
A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors

 , ,
, ,
Abstract
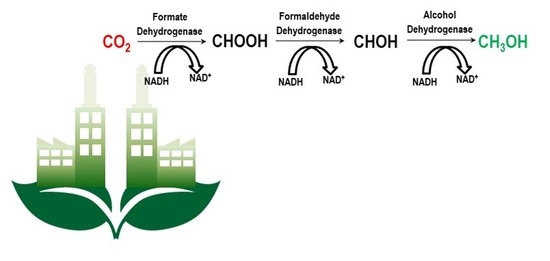
:
1. Introduction
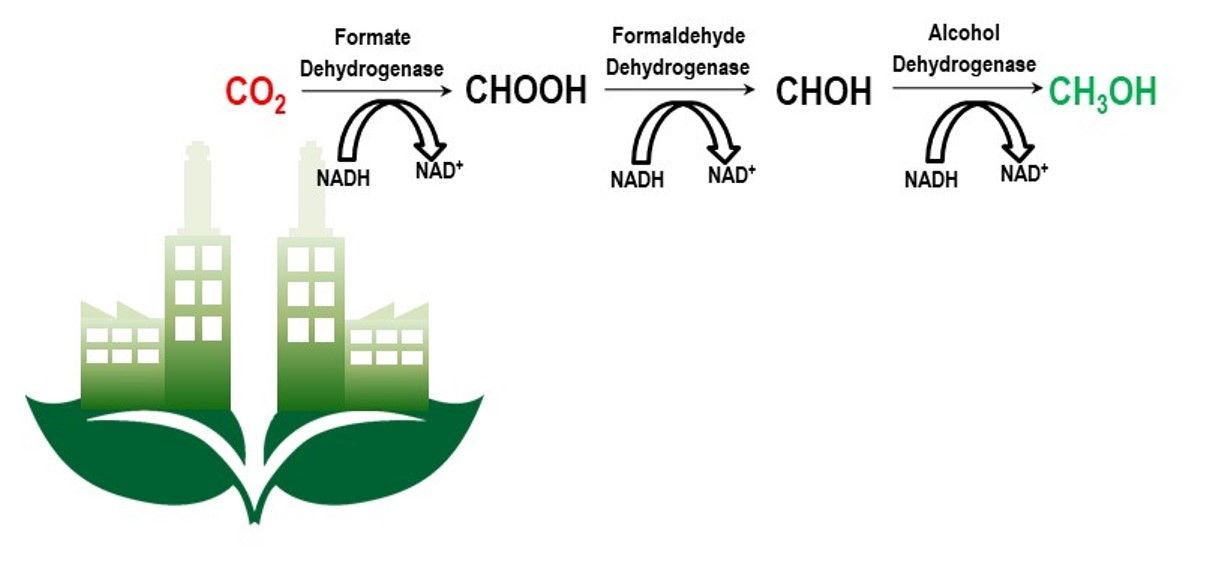
2. Formate Dehydrogenase Catalyzing CO2 Reduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FDH Sources | Classification | Efficiency towards CO2 Reduction | Ref. |
|---|---|---|---|
| FDH from Candida boidinii (CbFDH) | Metal-independent/NADH-dependent | The CbFDH was cloned and produced in E. coli BL21 (DE3). The kcat values towards CO2 reduction reported for the soluble and immobilized recombinant CbFDH in polyvinyl alcohol (PVA) hydrogel were about 0.3183 and 0.0367 s−1, respectively. | [28] |
| FDH from Myceliophthora thermophila (MtFDH) | Metal-independent/NADH-dependent | The gene of MtFDH was produced, cloned, and expressed in E. coli. The kcat obtained for the purified recombinant MtFDH when NaHCO3 was used as substrate was approximately 0.1 s−1. | [29] |
| FDH from Chaetomium thermophilum (CtFDH) | Metal-independent/NADH-dependent | CtFDH variants were expressed by transforming the plasmid libraries (G93-I94, R259, N120, and H312) into E. coli BL21 (DE3) cells. The kcat values calculated were approximately 0.0317, 0.0867, 0.055, and 0.105 s−1 for CtFDH wild type, variant A1, variant A2, and variant B, respectively. | [30] |
| FDH from Desulfovibrio desulfuricans (DdFDH) | Mo-containing/NADH-independent | FDH was purified from Desulfovibrio desulfuricans under aerobic conditions [31]. The purified DdFDH was immobilized in a cellulose membrane on the surface of a pyrolytic graphite electrode. Using direct electrochemical method in the absence of mediators, the maximum current observed (via cyclic voltammetry) was around the potential of −250 mV during the first cycle for all the three methods applied to add CO2 into the solution, indicating that DdFDH could provide high electrocatalytic activity towards CO2 reduction. | [32] |
| FDH from Escherichia coli (EcFDH) | Mo-containing/NADH-independent | E. coli was immobilized on an iron phthalocyanine (FePc)-dispersed carbide-derive carbon (CDC) anode. The FePc–CDC-based microbial electrolysis system showed maximum HCOOH production and Faradaic efficiency (FE) of approximately 30 mg/L.h and 58%, respectively, at an applied potential of −1.0 V (Ag/AgCl) and continuous flow of CO2 at 120 mg/L.h. | [26] |
| FDH from Cupriavidus necator (CnFDH/FdsABG) | Mo-containing/NADH-dependent | To express the FdsABG FDH, the pTrc12HLB-FdsGBACD vector was transformed into E. coli DH5α cells. The FdsABG provided a kcat value of 4.8 s−1 for CO2 reduction. | [23] |
| FDH from Rhodobacter aestuarii (RaFDH) | Mo-containing/NADH-dependent | RaFDH was heterologously expressed in E. coli. The recombinant RaFDH provided a kcat value of approximately 0.805 s−1. | [33] |
| FDH from Methylobacterium extorquens AM1 (FoDH1) | W-containing/NADH-dependent | FoDH1 was absorbed on Ketjen Black (KB) modified with a glassy carbon electrode (GCE). The maximum current density recorded was approximately –0.30 mA cm−2. | [27] |
| FDH from Syntrophobacter fumaroxidans (SfFDH) | W-containing/NADH-independent | The isolated SfFDH was absorbed on the pyrolytic graphite electrode surface. The maximum current density recorded was approximately 0.08 mA cm−2 at pH 5.9, initial CO2 of 10 mM and applied potential of −0.8 V. The kcat value calculated for CO2 reduction was 112 s−1. | [22,34] |
3. Types of Enzymatic Reactor Systems Available for Biocatalytic Conversion of CO2
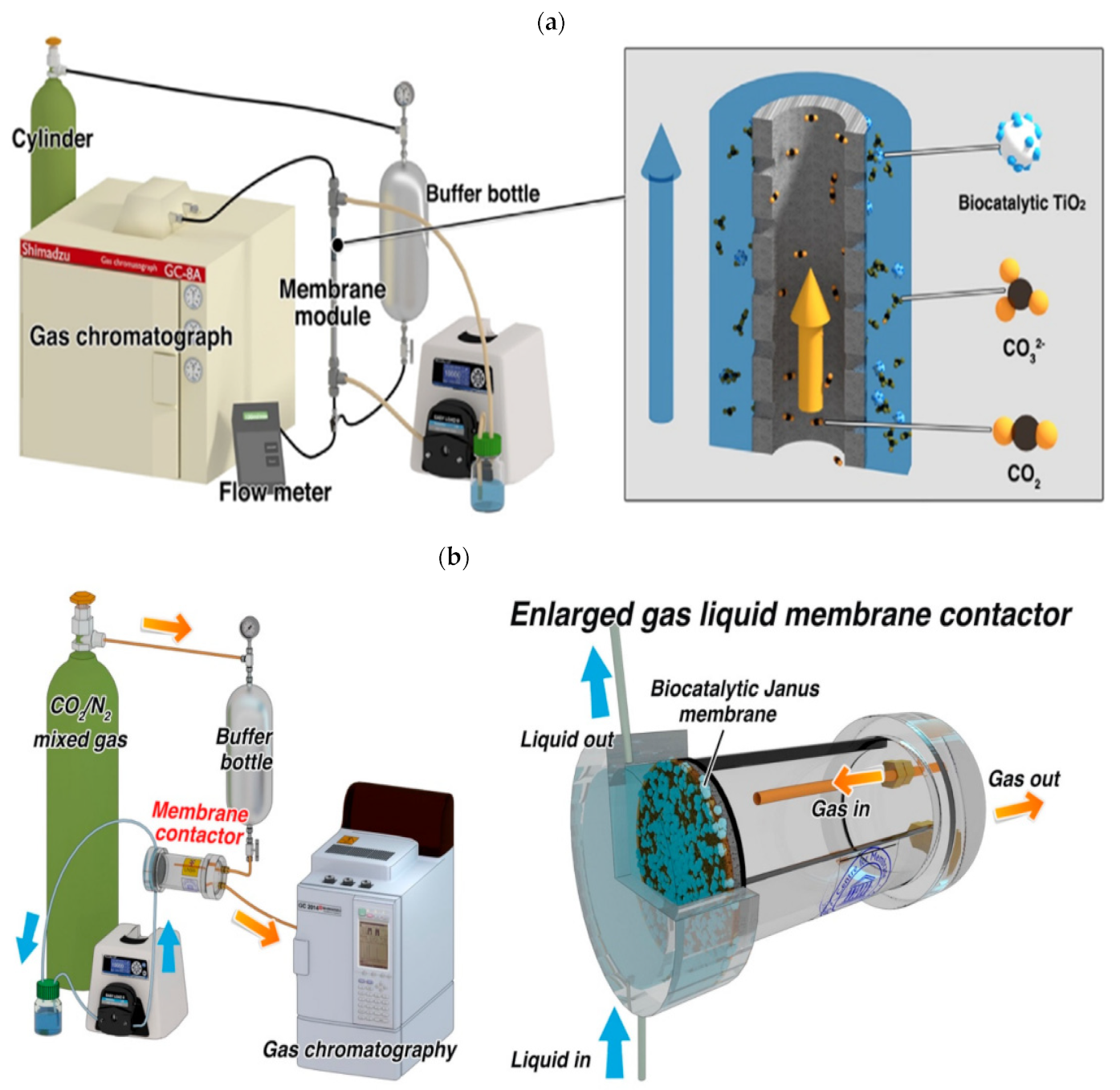
3.1. Enzyme Membrane Reactor (EMR) System
3.1.1. Types of Membrane Used in Reactor Setups
3.1.2. Enzyme Immobilization Techniques in EMRs
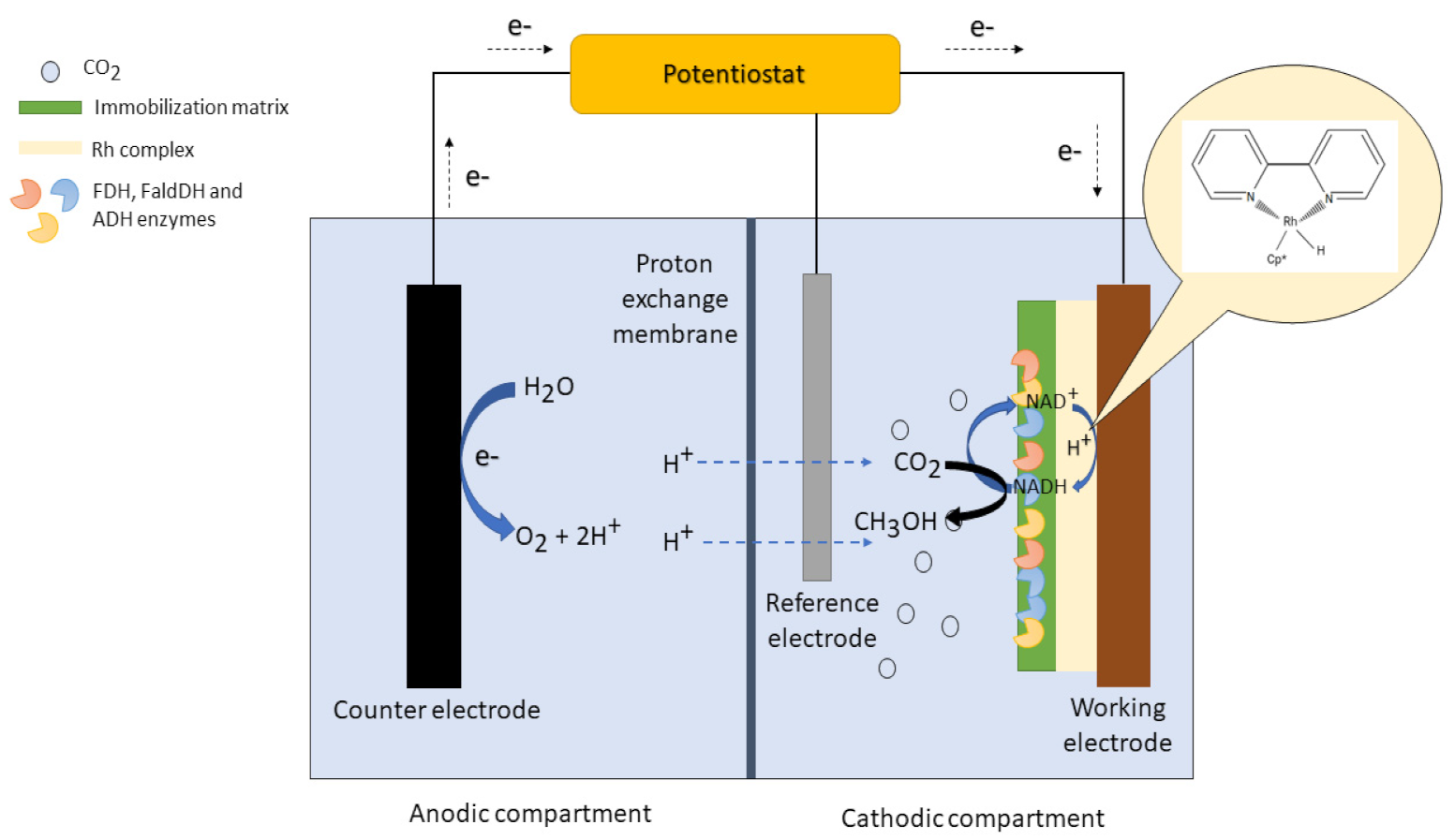
3.2. The Electrochemical Cell System
Challenges and Limitations of Biocatalytic CO2 Reduction in Electrochemical Cells
