1. Introduction
The separation of complex liquid mixtures by distillation is energy intensive due to the necessity of phase change. Membranes provide a low energy alternative to distillation by negating the requirement of phase change, which may help decarbonize the petrochemical industry via the debottlenecking of existing and proposed distillation columns [
1,
2]. For membranes to become widely adopted in the industry, there is a need to develop modeling platforms that allow for the prediction of separations of complex liquid mixtures over a wide range of temperature and pressure.
Glassy polymer membranes are beginning to be used in hydrocarbon separations [
3]. Glassy polymers are polymers at a temperature less than their glass transition temperature. In this state, the polymer chains are arrested in an out of equilibrium configuration. Based on extensive analysis in the gas separation literature [
4,
5,
6], the membrane microstructure is often imagined as a transiently porous structure in which molecules sorb from the feed and diffuse through the porous membrane via transient-free volume elements that open and close via random thermal fluctuations of the glassy polymer chains. The selectivity between two species can be estimated as the product of their guest diffusivity ratio and solubility ratio in the membrane.
The microstructure and chain dynamics of the polymer membrane are very different in the case of organic liquid separations such as pervaporation and organic solvent reverse osmosis. Here, the membrane dilates due to the high solubility of common organic liquids in most glassy polymers. The organic liquids often induce plasticization of the polymer glasses, thus enhancing chain mobility. In situations with strong solvent–polymer interactions, these two phenomena can couple to result in the formation of continuum-like clusters of molecules permeating and diffusing through the polymer membrane. It is worth contrasting this against the case of gas membrane separation, in which the gases are often sufficiently dilute in the glassy polymer system such that the gas diffusivity can be calculated via activation rates of single polymer segment motions and individual gas molecule diffusive jumps.
For polymers containing aromatic backbones such as Matrimid, PIM-1, and SBAD-1, aromatic hydrocarbons swell the polymer to a much higher degree than saturated hydrocarbons [
7]. In extreme cases, the solvent-swollen polymer can behave more like a liquid phase rather than a glassy polymer. Hence, the character of the membrane, glass-like or liquid-like, really depends on the sorbed liquid composition.
