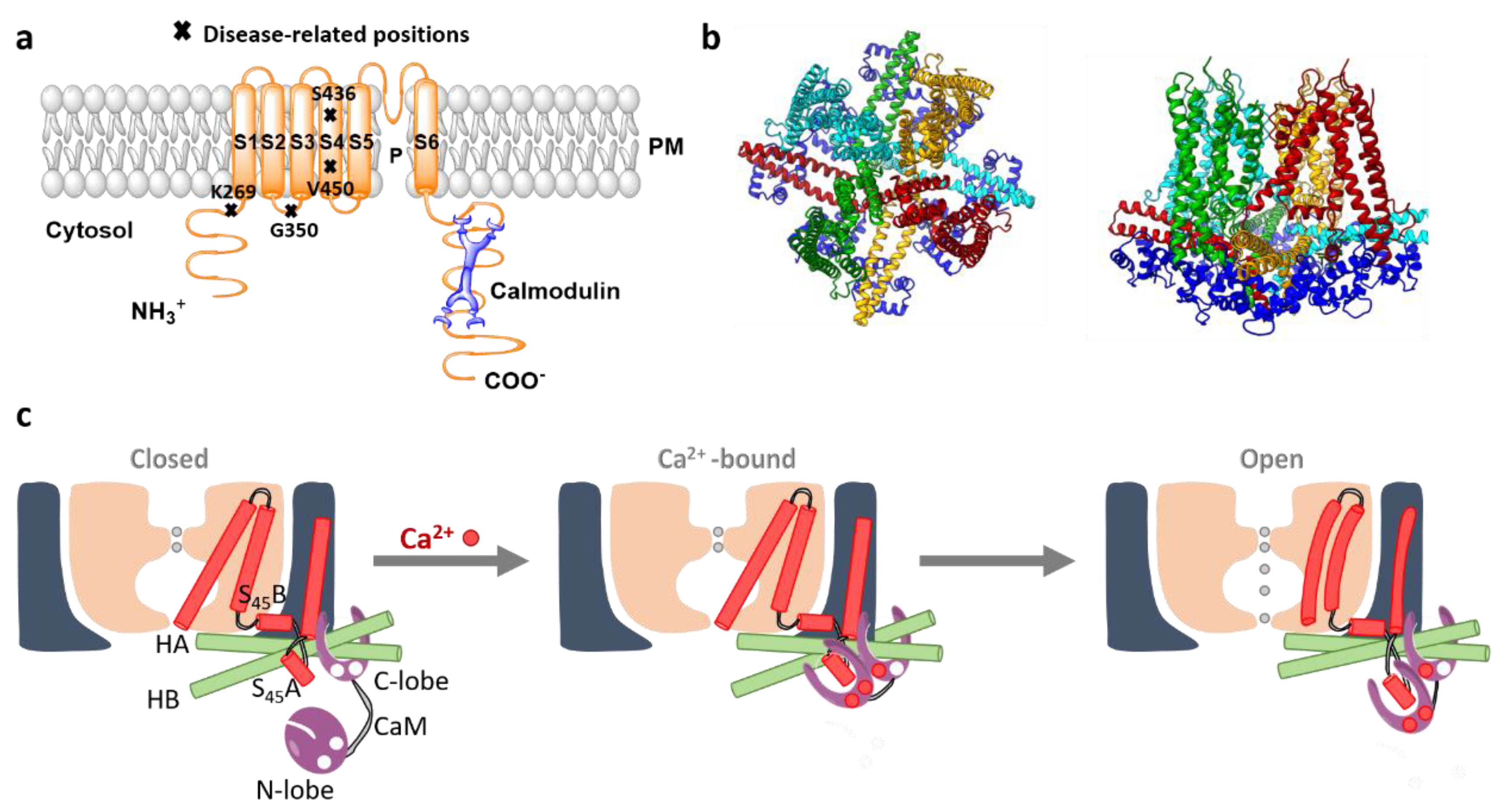{kind=link}
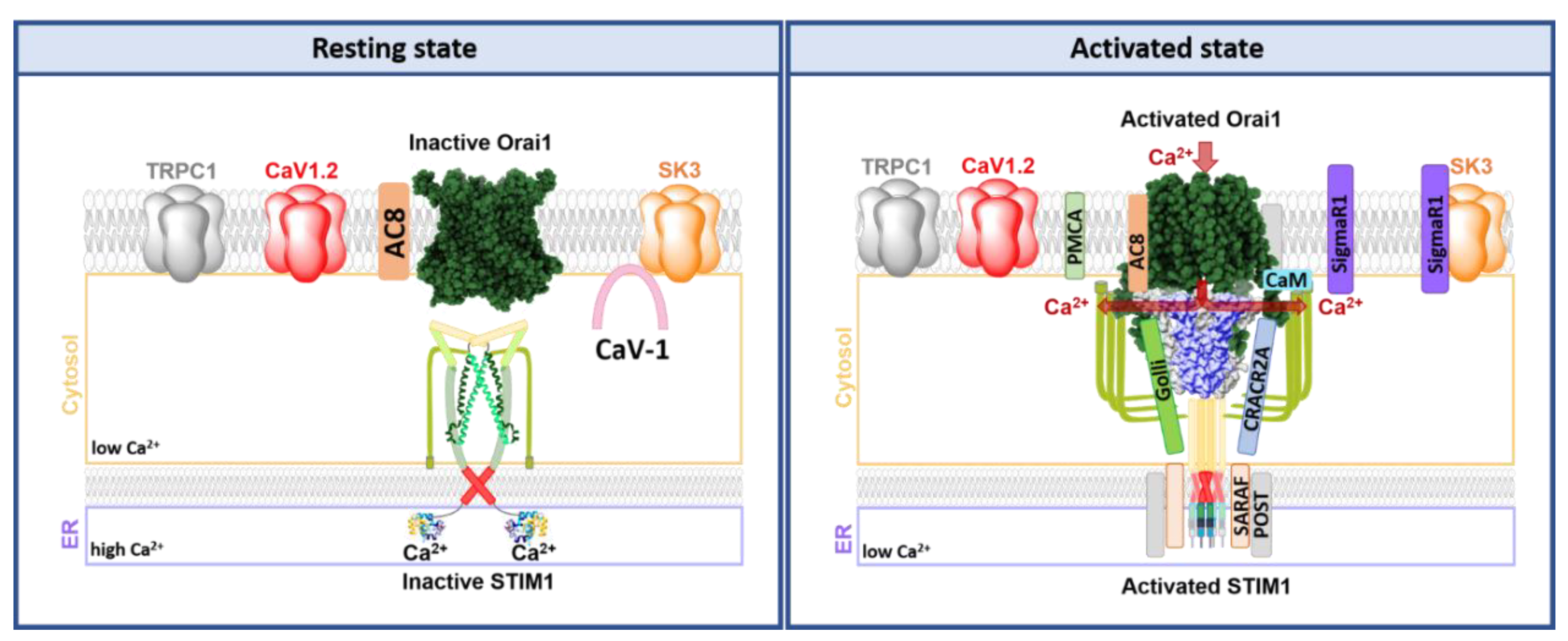{kind=link}
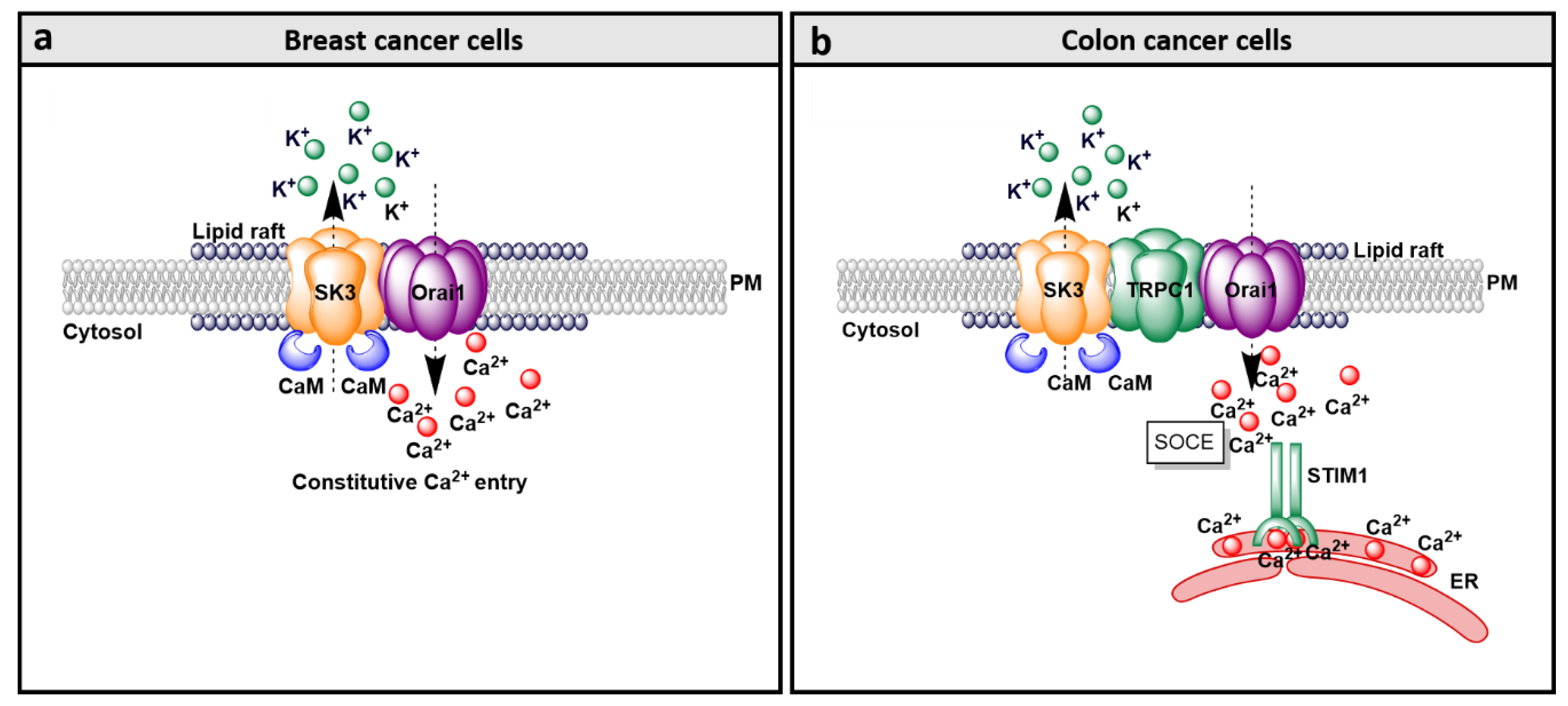{kind=link}
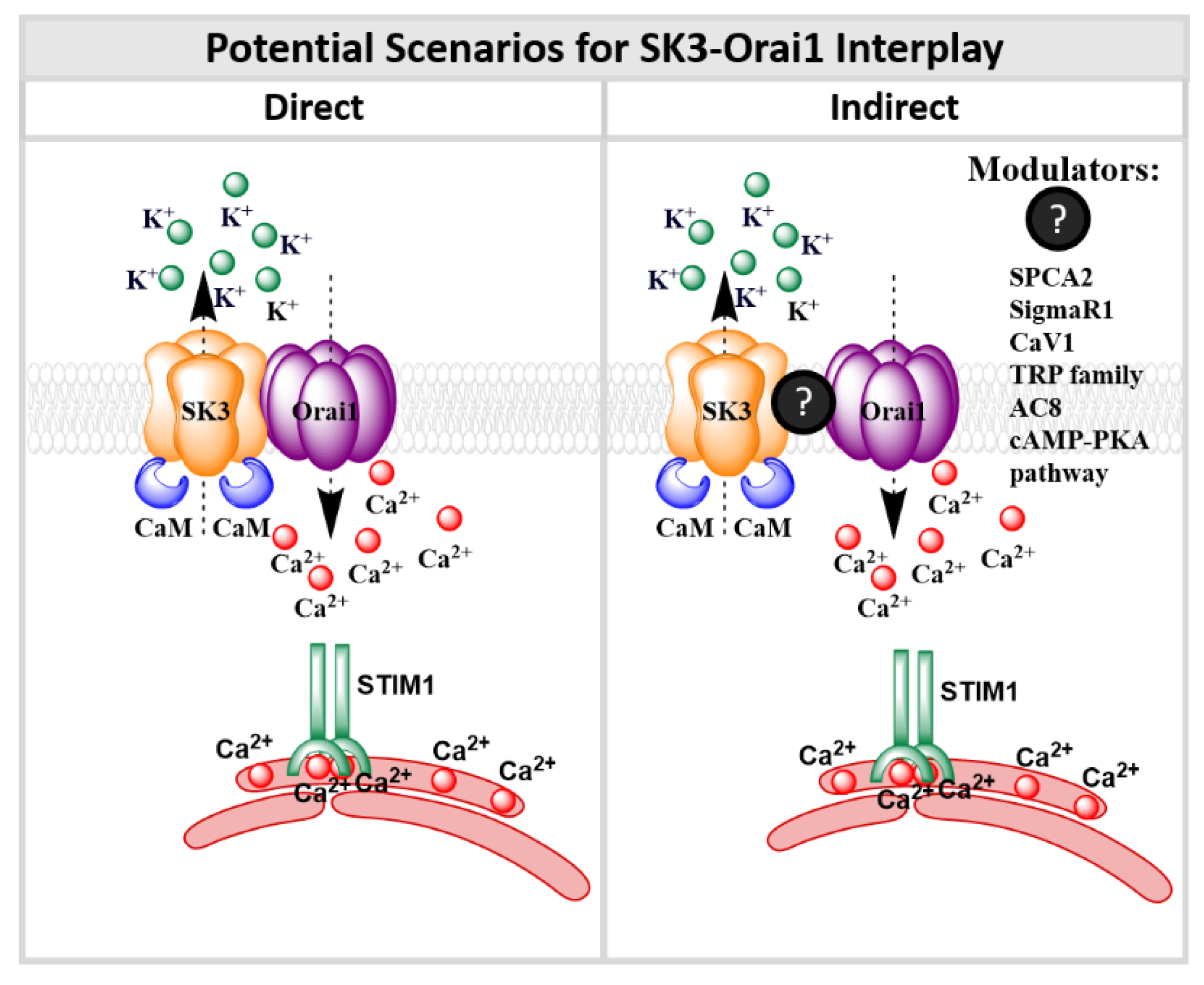{kind=link}
| Name | Empirical Formula | Function of Interest | Additional Information | References |
|---|---|---|---|---|
| 2-APB | (C6H5)2BOCH2CH2NH2 | Modulates: SOCE function in a concentration dependent manner | Activates: CRAC/STIM1 + Orai1 < 10 µM and Orai3 Blocks: CRAC/STIM1 + Orai1 > 50 µM Modulates: TRPV and TRPM channels | [206] |
| BTP2 | C15H9F6N5OS | Blocks: CRAC Channel | Blocks: TPRC3 channel inhibitor and reduces NFAT nuclear translocation and cytokine production | [234] |
| SKF-96365 | C22H26N2O3 · HCl | Blocks: receptor-mediated Ca2+ entry | Blocks: voltage-gated Ca2+ entry; TRPC channel | [235] |
| GSK-5498A | C18H11F6N3O | Blocks: CRAC channel | - | [223] |
| GSK-5503A | C23H17F2N3O2 | Blocks: CRAC channel | - | [224] |
| GSK-7975A | C18H12F5N3O2 | Blocks: Orai1 | - | [222] |
| Synta66 | C20H17FN2O3 | Blocks: CRAC channel | No effect on L-type Ca2+ channel either TRPC1/5 | [236] |
| Pyr6 | C17H9F7N4O | Blocks: SOCE | Effect on carbachol-induced, TRPC3-mediated calcium entry inhibits the typical Orai downstream signaling cascades in RBL mast cells (NFAT activation and degranulation) | [219] |
