
Imaging Endocytosis Dynamics in Health and Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
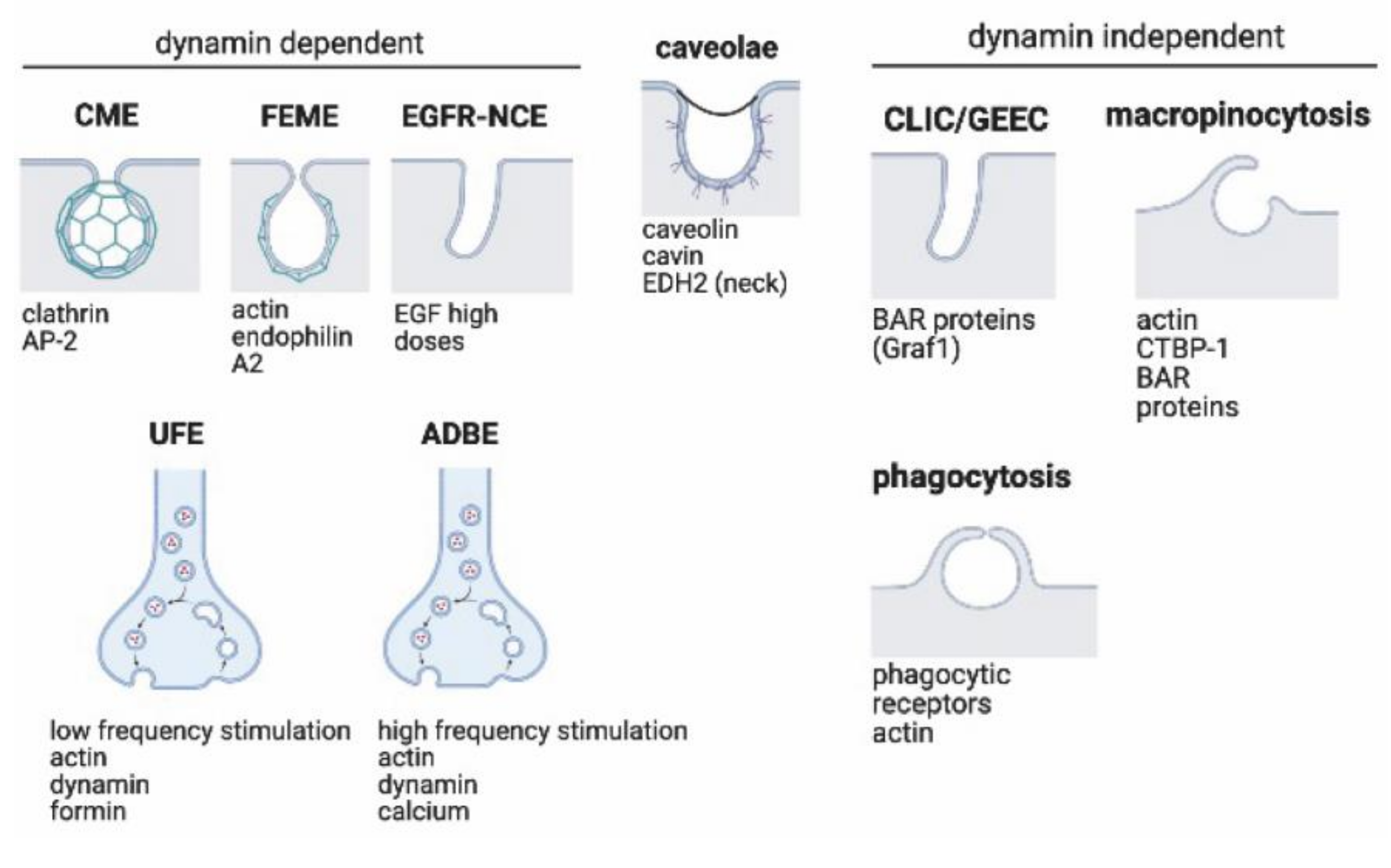
2. Endocytic Pathways
2.1. Dynamin-Dependent Pathways
2.1.1. Clathrin-Dependent Endocytosis
2.1.2. Fast Endophilin-Mediated Endocytosis (FEME)
2.1.3. EGFR Non-Clathrin Endocytosis
2.1.4. Ultrafast Endocytosis (UFE)
2.1.5. Activity-Dependent Bulk Endocytosis (ADBE)
2.2. Dynamin-Independent Pathways
2.2.1. Clathrin-Independent/Dynamin-Independent Endocytosis, CLICs/GEEC
2.2.2. IL2Rβ Uptake
2.2.3. Macropinocytosis and Phagocytosis
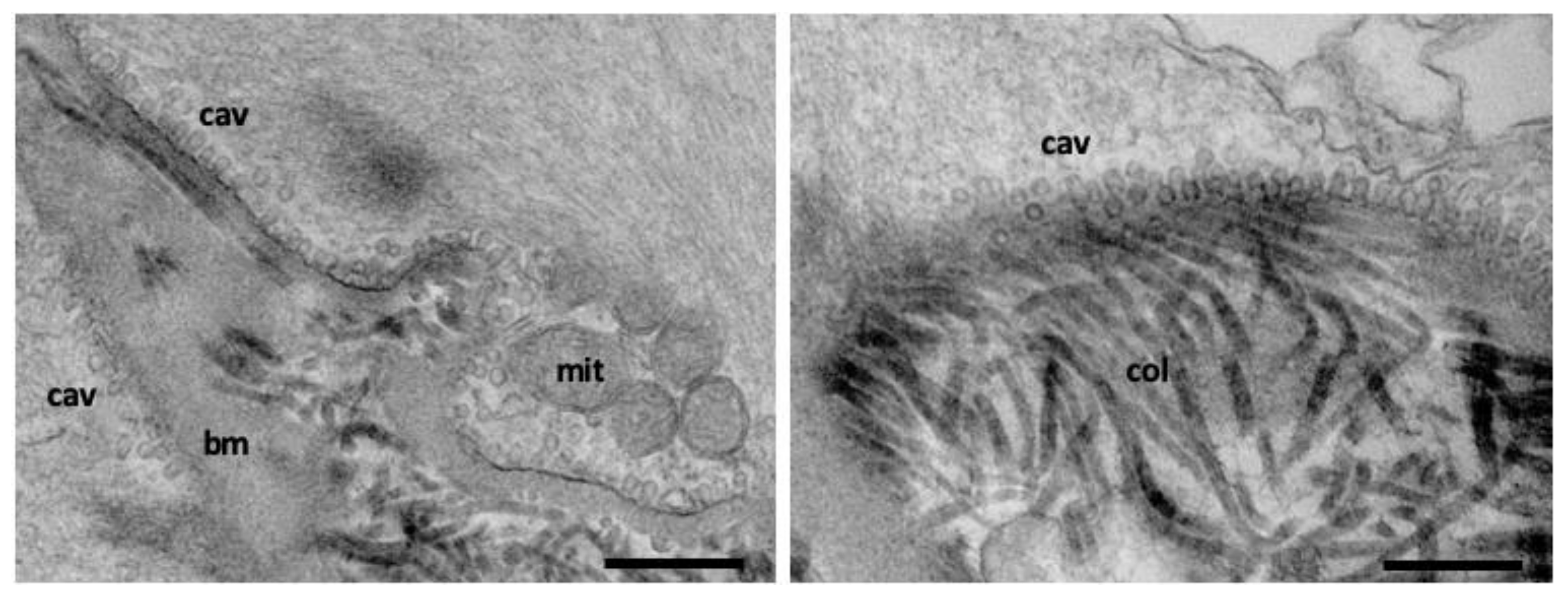
2.3. Caveolar Endocytosis
3. Diseases Caused by Defective Mechanisms of Endocytosis
3.1. Neurological Diseases
3.2. Cancer
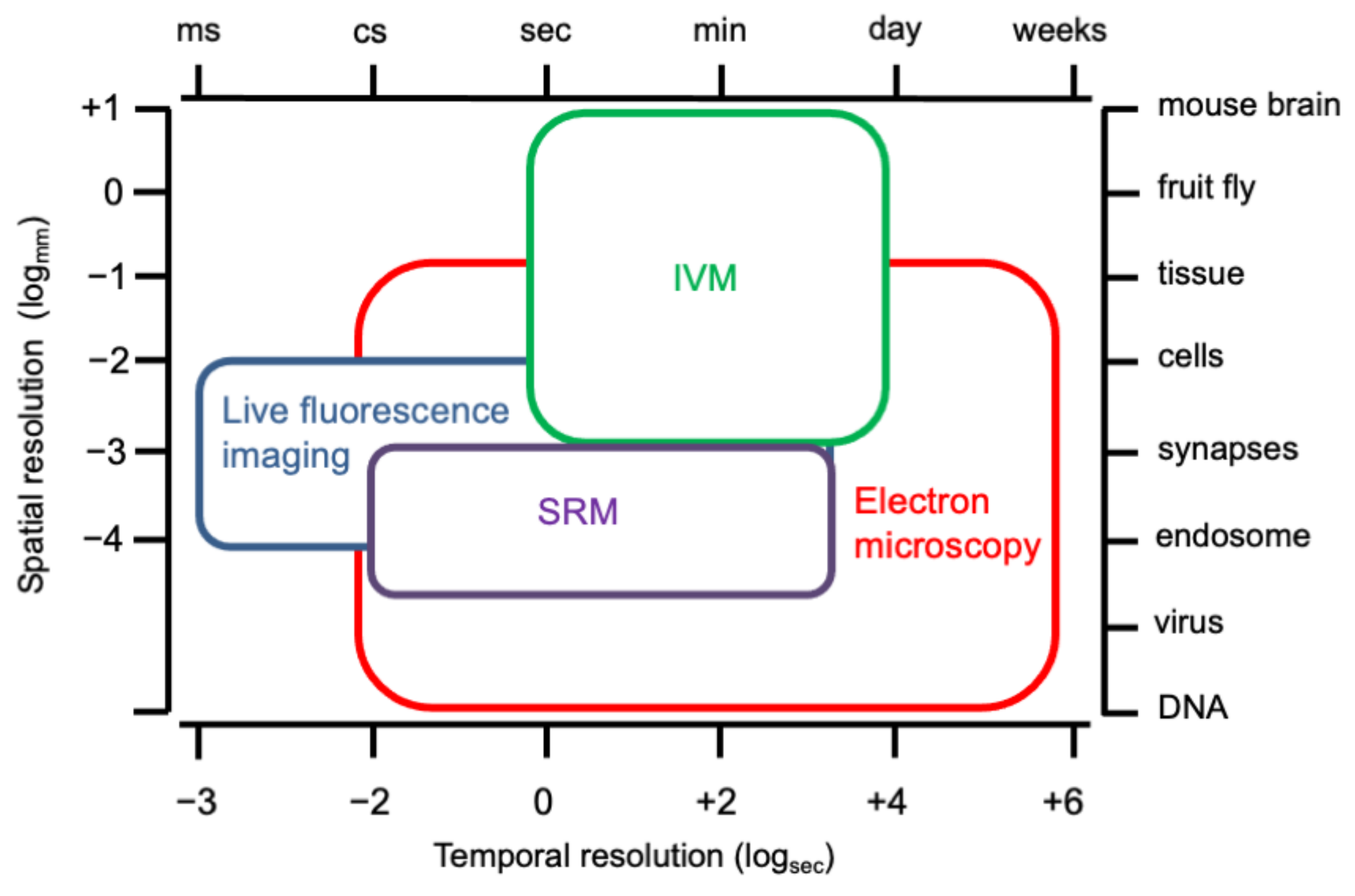
4. Current Imaging Tools and Techniques to Study Endocytosis in Living Cells and Tissues
4.1. Fluorescence Microscopy (FM)
4.1.1. Internalization Assay by Fluorescence Microscopy
4.1.2. Fluorescent Probes for Live Fluorescence Imaging
4.2. Super-Resolution Microscopy (SRM) Techniques
In Vivo Imaging and Its Applications to Visualize Membrane Trafficking
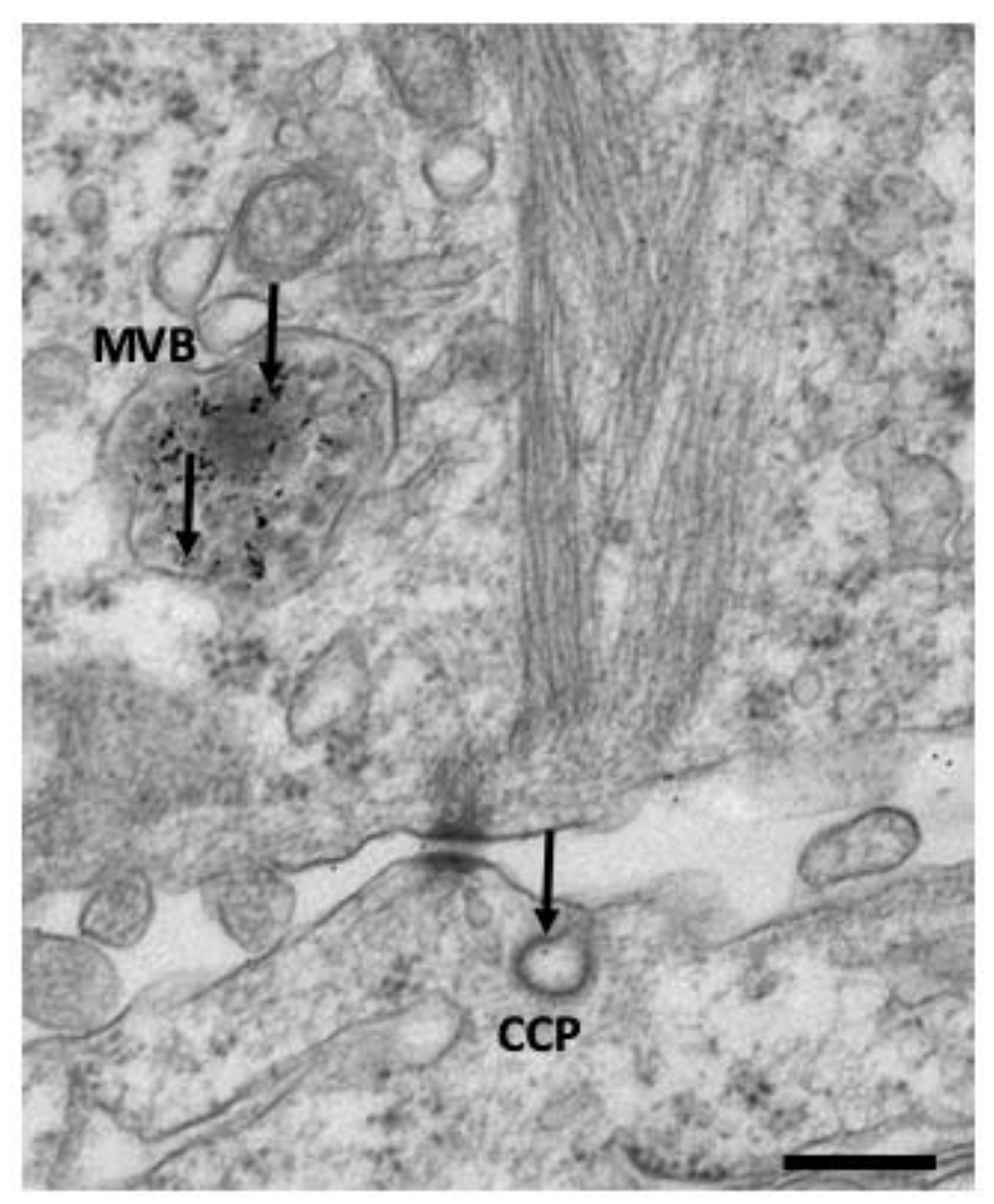
4.3. Electron Microscopy Techniques to Study Endocytosis
4.4. Correlative Microscopy Techniques to Study Membrane Trafficking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellman, I.; Yarden, Y. Endocytosis and Cancer. Cold Spring Harb. Perspect. Biol. 2013, 5, a016949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnycastle, K.; Davenport, E.C.; Cousin, M.A. Presynaptic Dysfunction in Neurodevelopmental Disorders: Insights from the Synaptic Vesicle Life Cycle. J. Neurochem. 2021, 157, 179–207. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, A.; Gieselmann, V. Lysosomal Disorders: From Storage to Cellular Damage. Biochim. Biophys. Acta Mol. Cell Res. 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarwood, R.; Hellicar, J.; Woodman, P.G.; Lowe, M. Membrane Trafficking in Health and Disease. DMM Dis. Models Mech. 2020, 13, dmm043448. [Google Scholar] [CrossRef] [PubMed]
- Kaźmierczak, Z.; Szostak-Paluch, K.; Przybyło, M.; Langner, M.; Witkiewicz, W.; Jędruchniewicz, N.; Dąbrowska, K. Endocytosis in Cellular Uptake of Drug Delivery Vectors: Molecular Aspects in Drug Development. Bioorg. Med. Chem. 2020, 28, 115556. [Google Scholar] [CrossRef]
- Steven, J.L. Metchnikoff on the Comparative Pathology of Inflammation. Glasg. Med. J. 1892, 38, 195–205. [Google Scholar]
- Jamieson, J.D. A Tribute to George E. Palade. J. Clin. Investig. 2008, 118, 3517–3518. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.G.W.; Brown, M.S.; Goldstein, J.L. Role of the Coated Endocytic Vesicle in the Uptake of Receptor-Bound Low Density Lipoprotein in Human Fibroblasts. Cell 1977, 10, 351–364. [Google Scholar] [CrossRef]
- Rosendale, M.; Perrais, D. Imaging in Focus: Imaging the Dynamics of Endocytosis. Int. J. Biochem. Cell Biol. 2017, 93, 41–45. [Google Scholar] [CrossRef]
- Baranov, M.V.; Olea, R.A.; van den Bogaart, G. Chasing Uptake: Super-Resolution Microscopy in Endocytosis and Phagocytosis. Trends Cell Biol. 2019, 29, 727–739. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular Mechanism and Physiological Functions of Clathrin-Mediated Endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517–533. [Google Scholar] [CrossRef] [PubMed]
- Benmerah, A.; Lamaze, C. Clathrin-Coated Pits: Vive La Différence? Traffic 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular Structure, Function, and Dynamics of Clathrin-Mediated Membrane Traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.M.; Baker, M.; Halebian, M.; Smith, C.J. Weak Molecular Interactions in Clathrin-Mediated Endocytosis. Front. Mol. Biosci. 2017, 4, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrlich, M.; Boll, W.; van Oijen, A.; Hariharan, R.; Chandran, K.; Nibert, M.L.; Kirchhausen, T. Endocytosis by Random Initiation and Stabilization of Clathrin-Coated Pits. Cell 2004, 118, 591–605. [Google Scholar] [CrossRef] [Green Version]
- Puthenveedu, M.A.; von Zastrow, M. Cargo Regulates Clathrin-Coated Pit Dynamics. Cell 2006, 127, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; di Fiore, P.P.; Polo, S. Clathrin-Independent Endocytosis of Ubiquitinated Cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Mamer, L.E.; Raychaudhuri, S.; Luvsanjav, D.; Eisen, J.; Trimbuch, T.; Söhl-Kielczynski, B.; Fenske, P.; Milosevic, I.; Rosenmund, C.; et al. Synaptojanin and Endophilin Mediate Neck Formation during Ultrafast Endocytosis. Neuron 2018, 98, 1184–1197.e6. [Google Scholar] [CrossRef] [Green Version]