Therefore, guest transport behaviors through the same glassy polymer membrane can span the range from individual molecular motions (e.g., light gas permeation) to the continuum-level convection of fluids through a dilated polymer such that the solvent–polymer system behaves as an equilibrium liquid polymer (
Figure 1).
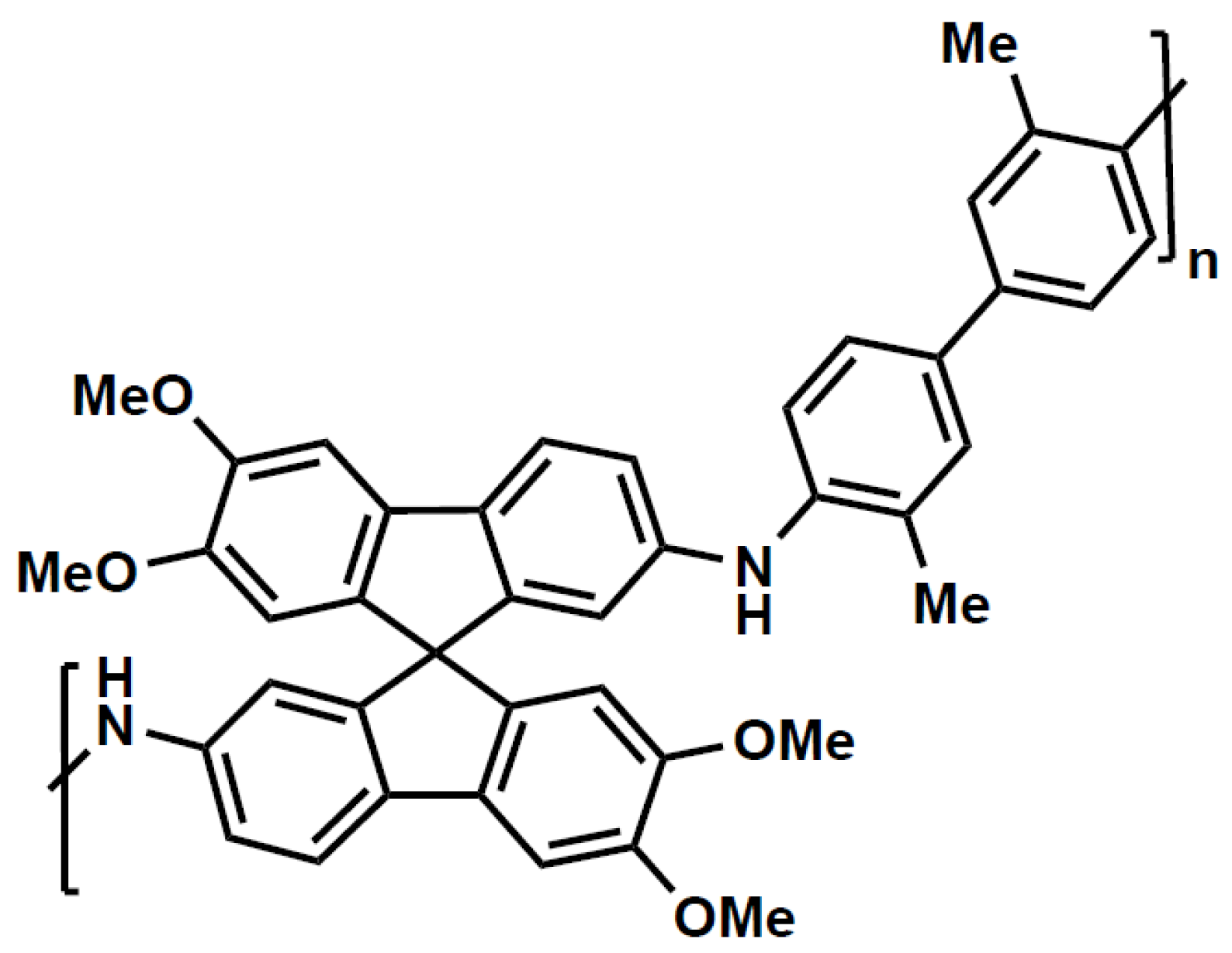
In this work we restrict our attention to the glassy polymer SBAD-1 [
3], which is a spirocyclic polymer with N-aryl bonds (
Figure 2). This membrane was chosen because there has been an extensive set of pure component sorption and diffusion measurements as well as an extensive set of membrane separations of liquid hydrocarbons using this polymer.
Mathias et al. [
8] measured the pure component vapor solubility as well as hydraulic permeability of nine hydrocarbon species (
Table 1) in SBAD-1, while Thompson et al. [
3] measured the temperature and pressure dependence of the separation of a liquid mixture composed of these nine species. In addition, the membrane separation of a light shale crude oil was also reported [
3].
In a recent publication, Marshall et al. [
9] applied the Maxwell-Stefan [
10] diffusion equation to the pure hydraulic liquid permeation data of Mathias et al. [
8]. The Maxwell–Stefan equation was applied in the mole fraction representation so that the extracted diffusivities have a clear molecular interpretation. For a pure species
i diffusing through an immobile polymer membrane along the dimension
z, the Maxwell–Stefan equation simplifies to,
where
Ni is the molecular flux of component
i,
ρi is the number density of species
i sorbed in the membrane,
Ði,p is the Maxwell–Stefan diffusivity of
i in polymer
p,
xp is the polymer mole fraction in the membrane,
kB is Boltzmann constant,
T is temperature in Kelvin, and
μi is chemical potential of
i. As written, Equation (1) is formulated for a flux of units (molecules/area/time). The Maxwell–Stefan diffusivities were extracted from the pure liquid hydraulic permeation data of Mathias et al. [
8] via data fitting to Equation (1).
Recently, Thompson et al. [
3] measured the membrane-based separation of a nine-component mixture (
Table 1) at a feed composition of
. The separation coefficient
Rj is defined as the ratio of permeate mole fraction
to the feed or retentate mole fraction
(these experiments were conducted at low stage cuts), see Equation (2) below.