| Pyr10 | C18H13F6N3O2S | Blocks: CRAC channel | TRP cation 3 (TRPC3) inhibitor | [219] |
| RO2959 | C21H19F2N5OS.xHCl | Blocks: SOCE | Potent inhibitor of human IL-2 production, potently blocks T-cell receptor-triggered gene expression and T-cell functional pathways | [231] |
| Channel Type or Channel-Protein Complexes | Expression Level | Localization | Associated Mutation | Biological Function | Ref | |||
|---|---|---|---|---|---|---|---|---|
| Normal Tissue | Cancer Tissue | Normal Tissue | Cancer Tissue | Normal Tissue | Cancer Tissue | |||
| Orai1 channels | low | high (reduced in prostate cancer) | Ubiquitously expressed in a diversity of tissues Heart brain kidney lung skeletal muscle and other organs | Renal carcinoma Breast Melanoma Glioma Esophageal squamous cell carcinoma Pancreatic adenocarcinoma Prostate | A137V (colorectal tumor) M139V (stomach carcinoma) S159L (uterine carcinoma) G183D (glioblastoma) G247S (neck carcinoma) | Involved in Ca2+ signaling indispensable role in the immune system | Proliferation Migration Increased cell survival Tumor growth progression metastasis | [61,132,251,252,253] |