3.3. Photocatalytic Reactor System
Challenges and Limitations of Biocatalytic CO2 Reduction in Photocatalytic Reactors
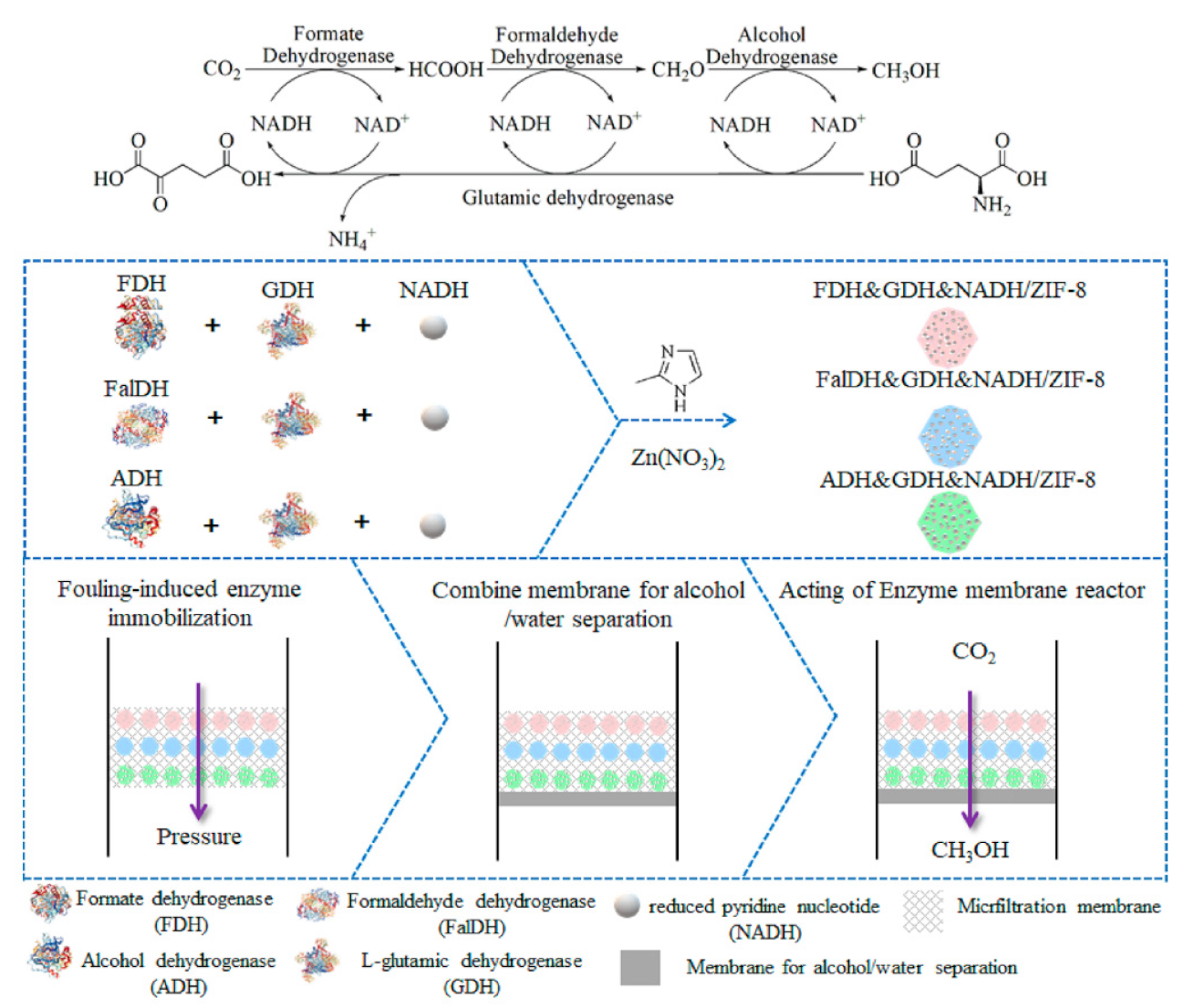
4. Performance of the Enzymatic Reactor Systems towards the Multi-Enzymatic Conversion of CO2 to CH3OH
5. Factors Affecting the Biocatalytic Productivity of Multi-Enzymatic Cascade Systems
5.1. Optimum Reaction Conditions
5.2. Immobilization of Enzymes and Cofactors
5.3. Cofactor Regeneration
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Le Quéré, C.; Jackson, R.B.; Jones, M.W.; Smith, A.J.P.; Abernethy, S.; Andrew, R.M.; De-Gol, A.J.; Willis, D.R.; Shan, Y.; Canadell, J.G.; et al. Temporary reduction in daily global CO2 emissions during the COVID-19 forced confinement. Nat. Clim. Chang. 2020, 10, 647–653. [Google Scholar] [CrossRef]
- UNEP. Emissions Gap Report 2021: The Heat Is On—A World of Climate Promises Not Yet Delivered; United Nations Environment Programme (UNEP): Nairobi, Kenya, 2021. [Google Scholar]
- Marpani, F.; Pinelo, M.; Meyer, A.S. Enzymatic conversion of CO2 to CH3OH via reverse dehydrogenase cascade biocatalysis: Quantitative comparison of efficiencies of immobilized enzyme systems. Biochem. Eng. J. 2017, 127, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Hailu, A.; Tamijani, A.A.; Mason, S.E.; Shaw, S.K. Efficient Conversion of CO2 to Formate Using Inexpensive and Easily Prepared Post-Transition Metal Alloy Catalysts. Energy Fuels 2020, 34, 3467–3476. [Google Scholar] [CrossRef]
- Schlager, S.; Dibenedetto, A.; Aresta, M.; Apaydin, D.H.; Dumitru, L.M.; Neugebauer, H.; Sariciftci, N.S. Biocatalytic and Bioelectrocatalytic Approaches for the Reduction of Carbon Dioxide using Enzymes. Energy Technol. 2017, 5, 812–821. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, C.; Peng, G.; Wang, H.; Li, X.; Zhong, L.; Wei, W.; Sun, Y. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Whang, H.S.; Lim, J.; Choi, M.S.; Lee, J.; Lee, H. Heterogeneous catalysts for catalytic CO2 conversion into value-added chemicals. BMC Chem. Eng. 2019, 1, 9. [Google Scholar] [CrossRef]
- Long, N.; Lee, J.; Koo, K.-K.; Luis, P.; Lee, M. Recent Progress and Novel Applications in Enzymatic Conversion of Carbon Dioxide. Energies 2017, 10, 473. [Google Scholar] [CrossRef] [Green Version]
- Salehmin, M.N.I.; Mohamed, M.A.; Shah, R.M.; Yunus, R.M.; Hir, Z.A.M. Application of Self-supported Materials for Photo and Photoelectrocatalysis. In Self-Standing Substrates; Rajender, I., Abdullah, B., Asiri, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; ISBN 9783030295219. [Google Scholar]
- Yaashikaa, P.R.; Kumar, P.S.; Varjani, S.J.; Saravanan, A. A review on photochemical, biochemical and electrochemical transformation of CO2 into value-added products. J. CO2 Util. 2019, 33, 131–147. [Google Scholar] [CrossRef]
- Xu, L.; Xiu, Y.; Liu, F.; Liang, Y.; Wang, S. Research progress in conversion of CO2 to valuable fuels. Molecules 2020, 25, 3653. [Google Scholar] [CrossRef]
- Obert, R.; Dave, B.C. Enzymatic conversion of carbon dioxide to methanol: Enhanced methanol production in silica sol-gel matrices. J. Am. Chem. Soc. 1999, 121, 12192–12193. [Google Scholar] [CrossRef]
- Ricca, E.; Brucher, B.; Schrittwieser, J.H. Multi-Enzymatic Cascade Reactions: Overview and Perspectives. Adv. Synth. Catal. 2011, 353, 2239–2262. [Google Scholar] [CrossRef]
- Sperl, J.M.; Sieber, V. Multienzyme Cascade Reactions—Status and Recent Advances. ACS Catal. 2018, 8, 2385–2396. [Google Scholar] [CrossRef]
- Calzadiaz-Ramirez, L.; Meyer, A.S. Formate dehydrogenases for CO2 utilization. Curr. Opin. Biotechnol. 2022, 73, 95–100. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Zhao, Z.; Liu, W. Effect of carbonic anhydrase on enzymatic conversion of CO2 to formic acid and optimization of reaction conditions. J. Mol. Catal. B Enzym. 2015, 116, 89–94. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Mateiu, R.V.; Pinelo, M. Cascade catalysis in membranes with enzyme immobilization for multi-enzymatic conversion of CO2 to methanol. New Biotechnol. 2015, 32, 319–327. [Google Scholar] [CrossRef]