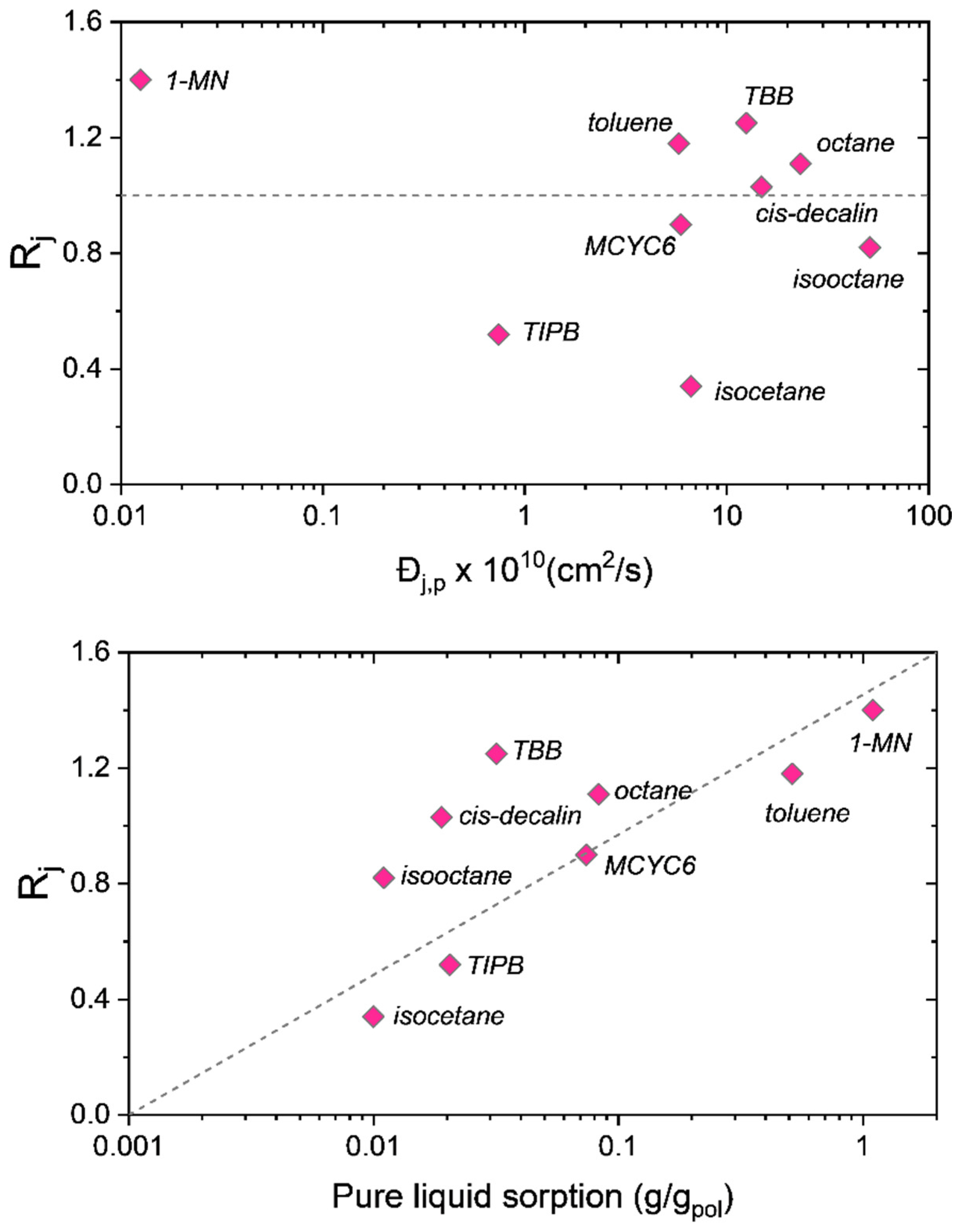
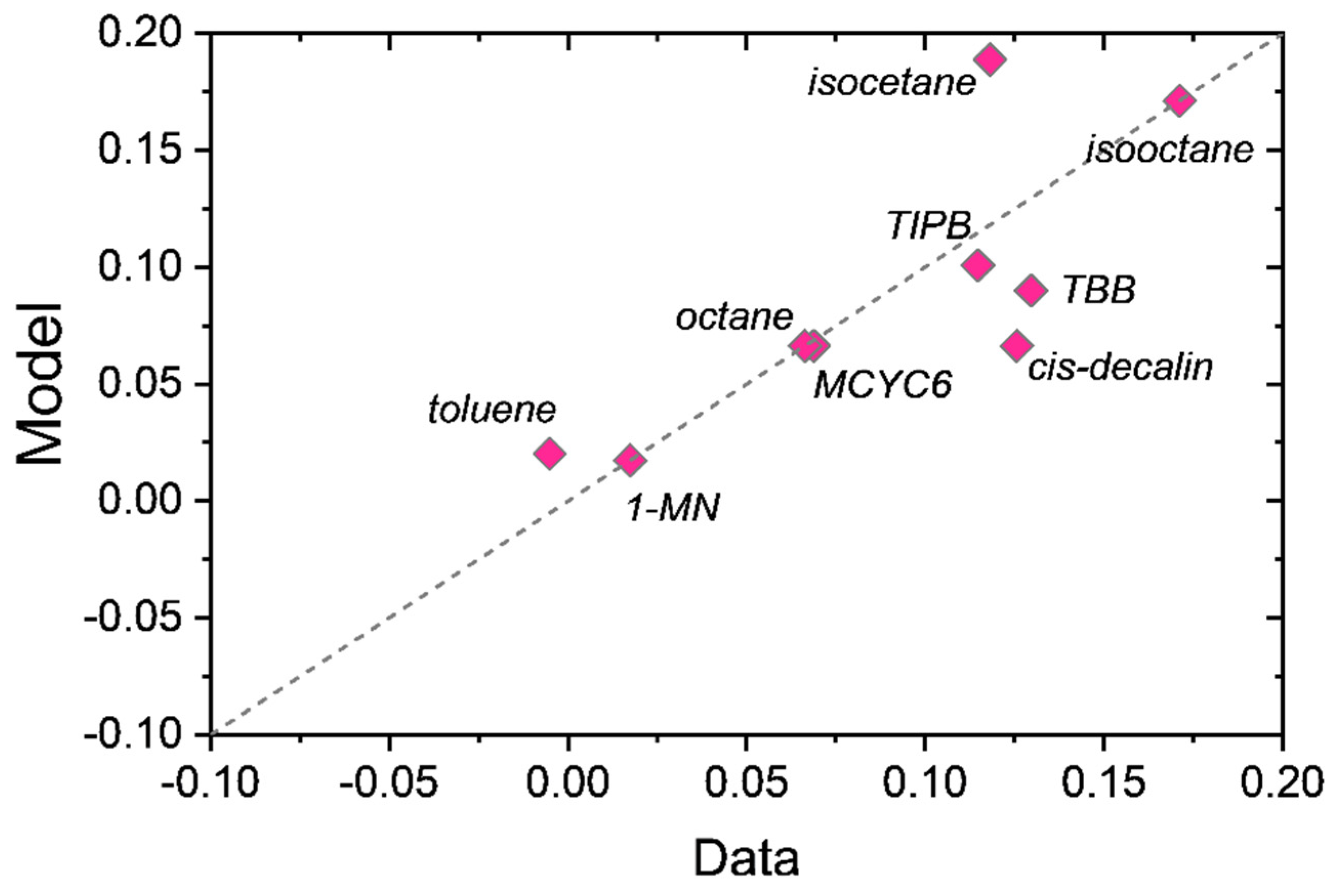
The top panel of
Figure 3 plots the separation coefficients of the nine-component mixture versus the pure liquid diffusivities in the membrane, while the bottom panel plots the separation coefficients versus the pure liquid solubility (sorption) in the membrane. The top panel of
Figure 3 shows no functional relationship between the separation coefficients and the pure component diffusivities. In fact, the species with the smallest diffusivity—1-MN, being nearly two orders of magnitude smaller—is the most purified in the permeate with an
Rj of 1.4. On the other hand, there is a clear functional dependence of
Rj on the pure liquid solubility. We note here that these diffusivities are on a molar basis, not a volumetric basis. We prefer the molar basis as it allows for a clear interpretation of the diffusivities, whereas the volumetric diffusivities commonly employed in polymer-based Maxwell–Stefan formulations require knowledge of solvent-averaged molecular weights of the membrane system [
11].
Instead of acting as a semi-rigid material with selective molecule-by-molecule diffusion (as in the case of diffusion-selective gas separation), the polymer membrane in the organic liquid separation can instead be approximated as an extracting phase (e.g., liquid–solid–liquid extraction). To capitalize on this observation, Marshall et al. [
9] proposed the following simple extension of Equation (1) to multi-component fluids containing
n species,
where
Ðavg is the average diffusivity of guest species in the membrane evaluated through the relation
