
The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
2.2. Fuzzy Oil Drop Model and Its Modification Taking into Account the Influence of Factors Other Than the Aqueous Environment
3. Results
- The RD value for the T–O–R relationship determines the degree of hydrophobic core presence compared to a system completely devoid of this presence. RD values <0.5 for this relationship suggest the presence of a hydrophobic core.
- The K values indicate the degree of participation of a factor other than polar in shaping the structure. The higher the value of K, the greater the proportion of the factor different from the aqueous environment, including the hydrophobic environment of the membrane in particular.
- The RD values for the M–O–T relationship express the degree of adjustment of the O distribution to the modified T distribution called M with the reference T distribution. Low RD values for M–O–T indicate that O is “approaching” the modified distribution, where the degree of modification is expressed by the value of parameter K.
- The value of K is determined using the step-wise procedure for successive K values, which involves looking for the minimum DKL for the O|M relationship.
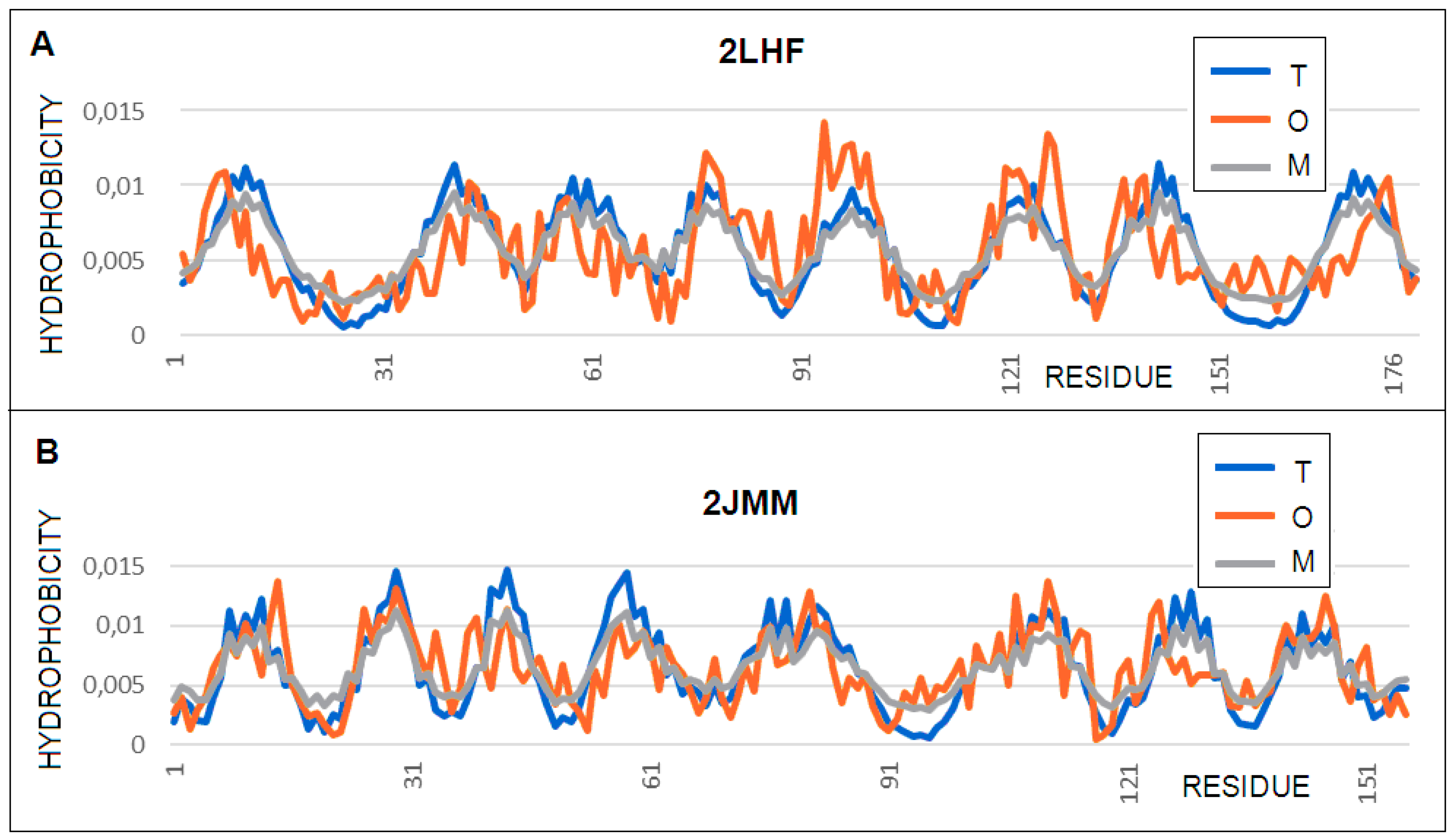
3.1. Proteins Implicated in Antibiotic Resistance (OprH and OmpA)
3.2. Oil Transport (6QAM, 6QWR)
3.3. Beta Barrel Proteins of Higher Diameter (OmpA, BamA)
3.4. Experimentally Modified Proteins (OprF, CarO1, and CarO2) (PDB: 4RLC, 4RL9I, and 4RLB)
3.5. Autotransporters (Hbp and EspP) (PDB: 3AEH + 2QOM)
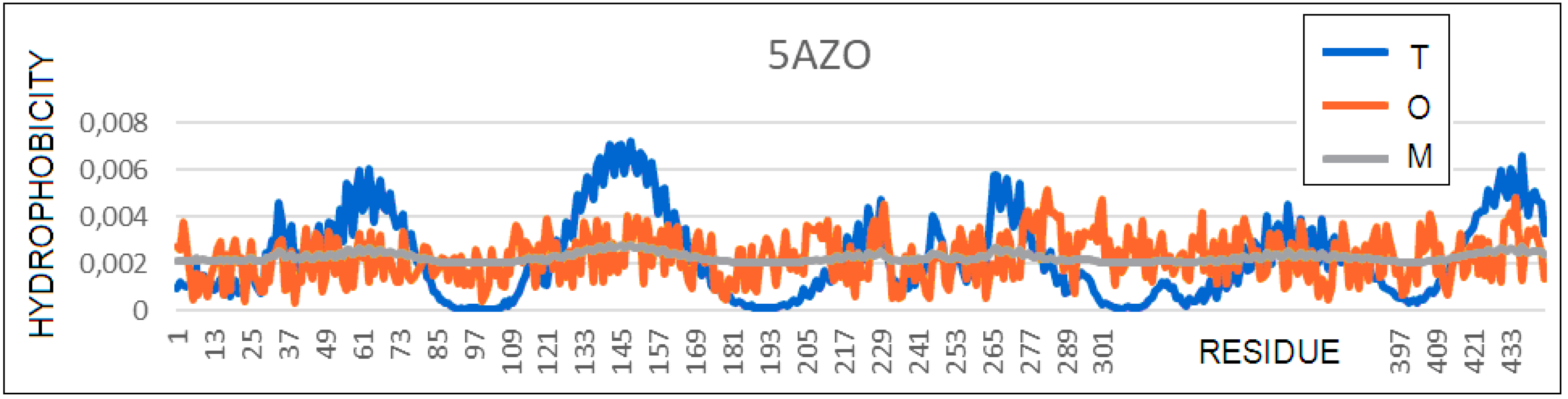
3.6. A Transmembrane Protein with a Helical Bundle (PAO1) (PDB 5AZO)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Plantinga, N.L.; Wittekamp, B.H.J.; Van Duijn, P.J.; Bonten, M.J.M. Fighting antibiotic resistance in the intensive care unit using antibiotics. Future Microbiol. 2015, 10, 391–406. [Google Scholar] [CrossRef]
- Zinner, S.H. Antibiotic use: Present and future. New Microbiol. 2007, 30, 321–325. [Google Scholar]
- Mohr, K.I. History of Antibiotics Research. How Overcome Antibiot. Crisis 2016, 398, 237–272. [Google Scholar] [CrossRef]
- Yoneyama, H.; Katsumata, R. Antibiotic Resistance in Bacteria and Its Future for Novel Antibiotic Development. Biosci. Biotechnol. Biochem. 2006, 70, 1060–1075. [Google Scholar] [CrossRef]