- Luis, P.; Molina-Fern, C. Immobilization of carbonic anhydrase for CO2 capture and its industrial implementation: A review. J. CO2 Util. 2021, 47, 101475. [Google Scholar]
- Al-Saydeh, S.A.; Zaidi, S.J. Carbon Dioxide Conversion to Methanol: Opportunities and Fundamental Challenges. In Carbon Dioxide Chemistry, Capture and Oil Recovery; InTech: London, UK, 2018; pp. 41–61. [Google Scholar]
- Liu, Y.; Li, F.; Zhang, X.; Ji, X. Recent progress on electrochemical reduction of CO2 to methanol. Curr. Opin. Green Sustain. Chem. 2020, 23, 10–17. [Google Scholar] [CrossRef]
- Ji, X.; Kang, Y.; Fan, T.; Xiong, Q.; Zhang, S.; Tao, W.; Zhang, H. An antimonene/Cp∗Rh(phen)Cl/black phosphorus hybrid nanosheet-based Z-scheme artificial photosynthesis for enhanced photo/bio-catalytic CO2 reduction. J. Mater. Chem. A 2020, 8, 323–333. [Google Scholar] [CrossRef]
- Moon, M.; Park, G.W.; Lee, J.; Lee, J.; Min, K. Recent progress in formate dehydrogenase (FDH) as a non-photosynthetic CO2 utilizing enzyme: A short review. J. CO2 Util. 2020, 42, 101353. [Google Scholar] [CrossRef]
- Yu, X.; Niks, D.; Ge, X.; Liu, H.; Hille, R.; Mulchandani, A. Synthesis of Formate from CO2 Gas Catalyzed by an O2-Tolerant NAD-Dependent Formate Dehydrogenase and Glucose Dehydrogenase. Biochemistry 2019, 58, 1861–1868. [Google Scholar] [CrossRef]
- Yu, X.; Niks, D.; Mulchandani, A.; Hille, R. Efficient reduction of CO2 by the molybdenum-containing formate dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J. Biol. Chem. 2017, 292, 16872–16879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tunney, J.M.; McMaster, J.; Garner, C.D. Molybdenum and Tungsten Enzymes. Compr. Coord. Chem. II 2004, 8, 459–477. [Google Scholar] [CrossRef]
- Singh, S.; Noori, M.T.; Verma, N. Efficient bio-electroreduction of CO2 to formate on a iron phthalocyanine-dispersed CDC in microbial electrolysis system. Electrochim. Acta 2020, 338, 135887. [Google Scholar] [CrossRef]
- Sakai, K.; Sugimoto, Y.; Kitazumi, Y.; Shirai, O.; Takagi, K. Direct electron transfer-type bioelectrocatalytic interconversion of carbon dioxide/formate and NAD+/NADH redox couples with tungsten-containing formate dehydrogenase. Electrochim. Acta 2017, 228, 537–544. [Google Scholar] [CrossRef]
- Yildirim, D.; Alagöz, D.; Toprak, A.; Tükel, S.; Fernandez-Lafuente, R. Tuning dimeric formate dehydrogenases reduction/oxidation activities by immobilization. Process Biochem. 2019, 85, 97–105. [Google Scholar] [CrossRef]
- Altaş, N.; Aslan, A.S.; Karataş, E.; Chronopoulou, E.; Labrou, N.E. Heterologous production of extreme alkaline thermostable NAD +—Dependent formate dehydrogenase with wide-range pH activity from Myceliophthora thermophila. Process Biochem. 2017, 61, 110–118. [Google Scholar] [CrossRef]
- Çakar, M.M.; Ruupunen, J.; Mangas-Sanchez, J.; Birmingham, W.R.; Yildirim, D.; Turunen, O.; Turner, N.J.; Valjakka, J.; Binay, B. Engineered formate dehydrogenase from Chaetomium thermophilum, a promising enzymatic solution for biotechnical CO2 fixation. Biotechnol. Lett. 2020, 42, 2251–2262. [Google Scholar] [CrossRef]
- Costa, C.; Teixeira, M.; Legall, J.; Moura, J.J.G.; Moura, I. Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: Isolation and spectroscopic characterization of the active sites (heme, iron-sulfur centers and molybdenum). JBIC J. Biol. Inorg. Chem. 1997, 2, 198–208. [Google Scholar] [CrossRef]
- Cordas, C.M.; Campaniço, M.; Baptista, R.; Maia, L.B.; Moura, I. Direct electrochemical reduction of carbon dioxide by a molybdenum- containing formate dehydrogenase. J. Inorg. Biochem. 2019, 196, 110694. [Google Scholar] [CrossRef]