| Orai1-SPCA2 | - | high | - | Breast | n.d. | Separately involved in cell Ca2+ signaling | Activation of Ras–ERK pathway, involved in proliferation and cell cycle Tumorigenesis Cell growth | [254,255] |
| Orai2 channels | low | high | brain and at lower levels in the spleen, lung, and small intestine | Parathyroid tumors Prostate | n.d. | Involved in Ca2+ signaling | Proliferation | [256,257] |
| Orai3 channels | low | high | wide tissue expression including the heart, brain, kidney, lung, skeletal muscle, and other organs | Breast Prostate Renal carcinoma Lung adenocarcinoma | n.d. | Involved in Ca2+ signaling | Proliferation Invasion Increased cell survival Tumor growth progression Apoptosis | [59,258,259] |
| STIM1 | low | high | Ubiquitously expressed in a diversity of tissues, such as heart, skeletal muscle, and the central nervous system | Breast Lung adenocarcinoma Glioblastoma Colorectal, gastric, cervical cancer renal carcinoma hepatocellular carcinoma | A79T, E87Q, W350L, G446C/V (lung adenocarcinoma) S116N (Glioblastoma) | Involved in Ca2+ signaling indispensable role in the immune system | Migration Invasion Increased cell survival | [260,261,262,263,264] |
| STIM2 | low | high | diverse primary lymphocytes such as Th, TC, and B-cells | Glioblastoma Prostate Melanoma Colorectal cancer | n.d. | Involved in Ca2+ signaling | Migration Invasion | [265,266,267] |