- Planson, A.-G.; Carbonell, P.; Grigoras, I.; Faulon, J.-L. Engineering antibiotic production and overcoming bacterial resistance. Biotechnol. J. 2011, 6, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Bacterial resistance to antibiotics: Enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Power, E. Impact of antibiotic restrictions: The pharmaceutical perspective. Clin. Microbiol. Infect. 2006, 12, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barger, A.; Fuhst, C.; Wiedemann, B. Pharmacological indices in antibiotic therapy. J. Antimicrob. Chemother. 2003, 52, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.Y.; Ng, A.M.; Persaud, A.K.; Furmanek, S.; Kothari, Y.N.; Price, J.D.; Wiemken, T.L.; Saad, M.A.; Guardiola, J.J.; Cavallazzi, R.S. Antibiotic Timing and Outcomes in Sepsis. Am. J. Med. Sci. 2018, 355, 524–529. [Google Scholar] [CrossRef]
- Jordan, S.; Tait, M. Antibiotic therapy. Nurs. Stand 1999, 7, 33–38. [Google Scholar] [CrossRef]
- Olesen, S.W.; Lipsitch, M.; Grad, Y.H. The role of “spillover” in antibiotic resistance. Proc. Natl. Acad. Sci. USA 2020, 117, 29063–29068. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.; Greko, C. Antibiotic resistance—Consequences for animal health, welfare, and food production. Upsala J. Med. Sci. 2014, 119, 96–102. [Google Scholar] [CrossRef]
- Machowska, A.; Lundborg, C.S. Drivers of Irrational Use of Antibiotics in Europe. Int. J. Environ. Res. Public Health 2018, 16, 27. [Google Scholar] [CrossRef] [Green Version]
- Årdal, C.; Blix, H.S.; Plahte, J.; Røttingen, J.-A. An antibiotic’s journey from marketing authorization to use, Norway. Bull. World Health Organ. 2017, 95, 220–226. [Google Scholar] [CrossRef]
- Walsh, C.T. Where will new antibiotics come from? Nat. Rev. Genet. 2003, 1, 65–70. [Google Scholar] [CrossRef]
- Mohamed, N.S.; Wilkie, W.A.; Remily, E.; Nace, J.; Delanois, R.E.; Browne, J.A. Antibiotic Choice: The Synergistic Effect of Single vs Dual Antibiotics. J. Arthroplast. 2020, 35, S19–S23. [Google Scholar] [CrossRef] [Green Version]
- Moser, C.; Lerche, C.J.; Thomsen, K.; Hartvig, T.; Schierbeck, J.; Jensen, P.; Ciofu, O.; Høiby, N. Antibiotic therapy as personalized medicine—general considerations and complicating factors. APMIS 2019, 127, 361–371. [Google Scholar] [CrossRef]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef]
- Wenzel, M.; Bandow, J.E. Proteomic signatures in antibiotic research. Proteomics 2011, 11, 3256–3268. [Google Scholar] [CrossRef]