- Min, K.; Park, Y.; Woo, G.; Lee, J.; Moon, M.; Hyun, C.; Lee, J. Elevated conversion of CO2 to versatile formate by a newly discovered formate dehydrogenase from Rhodobacter aestuarii. Bioresour. Technol. 2020, 305, 123155. [Google Scholar] [CrossRef]
- Reda, T.; Plugge, C.M.; Abram, N.J.; Hirst, J. Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 10654–10658. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Goh, K.; Wang, R.; Bae, T.H. A review on polymer-based membranes for gas-liquid membrane contacting processes: Current challenges and future direction. Sep. Purif. Technol. 2019, 229, 115791. [Google Scholar] [CrossRef]
- Hou, J.; Zulkifli, M.Y.; Mohammad, M.; Zhang, Y.; Razmjou, A.; Chen, V. Biocatalytic gas-liquid membrane contactors for CO2 hydration with immobilized carbonic anhydrase. J. Membr. Sci. 2016, 520, 303–313. [Google Scholar] [CrossRef]
- Rasouli, H.; Iliuta, I.; Bougie, F.; Garnier, A.; Iliuta, M.C. Enzyme-immobilized flat-sheet membrane contactor for green carbon capture. Chem. Eng. J. 2021, 421, 129587. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, L.; Chen, H.; Zhang, H. Selective separation of low concentration CO2 using hydrogel immobilized CA enzyme based hollow fiber membrane reactors. Chem. Eng. Sci. 2010, 65, 3199–3207. [Google Scholar] [CrossRef]
- Gao, S.; Mohammad, M.; Yang, H.C.; Xu, J.; Liang, K.; Hou, J.; Chen, V. Janus Reactors with Highly Efficient Enzymatic CO2 Nanocascade at Air-Liquid Interface. ACS Appl. Mater. Interfaces 2017, 9, 42806–42815. [Google Scholar] [CrossRef]
- Chai, M.; Razavi Bazaz, S.; Daiyan, R.; Razmjou, A.; Ebrahimi Warkiani, M.; Amal, R.; Chen, V. Biocatalytic micromixer coated with enzyme-MOF thin film for CO2 conversion to formic acid. Chem. Eng. J. 2021, 426, 130856. [Google Scholar] [CrossRef]
- Chai, M.; Razmjou, A.; Chen, V. Metal-organic-framework protected multi-enzyme thin-film for the cascade reduction of CO2 in a gas-liquid membrane contactor. J. Membr. Sci. 2021, 623, 118986. [Google Scholar] [CrossRef]
- Zhu, D.; Ao, S.; Deng, H.; Wang, M.; Qin, C.; Zhang, J.; Jia, Y.; Ye, P.; Ni, H. Ordered Coimmobilization of a Multienzyme Cascade System with a Metal Organic Framework in a Membrane: Reduction of CO2 to Methanol. ACS Appl. Mater. Interfaces 2019, 11, 33581–33588. [Google Scholar] [CrossRef]
- Luo, J.; Marpani, F.; Brites, R.; Frederiksen, L.; Meyer, A.S.; Jonsson, G.; Pinelo, M. Directing filtration to optimize enzyme immobilization in reactive membranes. J. Membr. Sci. 2014, 459, 1–11. [Google Scholar] [CrossRef]
- Ismail, F.H.; Marpani, F.; Othman, N.H.; Nik Him, N.R. Simultaneous separation and biocatalytic conversion of formaldehyde to methanol in enzymatic membrane reactor. Chem. Eng. Commun. 2021, 208, 636–645. [Google Scholar] [CrossRef]
- Marpani, F.; Zulkifli, M.K.; Ismail, F.H.; Pauzi, S.M. Immobilization of Alcohol Dehydrogenase in Membrane: Fouling Mechanism at Different Transmembrane Pressure. J. Korean Chem. Soc. 2019, 63, 260–265. [Google Scholar]
- Ismail, F.H.; Marpani, F.; Him, N.R.N. Immobilization of Alcohol Dehydrogenase in membrane: Fouling mechanism at different enzyme concentration. In Proceedings of the IOP Conference Series: Materials Science and Engineering, Kuala Lumpur, Malaysia, 17–19 July 2019; IOP Publishing: Bristol, UK, 2020; Volume 736, pp. 1–8. [Google Scholar]
- Rahman, N.A.; Marpani, F.; Othman, N.H.; Alias, N.H.; Jai, J.; Him, N.R.N. Biocatalytic Reduction of Formaldehyde to Methanol: Effect of pH on Enzyme Immobilization and Reactive Membrane Performance. Bull. Chem. React. Eng. Catal. 2021, 16, 472–480. [Google Scholar] [CrossRef]
- Wu, Z.; Nan, Y.; Zhao, Y.; Wang, X.; Huang, S.; Shi, J. Immobilization of carbonic anhydrase for facilitated CO2 capture and separation. Chin. J. Chem. Eng. 2020, 28, 2817–2831. [Google Scholar] [CrossRef]