- Cossart, P.; Helenius, A. Endocytosis of Viruses and Bacteria. Cold Spring Harb. Perspect. Biol. 2014, 6, a016972. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus Entry by Endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veiga, E.; Cossart, P. Listeria InlB Takes a Different Route to Met. Cell 2007, 130, 218–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latomanski, E.A.; Newton, H.J. Taming the Triskelion: Bacterial Manipulation of Clathrin. Microbiol. Mol. Biol. Rev. 2019, 83, e00058-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casamento, A.; Boucrot, E. Molecular Mechanism of Fast Endophilin-Mediated Endocytosis. Biochem. J. 2020, 477, 2327–2345. [Google Scholar] [CrossRef]
- Ferreira, A.P.A.; Casamento, A.; Carrillo Roas, S.; Halff, E.F.; Panambalana, J.; Subramaniam, S.; Schützenhofer, K.; Chan Wah Hak, L.; McGourty, K.; Thalassinos, K.; et al. Cdk5 and GSK3β Inhibit Fast Endophilin-Mediated Endocytosis. Nat. Commun. 2021, 12, 2424. [Google Scholar] [CrossRef]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; di Fiore, P.P. Clathrin-Mediated Internalization Is Essential for Sustained EGFR Signaling but Dispensable for Degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Algisi, V.; Nappo, G.; Conte, A.; Pascolutti, R.; Cuomo, A.; Bonaldi, T.; Argenzio, E.; Verhoef, L.G.G.C.; Maspero, E.; et al. Threshold-Controlled Ubiquitination of the EGFR Directs Receptor Fate. EMBO J. 2013, 32, 2140–2157. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.P.A.; Boucrot, E. Mechanisms of Carrier Formation during Clathrin-Independent Endocytosis. Trends Cell Biol. 2018, 28, 188–200. [Google Scholar] [CrossRef]
- Caldieri, G.; Barbieri, E.; Nappo, G.; Raimondi, A.; Bonora, M.; Conte, A.; Verhoef, L.G.G.C.; Confalonieri, S.; Malabarba, M.G.; Bianchi, F.; et al. Reticulon 3-Dependent ER-PM Contact Sites Control EGFR Non-Clathrin Endocytosis Europe PMC Funders Group. Science 2017, 356, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Boucrot, E. Fast and Ultrafast Endocytosis. Curr. Opin. Cell Biol. 2017, 47, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Rost, B.R.; Camacho-Pérez, M.; Davis, M.W.; Söhl-Kielczynski, B.; Rosenmund, C.; Jorgensen, E.M. Ultrafast Endocytosis at Mouse Hippocampal Synapses. Nature 2013, 504, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Trimbuch, T.; Camacho-Pérez, M.; Rost, B.R.; Brokowski, B.; Söhl-Kielczynski, B.; Felies, A.; Davis, M.W.; Rosenmund, C.; Jorgensen, E.M. Clathrin Regenerates Synaptic Vesicles from Endosomes. Nature 2014, 515, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Manni, M.M.; Tiberti, M.L.; Pagnotta, S.; Barelli, H.; Gautier, R.; Antonny, B. Acyl Chain Asymmetry and Polyunsaturation of Brain Phospholipids Facilitate Membrane Vesiculation without Leakage. eLife 2018, 7, e34394. [Google Scholar] [CrossRef] [PubMed]
- Chanaday, N.L.; Cousin, M.A.; Milosevic, I.; Watanabe, S.; Morgan, J.R. The Synaptic Vesicle Cycle Revisited: New Insights into the Modes and Mechanisms. J. Neurosci. 2019, 39, 8209–8216. [Google Scholar] [CrossRef] [Green Version]
- Cheung, G.; Cousin, M.A. Synaptic Vesicle Generation from Activity-Dependent Bulk Endosomes Requires Calcium and Calcineurin. J. Neurosci. 2013, 33, 3370–3379. [Google Scholar] [CrossRef] [Green Version]
- Nicholson-Fish, J.C.; Kokotos, A.C.; Gillingwater, T.H.; Smillie, K.J.; Cousin, M.A. VAMP4 Is an Essential Cargo Molecule for Activity-Dependent Bulk Endocytosis. Neuron 2015, 88, 973–984. [Google Scholar] [CrossRef] [Green Version]
- Römer, W.; Berland, L.; Chambon, V.; Gaus, K.; Windschiegl, B.; Tenza, D.; Aly, M.R.E.; Fraisier, V.; Florent, J.C.; Perrais, D.; et al. Shiga Toxin Induces Tubular Membrane Invaginations for Its Uptake into Cells. Nature 2007, 450, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, M.; Fujita, A.; Chadda, R.; Nixon, S.J.; Kurzchalia, T.V.; Sharma, D.K.; Pagano, R.E.; Hancock, J.F.; Mayor, S.; Parton, R.G. Ultrastructural Identification of Uncoated Caveolin-Independent Early Endocytic Vehicles. J. Cell Biol. 2005, 168, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Parkyn, C.J.; Vermeulen, E.G.M.; Mootoosamy, R.C.; Sunyach, C.; Jacobsen, C.; Oxvig, C.; Moestrup, S.; Liu, Q.; Bu, G.; Jen, A.; et