| SK channels | low | high | Neuronal tissues, colon, corpus cavernosum, adrenal gland, brain, prostate, bladder, liver, and heart | Breast Colon Medulloblastoma Melanoma Glioma Leukemia cells | n.d. | Synaptic function. Firing in pacemaker neurons. Control the pattern of single spike firing of dopamine neurons | Migration, proliferation tumor cell dissemination, and metastasis | [38,268,269,270] |
| SK3-Orai1 | low | high | guinea pig gall bladder smooth muscle | Breast | n.d | Involved in Ca2+ signaling regulate muscle contraction | Migration Ca2+-dependent invasive process Bone metastasis Constitutive Ca2+ entry | [34,44] |
| SK3-SigmaR1-Orai1 | - | high | - | Breast Colorectal | n.d. | Involved in Ca2+ signaling | Migration Ca2+-dependent invasive process Bone metastasis Constitutive Ca2+ entry | [271] |
| SK3-TRPC1-Orai1 | - | high | - | Colon | n.d. | Involved in Ca2+ signaling | Migration activation of the Akt pathway SOCE amplification Constitutive Ca2+ entry | [272] |
| SK3-cAMP-Orai1 | - | high | - | Breast | n.d. | Involved in Ca2+ signaling | Migration Constitutive Ca2+ entry Bone metastasis | [36]. |