- Cacicedo, M.L.; Castro, M.C.; Servetas, I.; Bosnea, L.; Boura, K.; Tsafrakidou, P.; Dima, A.; Terpou, A.; Koutinas, A.; Castro, G.R. Progress in bacterial cellulose matrices for biotechnological applications. Bioresour. Technol. 2016, 213, 172–180. [Google Scholar] [CrossRef]
- Dai, Z.; Ottesen, V.; Deng, J.; Helberg, R.M.L.; Deng, L. A brief review of nanocellulose based hybrid membranes for CO2 separation. Fibers 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Zhai, T.; Wang, C.; Meng, Z.; Liu, W. Immobilization of formate dehydrogenase on polyethyleneimine modified carriers for the enhancement of catalytic performance. Catal. Commun. 2021, 149, 106259. [Google Scholar] [CrossRef]
- Sigurdardóttir, S.B.; Lehmann, J.; Ovtar, S.; Grivel, J.C.; Negra, M.D.; Kaiser, A.; Pinelo, M. Enzyme Immobilization on Inorganic Surfaces for Membrane Reactor Applications: Mass Transfer Challenges, Enzyme Leakage and Reuse of Materials. Adv. Synth. Catal. 2018, 360, 2578–2607. [Google Scholar] [CrossRef]
- Kujawa, J.; Głodek, M.; Li, G.; Al-gharabli, S.; Knozowska, K.; Kujawski, W. Highly effective enzymes immobilization on ceramics: Requirements for supports and enzymes. Sci. Total Environ. 2021, 149647. [Google Scholar] [CrossRef]
- Soltani, B.; Asghari, M. Effects of ZnO nanoparticle on the gas separation performance of polyurethane mixed matrix membrane. Membranes 2017, 7, 43. [Google Scholar] [CrossRef] [Green Version]
- Hosakun, Y.; Halász, K.; Horváth, M.; Csóka, L.; Djoković, V. ATR-FTIR study of the interaction of CO2 with bacterial cellulose-based membranes. Chem. Eng. J. 2017, 324, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.; Jiang, S.; Yan, X.; Chen, R.; Cui, H. Challenges and Opportunities: Porous Supports in Carbonic Anhydrase Immobilization. J. CO2 Util. 2020, 42, 101305. [Google Scholar] [CrossRef]
- Dong, L.; Chen, M.; Li, J.; Shi, D.; Dong, W.; Li, X.; Bai, Y. Metal-organic framework-graphene oxide composites: A facile method to highly improve the CO2 separation performance of mixed matrix membranes. J. Membr. Sci. 2016, 520, 801–811. [Google Scholar] [CrossRef]
- Rouf, S.; Greish, Y.E.; Al-zuhair, S. Immobilization of formate dehydrogenase in metal organic frameworks for enhanced conversion of carbon dioxide to formate. Chemosphere 2021, 267, 128921. [Google Scholar] [CrossRef]
- Luo, J.; Meyer, A.S.; Jonsson, G.; Pinelo, M. Fouling-induced enzyme immobilization for membrane reactors. Bioresour. Technol. 2013, 147, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Schlager, S.; Dumitru, L.M.; Haberbauer, M.; Fuchsbauer, A.; Neugebauer, H.; Hiemetsberger, D.; Wagner, A.; Portenkirchner, E.; Sariciftci, N.S. Electrochemical Reduction of Carbon Dioxide to Methanol by Direct Injection of Electrons into Immobilized Enzymes on a Modified Electrode. ChemSusChem 2016, 9, 631–635. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.Y.; Li, P.; Noh, H.; Kung, C.; Buru, C.T.; Wang, X.; Zhang, X.; Omar, K. Stabilization of Formate Dehydrogenase in a Metal-Organic Frame-work for Bioelectrocatalytic Reduction of CO2. Angew. Chem. 2019, 131, 7764–7768. [Google Scholar] [CrossRef]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Investigating the enzymatic CO2 reduction to formate with electrochemical NADH regeneration in batch and semi-continuous operations. Chem. Eng. Process.—Process Intensif. 2019, 140, 78–84. [Google Scholar] [CrossRef]
- Sreedhar, I.; Upadhyay, U.; Roy, P.; Madabusi, S.; Patel, C.M. Carbon Capture and Utilization by graphenes-path covered and ahead. J. Clean. Prod. 2021, 284, 124712. [Google Scholar] [CrossRef]
- Barin, R.; Biria, D.; Rashid-Nadimi, S.; Asadollahi, M.A. Enzymatic CO2 reduction to formate by formate dehydrogenase from Candida boidinii coupling with direct electrochemical regeneration of NADH. J. CO2 Util. 2018, 28, 117–125. [Google Scholar] [CrossRef]