- Rokem, J.S.; Lantz, A.E.; Nielsen, J. Systems biology of antibiotic production by microorganisms. Nat. Prod. Rep. 2007, 24, 1262–1287. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, J.L. General principles of antibiotic resistance in bacteria. Drug Discov. Today Technol. 2014, 11, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Dodds, D.R. Antibiotic resistance: A current epilogue. Biochem. Pharmacol. 2017, 134, 139–146. [Google Scholar] [CrossRef]
- Gilmore, M.S. The CRISPR–Antibiotic Resistance Connection. CRISPR J. 2019, 2, 199–200. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L.; Banach, M. Model of Environmental Membrane Field for Transmembrane Proteins. Int. J. Mol. Sci. 2021, 22, 3619. [Google Scholar] [CrossRef]
- Edrington, T.C.; Kintz, E.; Goldberg, J.B.; Tamm, L.K. Structural Basis for the Interaction of Lipopolysaccharide with Outer Membrane Protein H (OprH) from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 39211–39223. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.U.; Alioth, S.; Hu, K.; Walser, R.; Koebnik, R.; Pervushin, K. A Minimal Transmembrane β-Barrel Platform Protein Studied by Nuclear Magnetic Resonance. Biochemistry 2007, 46, 1128–1140. [Google Scholar] [CrossRef]
- Cross, T.A.; Murray, D.T.; Watts, A. Helical membrane protein conformations and their environment. Eur. Biophys. J. 2013, 42, 731–755. [Google Scholar] [CrossRef] [Green Version]
- Schubeis, T.; Le Marchand, T.; Daday, C.; Kopec, W.; Movellan, K.T.; Stanek, J.; Schwarzer, T.S.; Castiglione, K.; de Groot, B.L.; Pintacuda, G.; et al. A β-barrel for oil transport through lipid membranes: Dynamic NMR structures of AlkL. Proc. Natl. Acad. Sci. USA 2020, 117, 21014–21021. [Google Scholar] [CrossRef] [PubMed]
- Pautsch, A.; Schulz, G.E. High-resolution structure of the OmpA membrane domain. J. Mol. Biol. 2000, 298, 273–282. [Google Scholar] [CrossRef] [Green Version]
- Noinaj, N.; Kuszak, A.; Gumbart, J.C.; Lukacik, P.; Chang, H.; Easley, N.C.; Lithgow, T.; Buchanan, S.K. Structural insight into the biogenesis of β-barrel membrane proteins. Nat. Cell Biol. 2013, 501, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.-B.; Zahn, M.; Burmann, I.M.; Bibow, S.; Hiller, S. Sequence-Specific Solution NMR Assignments of the β-Barrel Insertase BamA to Monitor Its Conformational Ensemble at the Atomic Level. J. Am. Chem. Soc. 2018, 140, 11252–11260. [Google Scholar] [CrossRef] [PubMed]