al. LRP1 Controls Biosynthetic and Endocytic Trafficking of Neuronal Prion Protein. J. Cell Sci. 2008, 121, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnataro, D.; Caputo, A.; Casanova, P.; Puri, C.; Paladino, S.; Tivodar, S.S.; Campana, V.; Tacchetti, C.; Zurzolo, C. Lipid Rafts and Clathrin Cooperate in the Internalization of PrPC in Epithelial FRT Cells. PLoS ONE 2009, 4, e5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundmark, R.; Doherty, G.J.; Howes, M.T.; Cortese, K.; Vallis, Y.; Parton, R.G.; McMahon, H.T. The GTPase-Activating Protein GRAF1 Regulates the CLIC/GEEC Endocytic Pathway. Curr. Biol. 2008, 18, 1802–1808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howes, M.T.; Kirkham, M.; Riches, J.; Cortese, K.; Walser, P.J.; Simpson, F.; Hill, M.M.; Jones, A.; Lundmark, R.; Lindsay, M.R.; et al. Clathrin-Independent Carriers Form a High Capacity Endocytic Sorting System at the Leading Edge of Migrating Cells. J. Cell Biol. 2010, 190, 675–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sathe, M.; Muthukrishnan, G.; Rae, J.; Disanza, A.; Thattai, M.; Scita, G.; Parton, R.G.; Mayor, S. Small GTPases and BAR Domain Proteins Regulate Branched Actin Polymerisation for Clathrin and Dynamin-Independent Endocytosis. Nat. Commun. 2018, 9, 1835. [Google Scholar] [CrossRef]
- Rennick, J.J.; Johnston, A.P.R.; Parton, R.G. Key Principles and Methods for Studying the Endocytosis of Biological and Nanoparticle Therapeutics. Nat. Nanotechnol. 2021, 16, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Thottacherry, J.J.; Kosmalska, A.J.; Kumar, A.; Vishen, A.S.; Elosegui-Artola, A.; Pradhan, S.; Sharma, S.; Singh, P.P.; Guadamillas, M.C.; Chaudhary, N.; et al. Mechanochemical Feedback Control of Dynamin Independent Endocytosis Modulates Membrane Tension in Adherent Cells. Nat. Commun. 2018, 9, 4217. [Google Scholar] [CrossRef] [Green Version]
- Lamaze, C.; Dujeancourt, A.; Baba, T.; Lo, C.G.; Benmerah, A.; Dautry-Varsat, A. Interleukin 2 Receptors and Detergent-Resistant Membrane Domains Define a Clathrin-Independent Endocytic Pathway. Mol. Cell 2001, 7, 661–671. [Google Scholar] [CrossRef]
- Grassart, A.; Dujeancourt, A.; Lazarow, P.B.; Dautry-Varsat, A.; Sauvonnet, N. Clathrin-Independent Endocytosis Used by the IL-2 Receptor Is Regulated by Rac1, Pak1 and Pak2. EMBO Rep. 2008, 9, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, M.C.; Teasdale, R.D. Defining Macropinocytosis. Traffic 2009, 10, 364–371. [Google Scholar] [CrossRef]
- Lin, X.P.; Mintern, J.D.; Gleeson, P.A. Macropinocytosis in Different Cell Types: Similarities and Differences. Membranes 2020, 10, 177. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis: Our Current Understanding of a Universal Biological Process. Front. Immunol. 2020, 11, 1066. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Mg, S.; Mayor, S. Diversity of Endocytic Mechanisms 256 Endocytosis Unplugged: Multiple Ways to Enter the Cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinchen, J.M.; Ravichandran, K.S.S. Phagocytic Signaling: You Can Touch, but You Can’t Eat. Curr. Biol. 2008, 18, R521–R524. [Google Scholar] [CrossRef] [Green Version]
- Parton, R.G.; Tillu, V.; McMahon, K.A.; Collins, B.M. Key Phases in the Formation of Caveolae. Curr. Opin. Cell Biol. 2021, 71, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Kozlov, M.M.; Ariotti, N. Caveolae and Lipid Sorting: Shaping the Cellular Response to Stress. J. Cell Biol. 2020, 219, e201905071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, H.P.; Hall, T.E.; Parton, R.G. Mechanoprotection by Skeletal Muscle Caveolae. BioArchitecture 2016, 6, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelkmans, L.; Helenius, A. Endocytosis via Caveolae. Traffic 2002, 3, 311–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayer, A.; Stoeber, M.; Ritz, D.; Engel, S.; Meyer, H.H.; Helenius, A. Caveolin-1 Is Ubiquitinated and Targeted to Intralumenal Vesicles in Endolysosomes for Degradation. J. Cell Biol. 2010, 191, 615–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Wu, Y.; Xu, L.; Jiang, T.; Tang, C.; Yin, C. Caveolae-Mediated Endocytosis Drives Robust SiRNA Delivery of Polymeric Nanoparticles to Macrophages. ACS Nano 2021, 15, 8267–8282. [Google Scholar] [CrossRef] [PubMed]