- Immanuel, S.; Sivasubramanian, R.; Ahmad Dar, M.; Gul, R. Recent progress and perspectives on electrochemical regeneration of reduced nicotinamide adenine dinucleotide (NADH). Chem.—Asian J. 2020, 15, 4256–4270. [Google Scholar] [CrossRef]
- Song, H.; Ma, C.; Liu, P.; You, C.; Lin, J.; Zhu, Z. A hybrid CO2 electroreduction system mediated by enzyme-cofactor conjugates coupled with Cu nanoparticle-catalyzed cofactor regeneration. J. CO2 Util. 2019, 34, 568–575. [Google Scholar] [CrossRef]
- Ma, T.; Fan, Q.; Li, X.; Qiu, J.; Wu, T.; Sun, Z. Graphene-based materials for electrochemical CO2 reduction. J. CO2 Util. 2019, 30, 168–182. [Google Scholar] [CrossRef]
- Wu, D.; Chen, W.; Wang, X.; Fu, X.; Luo, J. Metal-support interaction enhanced electrochemical reduction of CO2 to formate between graphene and Bi nanoparticles. J. CO2 Util. 2020, 37, 353–359. [Google Scholar] [CrossRef]
- Dongare, S.; Singh, N.; Bhunia, H. Nitrogen-doped graphene supported copper nanoparticles for electrochemical reduction of CO2. J. CO2 Util. 2021, 44, 101382. [Google Scholar] [CrossRef]
- Soozanipour, A.; Taheri-Kafrani, A. Enzyme Immobilization on Functionalized Graphene Oxide Nanosheets: Efficient and Robust Biocatalysts. Methods Enzymol. 2018, 609, 371–403. [Google Scholar] [CrossRef]
- Willner, I.; Mandler, D. Enzyme-catalysed biotransformations through photochemical regeneration of nicotinamide cofactors. Enzyme Microb. Technol. 1989, 11, 467–483. [Google Scholar] [CrossRef]
- Gu, F.; Wang, Y.; Meng, Z.; Liu, W.; Qiu, L. A coupled photocatalytic/enzymatic system for sustainable conversion of CO2 to formate. Catal. Commun. 2020, 136, 105903. [Google Scholar] [CrossRef]
- Ali, A.; Tahir, M. Recent advancements in engineering approach towards design of photo-reactors for selective photocatalytic CO2 reduction to renewable fuels. J. CO2 Util. 2019, 29, 205–239. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Baran, T.; Angelini, A.; Macyk, W. An integrated photocatalytic/enzymatic system for the reduction of CO2 to methanol in bioglycerol—Water. Beilstein J. Org. Chem. 2014, 10, 2556–2565. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Su, Z.; Wang, P.; Ma, G.; Zhang, S. Tethering of nicotinamide adenine dinucleotide inside hollow nanofibers for high-yield synthesis of methanol from carbon dioxide catalyzed by coencapsulated multienzymes. ACS Nano 2015, 9, 4600–4610. [Google Scholar] [CrossRef]
- Jiang, Z.; Wu, H.; Xu, S.; Huang, S. Enzymatic conversion of carbon dioxide to methanol by dehydrogenases encapsulated in sol-gel matrix. In Utilization of Greenhouse Gases; American Chemical Society: Washington, DC, USA, 2002; Volume 47, p. 306. [Google Scholar] [CrossRef]
- Ren, S.; Wang, Z.; Bilal, M.; Feng, Y.; Jiang, Y.; Jia, S.; Cui, J. Co-immobilization multienzyme nanoreactor with co-factor regeneration for conversion of CO2. Int. J. Biol. Macromol. 2020, 155, 110–118. [Google Scholar] [CrossRef]
- Li, Y.; Wen, L.; Tan, T.; Lv, Y. Sequential Co-immobilization of Enzymes in Metal-Organic Frameworks for Efficient Biocatalytic Conversion of Adsorbed CO2 to Formate. Front. Bioeng. Biotechnol. 2019, 7, 394. [Google Scholar] [CrossRef] [Green Version]
- El-Zahab, B.; Donnelly, D.; Wang, P. Particle-Tethered NADH for Production of Methanol from CO2 Catalyzed by Coimmobilized Enzymes. J. Anat. 2007, 99, 508–514. [Google Scholar] [CrossRef]
- Cazelles, R.; Drone, J.; Fajula, F.; Ersen, O.; Moldovan, S.; Galarneau, A. Reduction of CO2 to methanol by a polyenzymatic system encapsulated in phospholipids-silica nanocapsules. New J. Chem. 2013, 37, 3721–3730. [Google Scholar] [CrossRef]
- Sun, Q.; Jiang, Y.; Jiang, Z.; Zhang, L.; Sun, X.; Li, J. Green and efficient conversion of CO2 to methanol by biomimetic coimmobilization of three dehydrogenases in protamine-templated titania. Ind. Eng. Chem. Res. 2009, 48, 4210–4215. [Google Scholar] [CrossRef]