- Woodard, J.; Srivastava, K.R.; Rahamim, G.; Grupi, A.; Hogan, S.; Witalka, D.J.; Nawrocki, G.; Haas, E.; Feig, M.; Lapidus, L.J. Intramolecular Diffusion in α-Synuclein: It Depends on How You Measure It. Biophys. J. 2018, 115, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Zahn, M.; D’Agostino, T.; Eren, E.; Baslé, A.; Ceccarelli, M.; Berg, B.V.D. Small-Molecule Transport by CarO, an Abundant Eight-Stranded β-Barrel Outer Membrane Protein from Acinetobacter baumannii. J. Mol. Biol. 2015, 427, 2329–2339. [Google Scholar] [CrossRef]
- Tajima, N.; Kawai, F.; Park, S.-Y.; Tame, J.R. A Novel Intein-Like Autoproteolytic Mechanism in Autotransporter Proteins. J. Mol. Biol. 2010, 402, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Barnard, T.J.; Dautin, N.; Lukacik, P.; Bernstein, H.D.; Buchanan, S.K. Autotransporter structure reveals intra-barrel cleavage followed by conformational changes. Nat. Struct. Mol. Biol. 2007, 14, 1214–1220. [Google Scholar] [CrossRef]
- Yonehara, R.; Yamashita, E.; Nakagawa, A. Crystal structures of OprN and OprJ, outer membrane factors of multidrug tripartite efflux pumps of P seudomonas aeruginosa. Proteins Struct. Funct. Bioinform. 2016, 84, 759–769. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef] [Green Version]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. The possible molecular basis for differentiation of COVID-19 pandemy. 2021; submitted. [Google Scholar]
- Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. Characteristics of amyloids in context of environment. 2021; submitted. [Google Scholar]
- Levitt, M. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On Information and Sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banach, M.; Konieczny, L.; Roterman, I. Ligand binding cavity encoded as a local hydrophobicity deficiency. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 91–95. [Google Scholar]
- Banach, M.; Roterman, I. Recognition of protein complexation based on hydrophobicity distribution. Bioinformation 2009, 4, 98–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Konieczny, L.; Roterman, I. Why do antifreeze proteins require a solenoid? Biochimie 2018, 144, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Adak, A.; Khan, M.R. An insight into gut microbiota and its functionalities. Cell. Mol. Life Sci. 2018, 76, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Tarasiuk, A.; Fichna, J. Gut microbiota: What is its place in pharmacology? Expert Rev. Clin. Pharmacol. 2019, 12, 921–930. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Genet. 2020, 19, 55–71. [Google Scholar] [CrossRef]