- Damm, E.M.; Pelkmans, L.; Kartenbeck, J.; Mezzacasa, A.; Kurzchalia, T.; Helenius, A. Clathrin- and Caveolin-1-Independent Endocytosis: Entry of Simian Virus 40 into Cells Devoid of Caveolae. J. Cell Biol. 2005, 168, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Renard, H.F.; Boucrot, E. Unconventional Endocytic Mechanisms. Curr. Opin. Cell Biol. 2021, 71, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Wen, Z.; Gao, W.; Lin, Z.; Zhong, J.; Jiu, Y. Multifaceted Functions of Host Cell Caveolae/Caveolin-1 in Virus Infections. Viruses 2020, 12, 487. [Google Scholar] [CrossRef]
- Schreij, A.M.A.; Fon, E.A.; McPherson, P.S. Endocytic Membrane Trafficking and Neurodegenerative Disease. Cell. Mol. Life Sci. 2016, 73, 1529–1545. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.; Naj, A.C.; Beecham, G.W.; Wang, L.S.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Ertekin-Taner, N.; Fallin, M.D.; Friedland, R.; et al. Meta-Analysis Confirms CR1, CLU, and PICALM as Alzheimer Disease Risk Loci and Reveals Interactions with APOE Genotypes. Arch. Neurol. 2010, 67, 1473–1484. [Google Scholar] [CrossRef]
- Nelson, P.T.; Fardo, D.W.; Katsumata, Y. The MUC6/AP2A2 Locus and Its Relevance to Alzheimer’s Disease: A Review. J. Neuropathol. Exp. Neurol. 2021, 79, 568–584. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Pickering, E.; Liu, Y.C.; Hall, S.; Fournier, H.; Katz, E.; Dechairo, B.; John, S.; van Eerdewegh, P.; Soares, H. Meta-Analysis for Genome-Wide Association Study Identifies Multiple Variants at the BIN1 Locus Associated with Late-Onset Alzheimer’s Disease. PLoS ONE 2011, 6, e16616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common Variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP Are Associated with Alzheimer’s Disease. Nat. Genet. 2011, 43, 429–436. [Google Scholar] [CrossRef] [Green Version]
- Choudhry, H.; Aggarwal, M.; Pan, P.Y. Mini-Review: Synaptojanin 1 and Its Implications in Membrane Trafficking. Neurosci. Lett. 2021, 765, 136288. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A. Amyloid Precursor Protein & Endosomal-Lysosomal Dysfunction in Alzheimer’s Disease: Inseparable Partners in a Multifactorial Disease. FASEB J. 2017, 31, 2729–2743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagle, M.W.; Latourelle, J.C.; Labadorf, A.; Dumitriu, A.; Hadzi, T.C.; Beach, T.G.; Myers, R.H. The 4p16.3 Parkinson Disease Risk Locus Is Associated with GAK Expression and Genes Involved with the Synaptic Vesicle Membrane. PLoS ONE 2016, 11, e0160925. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Quadri, M.; Fang, M.; Rood, J.P.M.A.; Saute, J.A.; Chien, H.F.; Bouwkamp, C.G.; Graafland, J.; Minneboo, M.; Breedveld, G.J.; et al. DNAJC6 Mutations Associated with Early-Onset Parkinson’s Disease. Ann. Neurol. 2016, 79, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.E.; Karkheiran, S.; Powell, J.C.; Cao, M.; Makarov, V.; Darvish, H.; di Paolo, G.; Walker, R.H.; Shahidi, G.A.; Buxbaum, J.D.; et al. The Sac1 Domain of SYNJ1 Identified Mutated in a Family with Early-Onset Progressive Parkinsonism with Generalized Seizures. Hum. Mutat. 2013, 34, 1200–1207. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.; Spannl, S.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin Regulate Stress-Activated Cytoprotective Signaling on Mitochondria. eLife 2018, 7, e32282. [Google Scholar] [CrossRef] [PubMed]
- Kononenko, N.L.; Haucke, V. Neuronal Functions of Clathrin-Associated Endocytic Sorting Adaptors—From Molecules to Disease. Neuroforum 2020, 26, 209–217. [Google Scholar] [CrossRef]
- Nesbit, M.A.; Hannan, F.M.; Howles, S.A.; Reed, A.A.C.; Cranston, T.; Thakker, C.E.; Gregory, L.; Rimmer, A.J.; Rust, N.; Graham, U.; et al. Mutations in AP2S1 Cause Familial Hypocalciuric Hypercalcemia Type 3. Nat. Genet. 2013, 45, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, I.; Lopez-Hernandez, T.; Shor, O.; Galer, P.; Ganesan, S.; Pendziwiat, M.; Rademacher, A.; Ellis, C.A.; Hümpfer, N.; Schwarz, N.; et al. A Recurrent Missense Variant in AP2M1 Impairs Clathrin-Mediated Endocytosis and Causes Developmental and Epileptic Encephalopathy. Am. J. Hum. Genet. 2019, 104, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Steeg, P.S. Endocytosis: A Pivotal Pathway for Regulating Metastasis. Br. J. Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef]
- Hynes, N.E.; MacDonald, G. ErbB Receptors and Signaling Pathways in Cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef]