- Wang, X.; Li, Z.; Shi, J.; Wu, H.; Jiang, Z.; Zhang, W. Bioinspired approach to multienzyme cascade system construction for efficient carbon dioxide reduction. ACS Catal. 2014, 4, 962–972. [Google Scholar] [CrossRef]
- Zhang, Z.; Muschiol, J.; Huang, Y.; Sigurdardóttir, S.B.; Von Solms, N.; Daugaard, A.E.; Wei, J.; Luo, J.; Xu, B.H.; Zhang, S.; et al. Efficient ionic liquid-based platform for multi-enzymatic conversion of carbon dioxide to methanol. Green Chem. 2018, 20, 4339–4348. [Google Scholar] [CrossRef] [Green Version]
- do Valle Gomes, M.Z.; Palmqvist, A.E.C. Influence of operating conditions and immobilization on activity of alcohol dehydrogenase for the conversion of formaldehyde to methanol. New J. Chem. 2017, 41, 11391–11397. [Google Scholar] [CrossRef]
- Marpani, F.; Sárossy, Z.; Pinelo, M.; Meyer, A.S. Kinetics based reaction optimization of enzyme catalyzed reduction of formaldehyde to methanol with synchronous cofactor regeneration. Biotechnol. Bioeng. 2017, 114, 2762–2770. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.; Huang, W.; Taghipour, F. Recent progress and perspectives in the photocatalytic CO2 reduction of Ti-oxide-based nanomaterials. Appl. Surf. Sci. 2017, 396, 1696–1711. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Zhou, R.; Liu, W. Optimization of a photoregeneration system for NADH using pristine TiO2 as a catalyst. J. Mol. Catal. B Enzym. 2017, 133, S188–S193. [Google Scholar] [CrossRef]







| Enzymatic Reactor Setup | Optimum Reactor Conditions | Immobilization Approach | Immobilization Matrix | Initial NADH Amount (mM) | Faradaic Efficiency | NADH Regeneration | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Enzyme membrane reactor | PBS, pH 7.4, 30 °C, 1 h | Encapsulation | Co-immobilized in ZIF-8 | n.a. | 460.0 | - | Co-immobilization of glutamate dehydrogenase (GluDH) and PEI | [77] |
| Free enzyme system | n.a. | 100.0 | - | |||||
| Enzyme membrane reactor | 18 mL, PBS, pH 7, 27–37 °C, 24 h | Physical adsorption | Co-immobilized in polystyrene particles | 0.05 | 50.0 | - | Co-immobilization of GDH | [79] |
| Enzymatic membrane reactor | 0.6 mL, PBS, pH 6.5, 37 °C, 3 h, 5 bar | Encapsulation | Phospholipid–silica nanocapsules (NPS) | 100 | 45.2 | - | Co-immobilizing phosphite dehydrogenase (PTDH) | [80] |
| Enzyme membrane reactor | 2 mL, Tris-HCl, pH 7, 27–37 °C, 4 h | Encapsulation | Co-immobilization in protamine-templated titania | 25 | 60.0 | - | - | [81] |
| Enzyme membrane reactor | 2 mL, PBS, pH 7 | Encapsulation | Silica sol–gel | 25 | 91.2 | - | - | [12] |
| Polyelectrolyte-doped hollow nanofibers membrane reactor | 2 mL, PBS, pH 7, 20 °C, 10 h | Encapsulation | Poly(allylamine hydrochloride) (PAH)-doped PU nanofibers | 0.2 | 103.2 | - | Co-immobilization of GluDH | [75] |
| Free enzyme system | 0.2 | 36.2 | - | |||||
| Flat-sheet polymeric membrane reactor | 4 mL, Tris-HCl, pH 7, 20 °C, 30 min, 2 bar | Free enzyme system | 50 | 3.2 | - | Co-immobilization of GluDH | [17] | |
| Fouling induced enzyme immobilization (involving entrapment and adsorption) | Co-immobilization system | 50 | 3.0 | - | ||||
| Sequential immobilization system | 50 | 4.2 | - | |||||
| Ultrathin hybrid enzyme membrane reactor | 1 mL, PBS, pH 7, 37 °C, 3 bar | Entrapment | Gelatin modified with catechol groups (GelC)–silica hybrid microcapsules | 50 | 71.6 | - | - | [82] |
| Free enzyme system | 50 | 35.5 | - | - | ||||
| Photo-enzymatic reactor | 20 mL, EDTA–NaOH buffer solution, pH 7, 37 °C, 4.5 h | Encapsulation | Polyethylene hollow fiber membrane (PE HFM) | 2 | 81.7 | - | Regenerated photochemically by utilizing TiO2 photocatalyst, EDTA as electron donor and [Cp*Rh(bpy)(H2O)]2+ as co-catalysis | [72] |