where
is the mole fraction of
i in the membrane on a guest species basis
Equation (3) assumes that the diffusion in the membrane is fully mutualized. When combined with an appropriate thermodynamic model to calculate the solubility and chemical potential gradients, Equation (3) was shown to accurately represent the separation of liquid mixtures in several glassy polymer membranes [
9].
A consequence of Equation (3) is that the permeate mole fraction is independent of the diffusivities.
where
z = 0 is the axial location of the membrane–retentate boundary. In the second step of Equation (6), it is assumed the chemical potential varies linearly across the membrane (constant gradient approximation) [
9].
is the chemical potential of
i in the permeate and
is the chemical potential in the retentate. The fraction
is the mole fraction of component
i in the membrane at the retentate boundary.
Equation (6) provides a set of
n equations that are solved iteratively to obtain the set of mole fractions
. Once this numerical solution has been obtained, the component flux of each species is easily calculated as follows (within the constant gradient approximation,
in the denominator is the membrane thickness),
Using Equation (6) it was demonstrated [
9] that, when combined with the appropriate thermodynamic model, accurate predictions of the permeate compositions in SBAD-1 membrane-based separations of liquids could be made. Specifically, it was demonstrated that Equation (6) accurately predicted the separation highlighted in
Figure 3, as well as the separation of a light shale crude oil on this same SBAD-1 membrane.