- Parachoniak, C.A.; Luo, Y.; Abella, J.V.; Keen, J.H.; Park, M. GGA3 Functions as a Switch to Promote Met Receptor Recycling, Essential for Sustained ERK and Cell Migration. Dev. Cell 2011, 20, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Lanzetti, L.; Scita, G.; di Fiore, P.P. Endocytosis in the Context-Dependent Regulation of Individual and Collective Cell Properties. Nat. Rev. Mol. Cell Biol. 2021, 22, 625–643. [Google Scholar] [CrossRef]
- Parachoniak, C.A.; Park, M. Dynamics of Receptor Trafficking in Tumorigenicity. Trends Cell Biol. 2012, 22, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Tretiakova, M.S.; Mackinnon, A.C.; Ramnath, N.; Johnson, C.; Dietrich, S.; Seiwert, T.; Christensen, J.G.; Jagadeeswaran, R.; Krausz, T.; et al. Expression and Mutational Analysis of MET in Human Solid Cancers. Genes Chromosomes Cancer 2008, 47, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.P.; Hastings, J.F.; Han, J.Z.R.; Croucher, D.R. The Under-Appreciated Promiscuity of the Epidermal Growth Factor Receptor Family. Front. Cell Dev. Biol. 2016, 4, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, C.D.; de Mazière, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and Sorting of ErbB2 and the Site of Action of Cancer Therapeutics Trastuzumab and Geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef] [PubMed]
- Castagnola, P.; Bellese, G.; Birocchi, F.; Gagliani, M.C.; Tacchetti, C.; Cortese, K. Identification of an HSP90 Modulated Multi-Step Process for ERBB2 Degradation in Breast Cancer Cells. Oncotarget 2016, 7, 85411–85429. [Google Scholar] [CrossRef] [Green Version]
- Worthylake, R.; Opresko, L.K.; Wiley, H.S. ErbB-2 Amplification Inhibits down-Regulation and Induces Constitutive Activation of Both ErbB-2 and Epidermal Growth Factor Receptors. J. Biol. Chem. 1999, 274, 8865–8874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust Detection of EGFR Copy Number Changes and EGFR Variant III: Technical Aspects and Relevance for Glioma Diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII Escapes Down-Regulation Due to Impaired Internalization and Sorting to Lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Renzo, M.F.; Olivero, M.; Martone, T.; Maffe, A.; Maggiora, P.; de Stefani, A.; Valente, G.; Giordano, S.; Cortesina, G.; Comoglio, P.M. Somatic Mutations of the MET Oncogene Are Selected during Metastatic Spread of Human HNSC Carcinomas. Oncogene 2000, 19, 1547–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Steen, N.; Giovannetti, E.; Pauwels, P.; Peters, G.J.; Hong, D.S.; Cappuzzo, F.; Hirsch, F.R.; Rolfo, C. CMET Exon 14 Skipping: From the Structure to the Clinic. J. Thorac. Oncol. 2016, 11, 1423–1432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF Receptor Gene Mutations Are Common in Lung Cancers from “Never Smokers” and Are Associated with Sensitivity of Tumors to Gefitinib and Erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, T.; Bilder, D. At the Crossroads of Polarity, Proliferation and Apoptosis: The Use of Drosophila to Unravel the Multifaceted Role of Endocytosis in Tumor Suppression. Mol. Oncol. 2009, 3, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Morishige, M.; Hashimoto, S.; Ogawa, E.; Toda, Y.; Kotani, H.; Hirose, M.; Wei, S.; Hashimoto, A.; Yamada, A.; Yano, H.; et al. GEP100 Links Epidermal Growth Factor Receptor Signalling to Arf6 Activation to Induce Breast Cancer Invasion. Nat. Cell Biol. 2008, 10, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Menju, T.; Hashimoto, S.; Hashimoto, A.; Otsuka, Y.; Handa, H.; Ogawa, E.; Toda, Y.; Wada, H.; Date, H.; Sabe, H. Engagement of Overexpressed Her2 with GEP100 Induces Autonomous Invasive Activities and Provides a Biomarker for Metastases of Lung Adenocarcinoma. PLoS ONE 2011, 6, e25301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, R. Imaging the Dynamics of Endocytosis in Live Mammalian Tissues. Cold Spring Harb. Perspect. Biol. 2014, 6, a017012. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Evangelinos, M.; Wernet, V.; Eckert, A.F.; Ishitsuka, Y.; Fischer, R.; Nienhaus, G.U.; Takeshita, N. Superresolution and Pulse-Chase Imaging Reveal the Role of Vesicle Transport in Polar Growth of Fungal Cells. Sci. Adv. 2018, 4, e1701798. [Google Scholar] [CrossRef] [Green Version]
- Klumperman, J.; Raposo, G. The Complex Ultrastructure of the Endolysosomal System. Cold Spring Harb. Perspect. Biol. 2014, 6, a016857. [Google Scholar] [CrossRef] [Green Version]