| Photocatalyic reactor | 10 mL, PBS, pH 7.0, 5 h | Physical adsorption | Antimonene (AM)–electron mediator (M, Cp*Rh(phen)Cl)–black phosphorus (BP) hybrid nanosheet (AM/M/BP HNS) | - | 89.0 | - | Regenerated photochemically by utilizing Z-scheme electron transfer in AM/M/BP HNS and TEOA as electron donor | [21] |
| Photocatalytic reactor | 10 mL, TEOS, pH 7.0, 1 h | Encapsulation | Ca alginate beads | - | n.a. | n.d. | Regenerated photochemically by utilizing [CrF5(H2O)]2−@TiO2 photocatalyst, [Cp*Rh(bpy)H2O]Cl2 as electron mediator and water (H2O) as electron donor | [74] |
| Electrochemical reactor | 25 mL per compartment cell, applied potential of −1.2 V, carbon felt as working electrode, PBS, pH 7.6, 4 h | Physcial adsorption | Alginate–silicate hybrid gel | - | n.d | 10.0 | - | [60] |
| Electrochemical H-shaped cell | 20 mL per half-cell, Cu foam electrode, Nafion 117 membrane, PBS, pH 7.0, 25 °C, 5 h | Physcial adsorption | Modified electrospun polystyrene fibers | - | n.d. | n.d. | Regenerated electrochemically by utilizing Cu foam electrode, 0.95 mM NAD+ and applying constant potential at −1.1 V | [62] |
| Electrochemical reactor | 5 mL, CuNPs/CF electrode, 0.1 M PBS, pH 6.0, 5 h | Physical adsorption | Cu nanoparticles (CuNPs) | 3 | n.d. | 22.8 | Regenerated electrochemically by utilizing CuNPs electrodeposited on CF electrode, 1.1 mM NAD+ and applied potential at −1.2 V | [66] |
| Electrochemical cell | 10 mL, Rh-FTO electrode, Tris buffer, pH 7.0, 1 h | Encapsulation | NU-1006 | - | 79.0 | n.d. | Regenerated electrochemically by utilizing Rh-FTO electrode, 1 mM NAD+ and applied potential at −1.1 V | [61] |
| Enzyme membrane reactor | 4 mL, Tris-HCl, pH 7, 30 min | Fouling-induced immobilization | Polypropylene modified cellulose membrane | 5 | 24.5 | - | Co-immobilization of glucose dehydrogenase (GDH) | [83] |
| 4 mL, mixture of choline and L-glutamic acid ([CH][Glu]) ionic liquid solution, pH 7, 30 min | 5 | 85.8 | - | |||||
| Enzyme membrane reactor | 250 mL, PBS, pH 7, 37 °C, 3 bar | Encapsulation | Silica sol–gel | 100 | 92.1 | - | - | [76] |
| Enzyme membrane reactor | 6 mL, PBS, 25 °C, 5 bar, 6 h | Encapsulation | HKUST-1@PEI(100)-MIL-101(Cr) | 0.1 | 353.9 | - | Co-immobilization of GluDH | [78] |
| Enzyme membrane reactor | 10 mL, PBS, 6 h | Fouling-induced immobilization | Ordered co-immobilization of enzymes and co-enzymes in ZIF-8@PVDF | 10 | 40.5 | - | Co-immobilization of GluDH | [42] |
| Disordered immobilization of enzymes in ZIF-8@PVDF | 10 | 19.8 | - | |||||
| Free enzymes and co-enzymes in solution | 10 | 18.0 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad Rizal Lim, F.N.; Marpani, F.; Anak Dilol, V.E.; Mohamad Pauzi, S.; Othman, N.H.; Alias, N.H.; Nik Him, N.R.; Luo, J.; Abd Rahman, N. A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors. Membranes 2022, 12, 28. https://doi.org/10.3390/membranes12010028
Ahmad Rizal Lim FN, Marpani F, Anak Dilol VE, Mohamad Pauzi S, Othman NH, Alias NH, Nik Him NR, Luo J, Abd Rahman N. A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors. Membranes. 2022; 12(1):28. https://doi.org/10.3390/membranes12010028
Chicago/Turabian StyleAhmad Rizal Lim, Fatin Nasreen, Fauziah Marpani, Victoria Eliz Anak Dilol, Syazana Mohamad Pauzi, Nur Hidayati Othman, Nur Hashimah Alias, Nik Raikhan Nik Him, Jianquan Luo, and Norazah Abd Rahman. 2022. "A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors" Membranes 12, no. 1: 28. https://doi.org/10.3390/membranes12010028
APA StyleAhmad Rizal Lim, F. N., Marpani, F., Anak Dilol, V. E., Mohamad Pauzi, S., Othman, N. H., Alias, N. H., Nik Him, N. R., Luo, J., & Abd Rahman, N. (2022). A Review on the Design and Performance of Enzyme-Aided Catalysis of Carbon Dioxide in Membrane, Electrochemical Cell and Photocatalytic Reactors. Membranes, 12(1), 28. https://doi.org/10.3390/membranes12010028