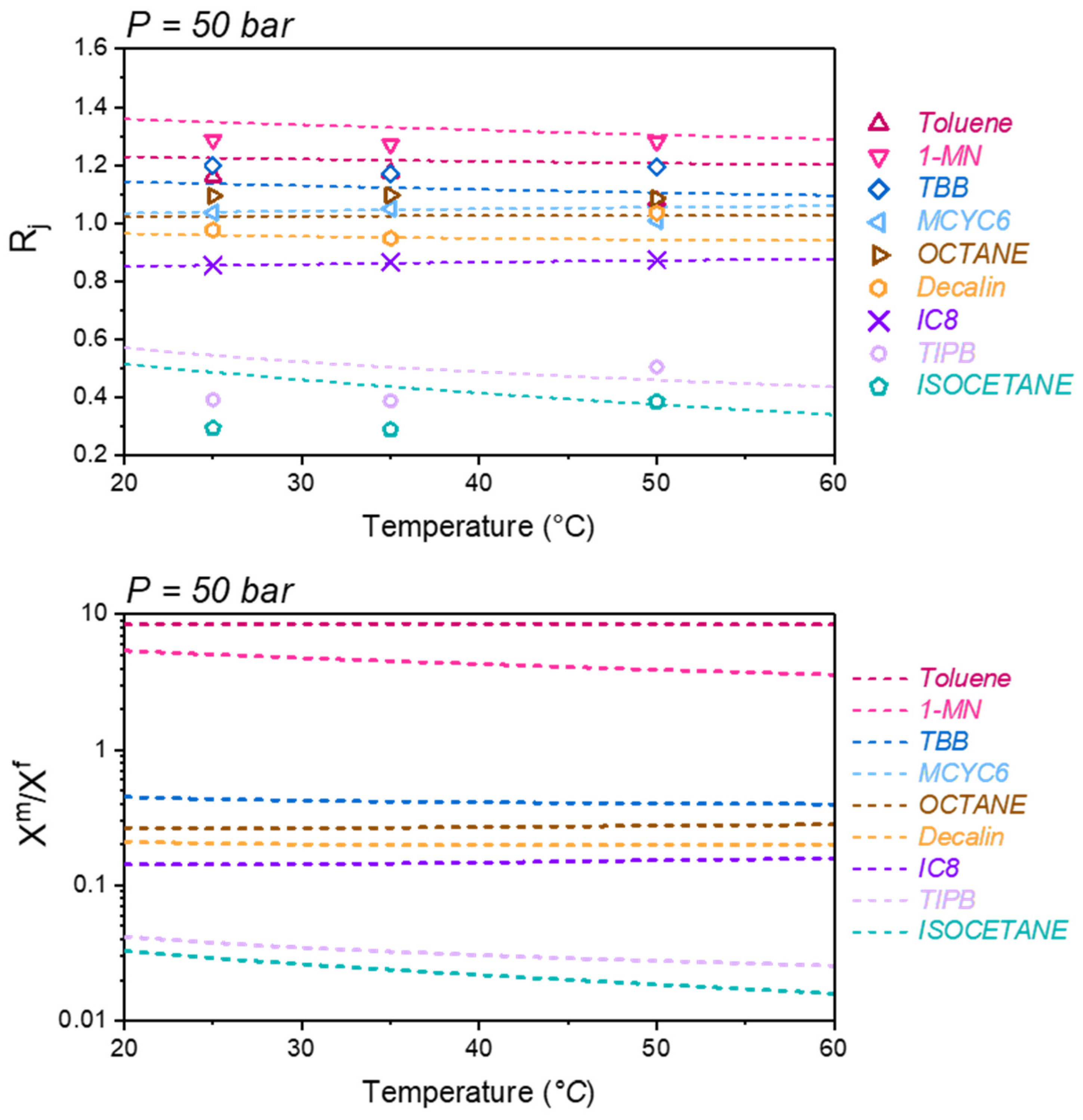
A question arises from this prior work: was this good agreement a coincidence? Can the model accurately predict the temperature and pressure dependence of
Rj without further tuning? We will explore this question in the current work. Of course, if it is assumed that Equation (6) is reasonably correct, then the accurate prediction of the temperature and pressure dependence of the membrane selectivity is necessarily a test of the underlying thermodynamic model. As in our previous application, we employed the dry glass reference perturbation theory [
12] (DGRPT) as our base thermodynamic model to calculate the solubility and chemical potential.