- Dygut, J.; Kalinowska, B.; Banach, M.; Piwowar, M.; Konieczny, L.; Roterman, I. Structural Interface Forms and Their Involvement in Stabilization of Multidomain Proteins or Protein Complexes. Int. J. Mol. Sci. 2016, 17, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Roterman, I. Solenoid—An amyloid under control. In From Globular Proteins to Amyloids; Roterman-Konieczna, I., Ed.; Elsevier: Amsterdam, The Netherlands; Oxford, UK; Cambridge, MA, USA, 2020; pp. 96–116. [Google Scholar]
- Roterman, I.; Banach, M.; Konieczny, L. Towards the design of anti-amyloid short peptide helices. Bioinformation 2018, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Vogt, J.; Schulz, G.E. The structure of the outer membrane protein OmpX from Escherichia coli reveals possible mechanisms of virulence. Structure 1999, 7, 1301–1309. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB–ID + Chain Length | Protein | Source Organism | Ref. |
|---|---|---|---|
| 2LHF–128 aa | Outer membr–oprh | Pseudomonas aeruginosa | [27] |
| 2JMM–165 aa | Outer membr | Escherichia coli | [28,29] |
| 6QWR–211 aa | Outer membr–alkl | Pseudomonas oleovorans | [30] |
| Oil transporter-lipid | |||
| 6QAM–211 aa | Outer membr–alkl | Pseudomonas oleovorans | [30] |
| Oil transporter-detergent | |||
| 1QJP–137 aa | Outer membr-ompa | Escherichia coli | [31] |
| 4K3C–532 aa | Outer membr-factor bama | Haemophilus ducreyi | [32] |
| 6FSU–388 aa | Outer membr-factor bama | Escherichia coli | [33] |
| 4N75–379 aa | Outer membr-biogenesis | Escherichia coli | [34] |
| 4RLC–135 aa | Outer membr-oprf | Pseudomonas aeruginosa | [35] |
| 4RL9–205 aa | Outer memb–carbapenem associated | Acinetobacter baumannii | [35] |
| 4RLB-213 aa | Outer memb–carbapenem associated | Acinetobacter baumannii | [35] |
| 3AEH–277 aa | Autotransporter-hydrolase | Escherichia coli | [36] |
| 2QOM–265 aa | Autotransporter-hydrolase | Escherichia coli | [37] |
| 5AZO–444 aa | Efflux pump–oprn | Pseudomonas aeruginosa | [38] |
| PDB-ID | Protein Characteristics | RD T–O–R | K | RD M–O–T | Length (aa) |
|---|---|---|---|---|---|
| HIGH RESISTANCE | |||||
| 2LHF | protein H (OprH) | 0.472 | 0.2 | 0.404 | 178 |
| β-barrel | 0.603 | 0.4 | 0.377 | 74 | |
| 2JMM | pr. A (OmpA)–modif. | 0.472 | 0.3 | 0.386 | 156 |
| β-barrel | 0.461 | 0.2 | 0.486 | 84 | |
| DIFFERENT EXTERNAL CONDITIONS | |||||
| 6QWR | Oil transport–lipid | 0.556 | 0.4 | 0.384 | 211 |
| β-barrel | AlkL | 0.537 | 0.4 | 0.166 | 110 |
| Loops | 0.599 | 0.7 | 0.213 | 101 | |
| 6QAM | 0.575 | 0.5 | 0.365 | 211 | |
| β-barrel | Oil transport–detergent | 0.582 | 0.4 | 0.415 | 75 |