- Martineau, M.; Somasundaram, A.; Grimm, J.B.; Gruber, T.D.; Choquet, D.; Taraska, J.W.; Lavis, L.D.; Perrais, D. Semisynthetic Fluorescent PH Sensors for Imaging Exocytosis and Endocytosis. Nat. Commun. 2017, 8, 1412. [Google Scholar] [CrossRef] [Green Version]
- Royle, S.J.; Granseth, B.; Odermatt, B.; Derevier, A.; Lagnado, L. Imaging PHluorin-Based Probes at Hippocampal Synapses. Methods Mol. Biol. 2008, 457, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adie, E.J.; Kalinka, S.; Smith, L.; Francis, M.J.; Marenghi, A.; Cooper, M.E.; Briggs, M.; Michael, N.P.; Milligan, G.; Game, S. A PH-Sensitive Fluor, CypHerTM 5, Used to Monitor Agonist-Induced G Protein-Coupled Receptor Internalization in Live Cells. BioTechniques 2002, 33, 1152–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Sinha, R.; Martineau, M.; Kahms, M.; Klingauf, J. A Common Origin of Synaptic Vesicles Undergoing Evoked and Spontaneous Fusion. Nat. Neurosci. 2010, 13, 1451–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierzyńska-Mach, A.; Janowski, P.A.; Dobrucki, J.W. Evaluation of Acridine Orange, LysoTracker Red, and Quinacrine as Fluorescent Probes for Long-Term Tracking of Acidic Vesicles. Cytom. Part A 2014, 85, 729–737. [Google Scholar] [CrossRef]
- Asanov, A.; Zepeda, A.; Vaca, L. A Novel Form of Total Internal Reflection Fluorescence Microscopy (LG-TIRFM) Reveals Different and Independent Lipid Raft Domains in Living Cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2010, 1801, 147–155. [Google Scholar] [CrossRef]
- Wang, J.; Richards, D.A. Segregation of PIP2 and PIP3 into Distinct Nanoscale Regions within the Plasma Membrane. Biol. Open 2012, 1, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Rappoport, J.Z.; Kemal, S.; Benmerah, A.; Simon, S.M. Dynamics of Clathrin and Adaptor Proteins during Endocytosis. Am. J. Physiol.-Cell Physiol. 2006, 291, C1072–C1081. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Marsland, R.; Upadhyayula, S.; Song, E.; Dang, S.; Capraro, B.R.; Wang, W.; Skillern, W.; Gaudin, R.; Ma, M.; et al. Dynamics of Phosphoinositide Conversion in Clathrin-Mediated Endocytic Traffic. Nature 2017, 552, 410–414. [Google Scholar] [CrossRef]
- Nozumi, M.; Nakatsu, F.; Katoh, K.; Igarashi, M. Coordinated Movement of Vesicles and Actin Bundles during Nerve Growth Revealed by Superresolution Microscopy. Cell Rep. 2017, 18, 2203–2216. [Google Scholar] [CrossRef] [Green Version]
- Kusumi, A.; Suzuki, K.G.N.; Kasai, R.S.; Ritchie, K.; Fujiwara, T.K. Hierarchical Mesoscale Domain Organization of the Plasma Membrane. Trends Biochem. Sci. 2011, 36, 604–615. [Google Scholar] [CrossRef]
- Freeman, S.A.; Goyette, J.; Furuya, W.; Woods, E.C.; Bertozzi, C.R.; Bergmeier, W.; Hinz, B.; van der Merwe, P.A.; Das, R.; Grinstein, S. Integrins Form an Expanding Diffusional Barrier That Coordinates Phagocytosis. Cell 2016, 164, 128–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sochacki, K.A.; Dickey, A.M.; Strub, M.P.; Taraska, J.W. Endocytic Proteins Are Partitioned at the Edge of the Clathrin Lattice in Mammalian Cells. Nat. Cell Biol. 2017, 19, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Picco, A.; Mund, M.; Ries, J.; Nédélec, F.; Kaksonen, M. Visualizing the Functional Architecture of the Endocytic Machinery. eLife 2015, 2015, e04535. [Google Scholar] [CrossRef]
- Mund, M.; van der Beek, J.A.; Deschamps, J.; Dmitrieff, S.; Hoess, P.; Monster, J.L.; Picco, A.; Nédélec, F.; Kaksonen, M.; Ries, J. Systematic Nanoscale Analysis of Endocytosis Links Efficient Vesicle Formation to Patterned Actin Nucleation. Cell 2018, 174, 884–896.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangadharan, V.; Nohe, A.; Caplan, J.; Czymmek, K.; Duncan, R.L. Caveolin-1 Regulates P2X7 Receptor Signaling in Osteoblasts. Am. J. Physiol.-Cell Physiol. 2015, 308, C41–C50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.; Baddeley, D.; Bushong, E.A.; Yu, Z.; Ellisman, M.H.; Hoshijima, M.; Soeller, C. Nanoscale Distribution of Ryanodine Receptors and Caveolin-3 in Mouse Ventricular Myocytes: Dilation of T-Tubules near Junctions. Biophys. J. 2013, 104, L22–L24. [Google Scholar] [CrossRef] [Green Version]
- Kreft, M.; Jorgačevski, J.; Stenovec, M.; Zorec, R. Ångstrom-Size Exocytotic Fusion Pore: Implications for Pituitary Hormone Secretion. Mol. Cell. Endocrinol. 2018, 463, 65–71. [Google Scholar] [CrossRef]