In our previous work [
9], it was demonstrated that when Equation (6) is combined with DGRPT [
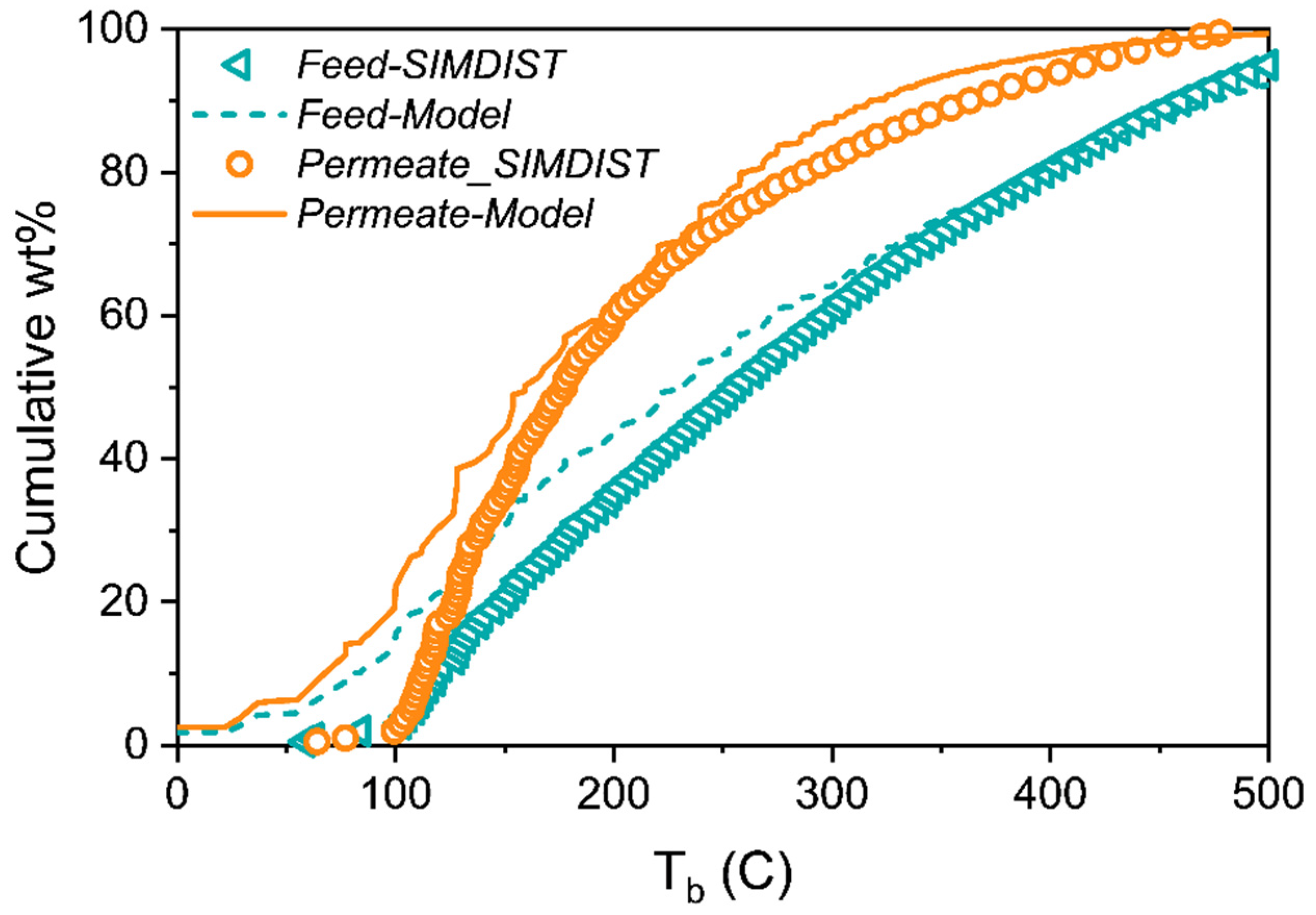
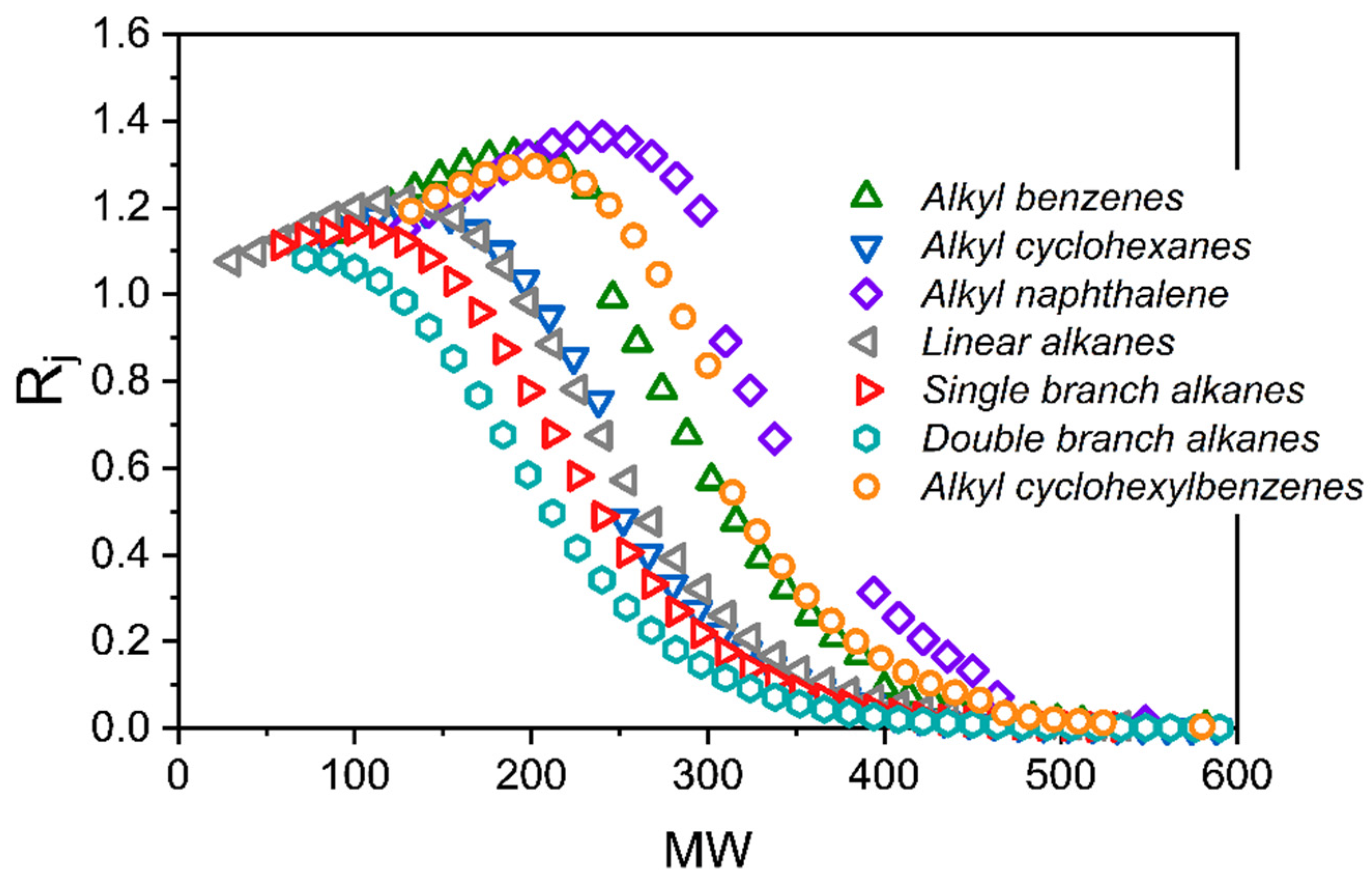
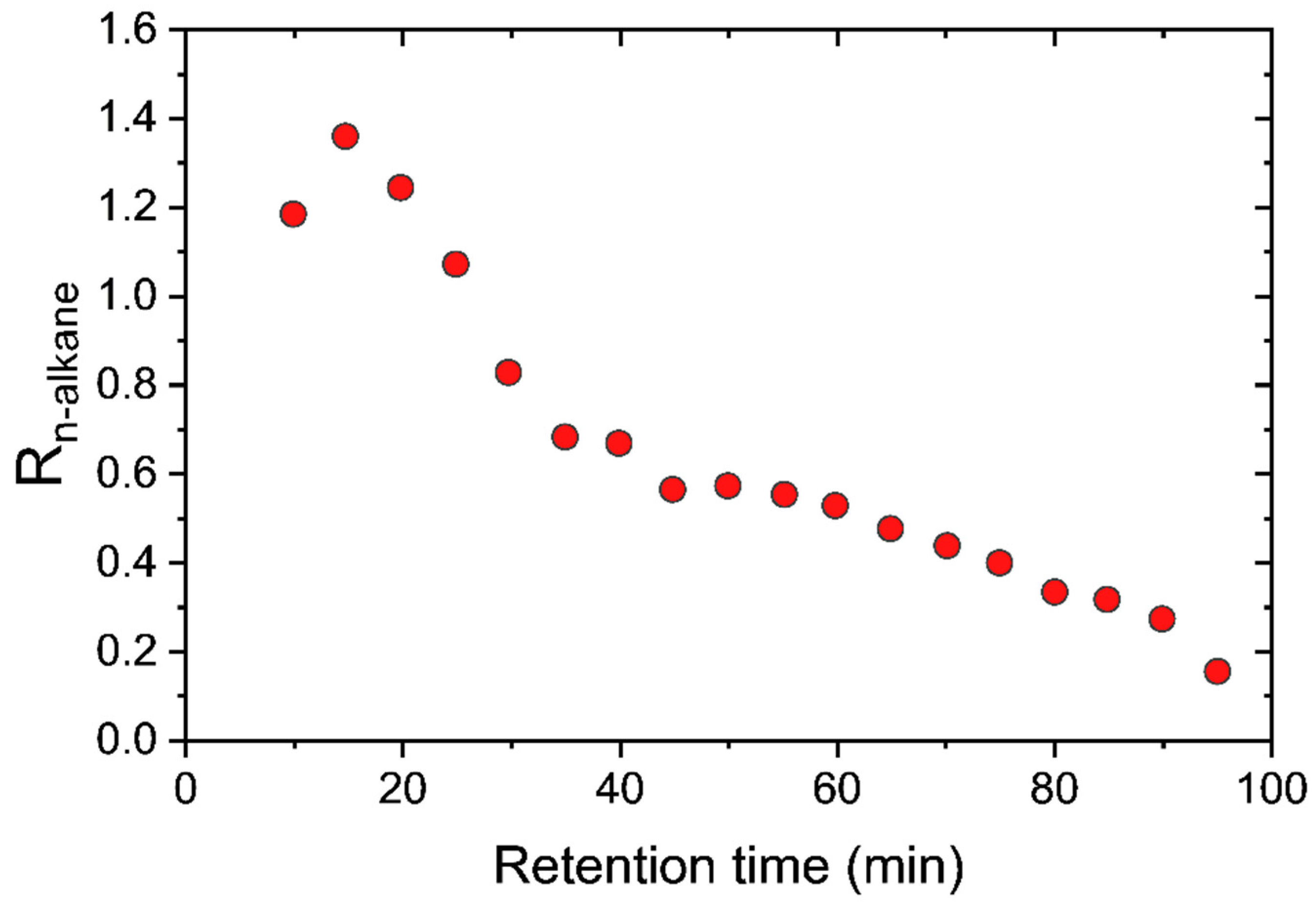
12], the separation of the nine-component liquid mixture can be accurately predicted using only pure component vapor sorption isotherms at room temperature to parameterize the model. Furthermore, this same approach was applied to predict the SBAD-1 membrane-based separation of a light shale crude, and it accurately predicted the distillation curve of the permeate using a petroleum fraction representation based on the boiling point. While being useful for predicting the distillation properties, this approach did not allow for the prediction of the partitioning of molecular classes. In this work we represent the light shale crude using a detailed compositional model based on structure-oriented lumping [
13], which allows for the prediction of the effect of molecular structures on enrichment in the permeate. We then demonstrated that the model predictions of the molecular class based separation are consistent with experimental data. As far as we are aware, this is the first example of such a model prediction and validation in the literature.
2. Theory and Model Development: Dry Glass Reference Perturbation Theory
As illustrated in