| Loops | AlkL | 0.676 | 1.2 | 0.318 | 136 |
| HIGHER DIAMETER BARREL | |||||
| 1QJP | 0.558 | 0.4 | 0.382 | 137 | |
| β-barrel | 0.643 | 0.6 | 0.349 | 107 | |
| 6FSU | 0.664 | 0.9 | 0.310 | 388 | |
| β-barrel | 0.718 | 0.9 | 0.281 | 197 | |
| 4K3C | 0.699 | 1.2 | 0.291 | 532 | |
| β-barrel | 0.726 | 1.3 | 0.273 | 195 | |
| 4N75 | 0.727 | 1.2 | 0.261 | 379 | |
| β-barrel | 0.743 | 1.3 | 0.255 | 191 | |
| DIFFERENT RESISTANCE | |||||
| 4RL9 | Small mol. transport | 0.745 | 1.2 | 0.239 | 205 |
| β-barrel | 0.818 | 1.1 | 0.180 | 76 | |
| β-sheet | 0.736 | 1.2 | 0.262 | 46 | |
| Helix | 0.448 | 0.5 | 0.448 | 19 | |
| 4RLB | Small mol. Transport | 0.741 | 1.3 | 0.245 | 213 |
| β-barrel | 0.801 | 1.1 | 0.196 | 78 | |
| β-sheet | 0.806 | 1.8 | 0.193 | 97 | |
| Helix | 0.651 | 1.8 | 0.348 | 21 | |
| 4RLC | Small mol. Transport | 0.503 | 0.3 | 0.409 | 135 |
| β-barrel | 0.503 | 0.3 | 0.409 | 135 | |
| AUTOTRANSPORTER | |||||
| 3AEH | autotransporter | 0.707 | 1.3 | 0.289 | 277 |
| β-barrel | 0.644 | 0.7 | 0.336 | 234 | |
| 2QOM | autotransporter | 0.696 | 1.4 | 0.298 | 265 |
| β-barrel | 0.641 | 0.8 | 0.346 | 186 | |
| EFFLUX PUMP | |||||
| 5AZO | efflux pump | 0.825 | 1.6 | 0.169 | 444 |
| Helices | 0.788 | 1.2 | 0.206 | 314 | |
| β-sheet | 0.837 | 1.5 | 0.154 | 57 | |
| Structure | Average Diameter (Å) | Protein Name |
|---|---|---|
| 2HLF | 14.2 | OprH |
| 6QAM | 18.4 | AlkL |
| 6QWR | 16.3 | AlkL |
| 1QJP | 15.9 | OmpA |
| 6FSU | 31.4 | BamA |
| 4K3C | 35.2 | BamA |
| 4N75 | 32.9 | BamA |
| 4RL9 | 18.3 | CarO1 |
| 4RLB | 12.6 | CarO2 |
| 4RLC | 17.1 | OprF |
| 3AEH | 24.9 | Hbp |
| 2QOM | 24.8 | EspP |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roterman, I.; Stapor, K.; Fabian, P.; Konieczny, L. The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins. Membranes 2021, 11, 580. https://doi.org/10.3390/membranes11080580
Roterman I, Stapor K, Fabian P, Konieczny L. The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins. Membranes. 2021; 11(8):580. https://doi.org/10.3390/membranes11080580
Chicago/Turabian StyleRoterman, Irena, Katarzyna Stapor, Piotr Fabian, and Leszek Konieczny. 2021. "The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins" Membranes 11, no. 8: 580. https://doi.org/10.3390/membranes11080580
APA StyleRoterman, I., Stapor, K., Fabian, P., & Konieczny, L. (2021). The Functional Significance of Hydrophobic Residue Distribution in Bacterial Beta-Barrel Transmembrane Proteins. Membranes, 11(8), 580. https://doi.org/10.3390/membranes11080580