| IK channels | low | high | Blood, microglial endothelial and epithelial cells trachea, prostate, placenta and salivary glands. presence in excitable cells such as central neurons and cardiomyocytes | Prostate Breast Glioblastoma Endometrial, hepatocellular, and cervical carcinoma | n.d. | Immune responses of B and T cells Secretion in epithelial tissues | Tumor cell signaling including cell cycle progression Proliferation, Migration and the Epithelial-Mesenchymal Transition | [19,42,273,274,275] |
| BK channels | low | high | Skeletal muscles Nervous system, Smooth muscle cells | Prostate Breast Glioblastoma Neuroblastoma | n.d. | Regulation of calcium signaling related processes | Proliferation, migration, metastasis, apoptosis | [276,277,278] |
| Name | Empirical Formula | Function of Interest | Additional Information | References |
|---|---|---|---|---|
| 1-EBIO | C9H10N2O | Activates: SK1, SK2, SK3, SK4 | - | [313,318,323] |
| NS309 | C8H4Cl2N2O2 | Activates: SK1, SK3, SK4 | Blocks: L-type channel | [314,316,318] |
| Riluzole | C8H5F3N2OS | Activates: SK1, SK2, SK3, SK4 | Activates: TRPC5 channel | [313,324] |
| CyPPA | C16H23N5 | Activates: SK2, SK3 | Blocks: TRPM7 | [314,318,325] |
| SKA-111 | C12H10N2S | Activates: SK4 | - | [326] |
| SKA-121 | C12H10N2O | Activates: SK4 | - | [326] |
| ICAGEN | C13H10N4S | Blocks: SK1, SK2, SK3 | - | [315] |