- Yu, C.C.; Barry, N.C.; Wassie, A.T.; Sinha, A.; Bhattacharya, A.; Asano, S.; Zhang, C.; Chen, F.; Hobert, O.; Goodman, M.B.; et al. Expansion Microscopy of c. Elegans. eLife 2020, 9, e46249. [Google Scholar] [CrossRef]
- Eilers, Y.; Ta, H.; Gwosch, K.C.; Balzarotti, F.; Hell, S.W. MINFLUX Monitors Rapid Molecular Jumps with Superior Spatiotemporal Resolution. Proc. Natl. Acad. Sci. USA 2018, 115, 6117–6122. [Google Scholar] [CrossRef] [Green Version]
- Pleiner, T.; Bates, M.; Trakhanov, S.; Lee, C.T.; Schliep, J.E.; Chug, H.; Böhning, M.; Stark, H.; Urlaub, H.; Görlich, D. Nanobodies: Site-Specific Labeling for Super-Resolution Imaging, Rapid Epitope- Mapping and Native Protein Complex Isolation. eLife 2015, 4, e11349. [Google Scholar] [CrossRef] [Green Version]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-Resolution Microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Gyparaki, M.T.; Beliu, G.; Schueder, F.; Griffié, J.; Manley, S.; Jungmann, R.; Sauer, M.; Lakadamyali, M.; Zimmer, C. Single-Molecule Localization Microscopy. Nat. Rev. Methods Primers 2021, 1, 39. [Google Scholar] [CrossRef]
- Schlichthaerle, T.; Eklund, A.S.; Schueder, F.; Strauss, M.T.; Tiede, C.; Curd, A.; Ries, J.; Peckham, M.; Tomlinson, D.C.; Jungmann, R. Site-Specific Labeling of Affimers for DNA-PAINT Microscopy. Angew. Chem. Int. Ed. 2018, 57, 11060–11063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strauss, S.; Nickels, P.C.; Strauss, M.T.; Sabinina, V.J.; Carter, J.D.; Gupta, S.; Janjic, N.; Jungmann, R. Modified Aptamers Enable Quantitative Sub-10-Nm Cellular DNA- PAINT Imaging. Nat. Methods 2019, 15, 685–688. [Google Scholar] [CrossRef]
- Yang, B.; Przybilla, F.; Mestre, M.; Trebbia, J.-B.; Lounis, B. Large Parallelization of STED Nanoscopy Using Optical Lattices. Opt. Express 2014, 22, 5581. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Zhang, C.; Yu, B.; Lin, D.; Qu, J. Super-Resolution Microscopy: Shedding New Light on In Vivo Imaging. Front. Chem. 2021, 9, 746900. [Google Scholar] [CrossRef]
- Booth, M.J. Adaptive Optical Microscopy: The Ongoing Quest for a Perfect Image. Light Sci. Appl. 2014, 3, 165. [Google Scholar] [CrossRef]
- Ebrahim, S.; Weigert, R. Intravital Microscopy in Mammalian Multicellular Organisms. Curr. Opin. Cell Biol. 2019, 59, 97–103. [Google Scholar] [CrossRef]
- Masedunskas, A.; Milberg, O.; Porat-Shliom, N.; Sramkova, M.; Wigand, T.; Amornphimoltham, P.; Weigert, R. Intravital Microscopy: A Practical Guide on Imaging Intracellular Structures in Live Animals. BioArchitecture 2012, 2, 143–157. [Google Scholar] [CrossRef]
- Bhirde, A.A.; Patel, V.; Gavard, J.; Zhang, G.; Sousa, A.A.; Masedunskas, A.; Leapman, R.D.; Weigert, R.; Gutkind, J.S.; Rusling, J.F. Targeted Killing of Cancer Cells in Vivo and in Vitro with EGF-Directed Carbon Nanotube-Based Drug Delivery. ACS Nano 2009, 3, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Amornphimoltham, P.; Rechache, K.; Thompson, J.; Masedunskas, A.; Leelahavanichkul, K.; Patel, V.; Molinolo, A.; Gutkind, J.S.; Weigert, R. Rab25 Regulates Invasion and Metastasis in Head and Neck Cancer. Clin. Cancer Res. 2013, 19, 1375–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llewellyn, M.E.; Barretto, R.P.J.; Delp, S.L.; Schnitzer, M.J. Minimally Invasive High-Speed Imaging of Sarcomere Contractile Dynamics in Mice and Humans. Nature 2008, 454, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Barretto, R.P.J.; Messerschmidt, B.; Schnitzer, M.J. In Vivo Fluorescence Imaging with High-Resolution Microlenses. Nat. Methods 2009, 6, 511–512. [Google Scholar] [CrossRef]
- Cao, L.; Kobayakawa, S.; Yoshiki, A.; Abe, K. High Resolution Intravital Imaging of Subcellular Structures of Mouse Abdominal Organs Using a Microstage Device. PLoS ONE 2012, 7, 33876. [Google Scholar] [CrossRef] [PubMed]
- Ritsma, L.; Steller, E.J.A.; Ellenbroek, S.I.J.; Kranenburg, O.; Borel Rinkes, I.H.M.; van Rheenen, J. Surgical Implantation of an Abdominal Imaging Window for Intravital Microscopy. Nat. Protoc. 2013, 8, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ROTH, T.F.; PORTER, K.R. Yolk Protein Uptake in the Oocyte of the Mosquito Aedes Aegypti. L. J. Cell Biol. 1964, 20, 313–332. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.; Mantell, J.; Carter, D.; Tilly, G.; Verkade, P. Studying Intracellular Transport Using High-Pressure Freezing and Correlative Light Electron Microscopy. Semin. Cell Dev. Biol. 2009, 20, 910–919. [Google Scholar] [CrossRef]