| NS8593 | C17H18ClN3 | Blocks: SK1, SK2, SK3 | Blocks: TRPM7 | [315,318,327] |
| BMB | C21H20BrNO6 | Blocks: SK3 | Blocks: gamma-aminobutyric acid (GABA)-gated Cl- channels | [314] |
| Apamin | C79H131N31O24S4 | Blocks: SK1, SK2, SK3 | - | [312,328] |
| TRAM34 | C22H17ClN2 | Blocks: SK4 | - | [318,329] |
| 4-AP | C5H6N2 | Blocks: SK3 | Blocks: Voltage gated potassium channels | [317] |
| RA-2 | C22H16F2O6 | Blocks: SK1, SK2, SK3, SK4 | - | [330] |
| Alkyl-ether-lipids | - | Blocks: SK3 | - | [38,317] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiffner, A.; Derler, I. Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes 2020, 10, 425. https://doi.org/10.3390/membranes10120425
Tiffner A, Derler I. Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes. 2020; 10(12):425. https://doi.org/10.3390/membranes10120425
Chicago/Turabian StyleTiffner, Adéla, and Isabella Derler. 2020. "Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer" Membranes 10, no. 12: 425. https://doi.org/10.3390/membranes10120425
APA StyleTiffner, A., & Derler, I. (2020). Molecular Choreography and Structure of Ca2+ Release-Activated Ca2+ (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes, 10(12), 425. https://doi.org/10.3390/membranes10120425