- Cortese, K.; Howes, M.T.; Lundmark, R.; Tagliatti, E.; Bagnato, P.; Petrelli, A.; Bono, M.; McMahon, H.T.; Parton, R.G.; Tacchetti, C. The HSP90 Inhibitor Geldanamycin Perturbs Endosomal Structure and Drives Recycling ErbB2 and Transferrin to Modified MVBs/Lysosomal Compartments. Mol. Biol. Cell 2013, 24, 129–144. [Google Scholar] [CrossRef]
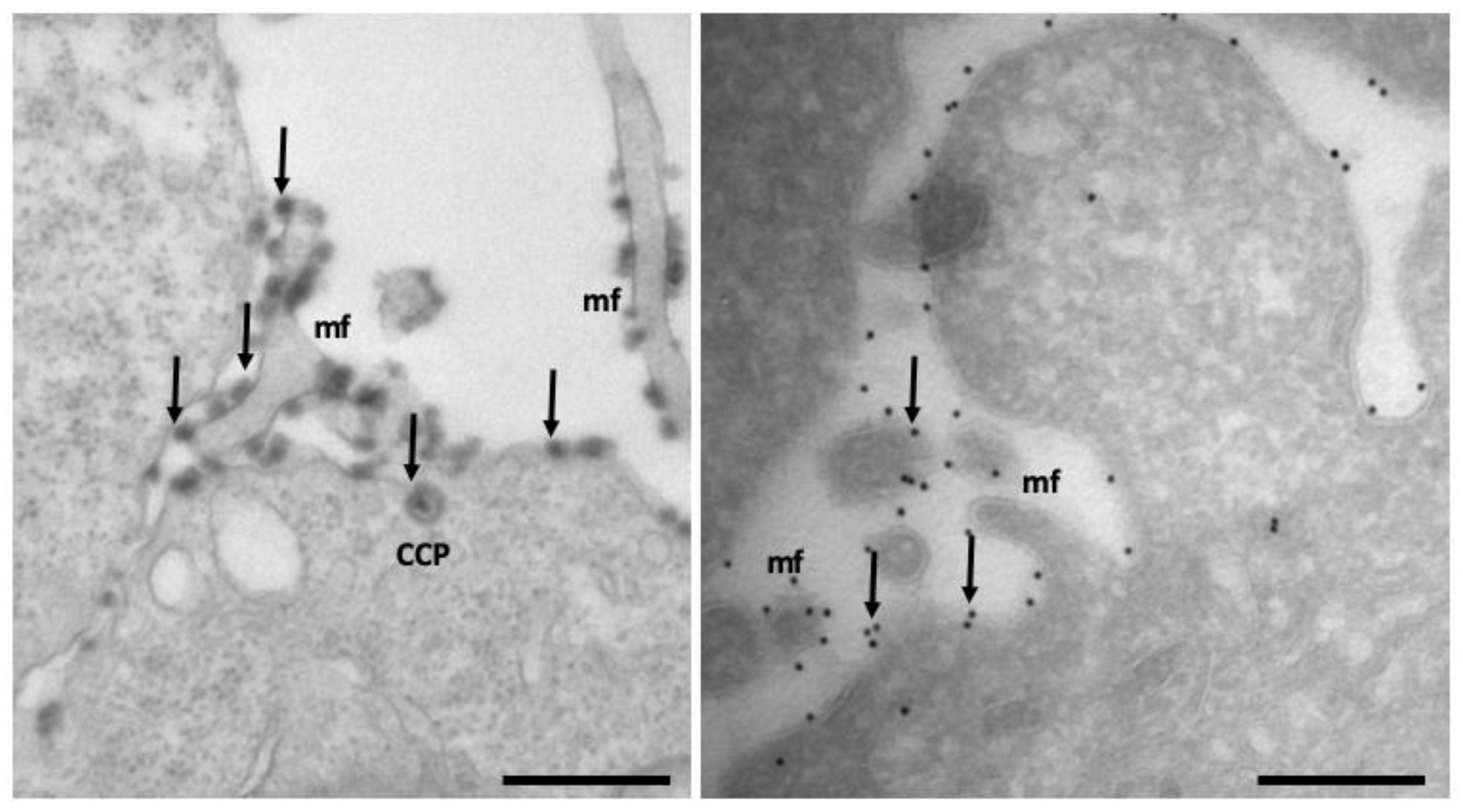
- Slot, J.W.; Geuze, H.J. Sizing of Protein A-Colloidal Gold Probes for Immunoelectron Microscopy. J. Cell Biol. 1981, 90, 533–536. [Google Scholar] [CrossRef] [PubMed]
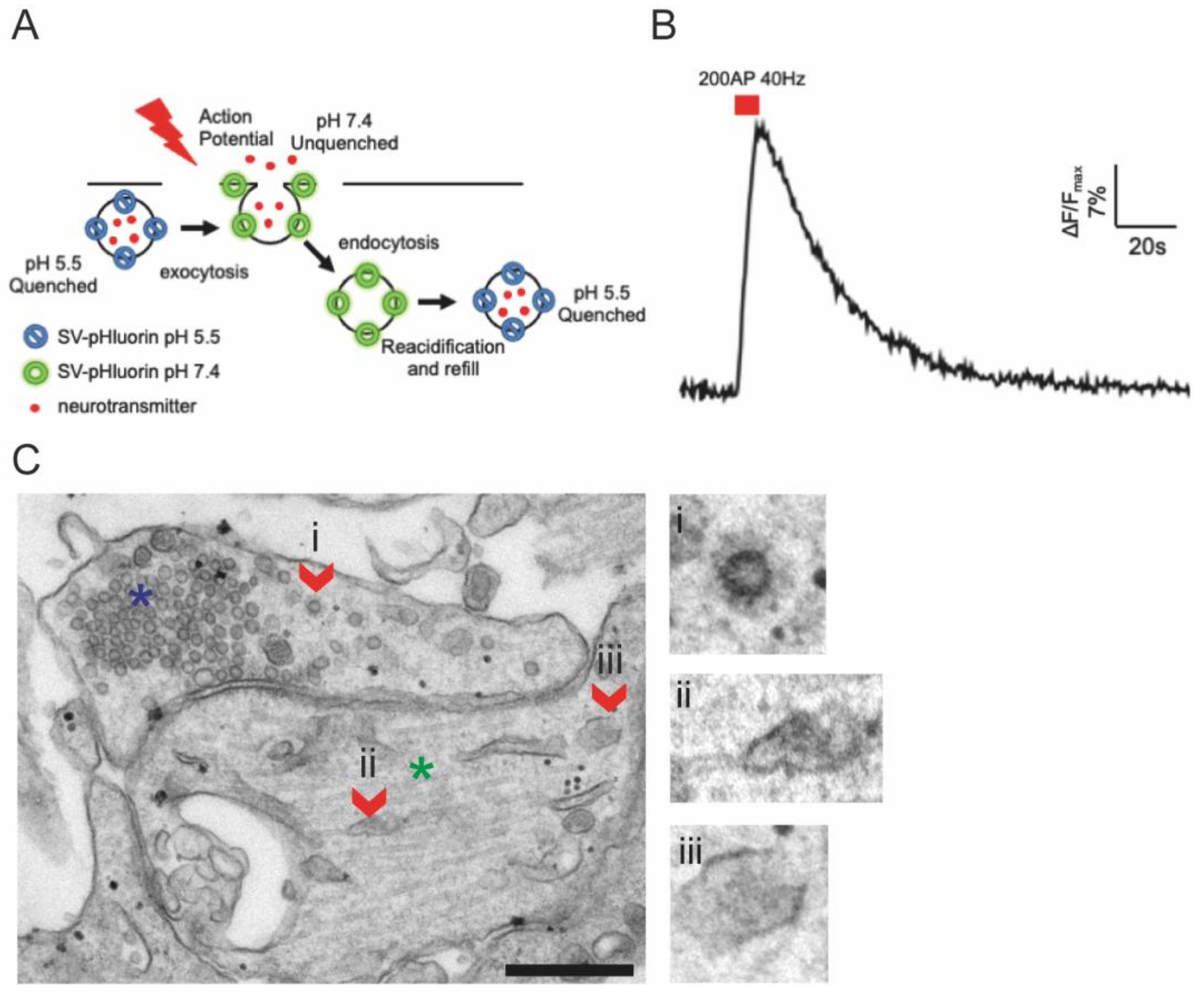
- Li, S.; Raychaudhuri, S.; Watanabe, S. Flash-and-Freeze: A Novel Technique to Capture Membrane Dynamics with Electron Microscopy. J. Vis. Exp. 2017, 2017, 55664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soykan, T.; Kaempf, N.; Sakaba, T.; Vollweiter, D.; Goerdeler, F.; Puchkov, D.; Kononenko, N.L.; Haucke, V. Synaptic Vesicle Endocytosis Occurs on Multiple Timescales and Is Mediated by Formin-Dependent Actin Assembly. Neuron 2017, 93, 854–866.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukulski, W.; Schorb, M.; Kaksonen, M.; Briggs, J.A.G. Plasma Membrane Reshaping during Endocytosis Is Revealed by Time-Resolved Electron Tomography. Cell 2012, 150, 508–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parlanti, P.; Cappello, V. Microscopes, Tools, Probes, and Protocols: A Guide in the Route of Correlative Microscopy for Biomedical Investigation. Micron 2022, 152, 103182. [Google Scholar] [CrossRef]
- Walter, A.; Kleywegt, G.J.; Verkade, P. Correlative Multimodal Imaging: Building a Community. Methods Cell Biol. 2021, 162, 417–430. [Google Scholar] [PubMed]
- Cortese, K.; Vicidomini, G.; Gagliani, M.C.; Boccacci, P.; Diaspro, A.; Tacchetti, C. 3D HDO-CLEM: Cellular Compartment Analysis by Correlative Light-Electron Microscopy on Cryosection. In Methods in Cell Biology; Academic Press: Cambridge, MA, USA, 2012; Volume 111, pp. 95–115. [Google Scholar]
- Franke, C.; Repnik, U.; Segeletz, S.; Brouilly, N.; Kalaidzidis, Y.; Verbavatz, J.M.; Zerial, M. Correlative Single-Molecule Localization Microscopy and Electron Tomography Reveals Endosome Nanoscale Domains. Traffic 2019, 20, 601–617. [Google Scholar] [CrossRef] [Green Version]
- van der Beek, J.; de Heus, C.; Liv, N.; Klumperman, J. Quantitative Correlative Microscopy Reveals the Ultrastructural Distribution of Endogenous Endosomal Proteins. J. Cell Biol. 2022, 221, e202106044. [Google Scholar] [CrossRef] [PubMed]
- Fermie, J.; de Jager, L.; Foster, H.; Veenendaal, T.; de Heus, C.; van Dijk, S.; ten Brink, C.; Oorschot, V.M.J.; Yang, L.; Li, W.; et al. Bimodal Endocytic Probe for Three-Dimensional Correlative Light and Electron Microscopy. SSRN Electron. J. 2021. [Google Scholar] [CrossRef]
- Sartori-Rupp, A.; Cordero Cervantes, D.; Pepe, A.; Gousset, K.; Delage, E.; Corroyer-Dulmont, S.; Schmitt, C.; Krijnse-Locker, J.; Zurzolo, C. Correlative Cryo-Electron Microscopy Reveals the Structure of TNTs in Neuronal Cells. Nat. Commun. 2019, 10, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baena, V.; Conrad, R.; Friday, P.; Fitzgerald, E.; Kim, T.; Bernbaum, J.; Berensmann, H.; Harned, A.; Nagashima, K.; Narayan, K. Fib-Sem as a Volume Electron Microscopy Approach to Study Cellular Architectures in SARS-CoV-2 and Other Viral Infections: A Practical Primer for a Virologist. Viruses 2021, 13, 611. [Google Scholar] [CrossRef] [PubMed]
- Speiser, A.; Müller, L.R.; Hoess, P.; Matti, U.; Obara, C.J.; Legant, W.R.; Kreshuk, A.; Macke, J.H.; Ries, J.; Turaga, S.C. Deep Learning Enables Fast and Dense Single-Molecule Localization with High Accuracy. Nat. Methods 2021, 18, 1082–1090. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tagliatti, E.; Cortese, K. Imaging Endocytosis Dynamics in Health and Disease. Membranes 2022, 12, 393. https://doi.org/10.3390/membranes12040393
Tagliatti E, Cortese K. Imaging Endocytosis Dynamics in Health and Disease. Membranes. 2022; 12(4):393. https://doi.org/10.3390/membranes12040393
Chicago/Turabian StyleTagliatti, Erica, and Katia Cortese. 2022. "Imaging Endocytosis Dynamics in Health and Disease" Membranes 12, no. 4: 393. https://doi.org/10.3390/membranes12040393
APA StyleTagliatti, E., & Cortese, K. (2022). Imaging Endocytosis Dynamics in Health and Disease. Membranes, 12(4), 393. https://doi.org/10.3390/membranes12040393