Figure 1, glassy polymer membranes are exceptionally challenging to model due to the fact that their properties may vary substantially depending on the feed composition. Consider the case of a binary liquid mixture containing a non-dilating “C1” and a dilating “C2” species. In pure C1, the dilation of the membrane will be limited, and guest transport through the glassy polymer will be somewhat similar to gas transport mechanisms. While in pure C2, the membrane will be highly swollen, behaving more like an equilibrium liquid polymer. In this regime, equilibrium theories such as Flory–Huggins or statistical associating fluid theory (SAFT) [
14] may be used. What happens for intermediate compositions? At what composition does the polymer transit from solid like to liquid like? Is it a smooth transition?
To answer these questions, an approach is needed which can bridge these two extremes. The non-equilibrium thermodynamics of glassy polymers [
15,
16,
17,
18,
19] (NETGP) of Sarti and Doghieri was designed to address this gap. NETGP maps the non-equilibrium solubility in glassy polymers onto equilibrium equations of state.
Consider a fluid mixture composed of
n species at temperature
T and pressure
P, in contact with a glassy polymer. The equilibrium relations for the
n sorbing species are similar to the equilibrium case,
where
is the chemical potential of
i in the fluid phase of mole fraction
. Since the fluid phase is considered an equilibrium fluid, the chemical potential is described as functions of
T,
P and
. Here,
is the chemical potential of
i in the glassy polymer phase. It is specified as functions of
T, the set of guest species number densities
, and the number density of the glassy polymer
ρg. In effect,
ρg has replaced
P as a specification to the chemical potential. This is necessary in glassy polymers due to the fact that the thermodynamics pressure
P, is not defined in a glassy polymer [
9]. Both chemical potentials in the fluid phase and the polymer phase, Equation (9), are evaluated with equilibrium-density-explicit equations of state.
As it is assumed that the composition of the bulk fluid phase at the feed side is known, there are n + 1 unknown variables corresponding to all species densities in the polymer phase. However, there are only n equations provided by Equation (9). The additional relation in NETGP is obtained by treating the polymer density as an order parameter that must be specified. That is, the polymer density ρg in the presence of sorbed guest species is not calculated self-consistently in the theory.
For non-swelling polymers, one can simply set
, where
is the measured density of the solvent-free glassy polymer. We refer to this density as the “dry glass density”. For vapors/solvents that swell the polymer, setting
gives poor results [
20]. To compensate, the following correlation is often used:
where
fi is the component fugacity (equal to the partial pressure in ideal gases), and
ks,i is an empirical coefficient which is adjusted to sorption data. Equations (9) and (10), when combined with a density explicit equilibrium equation of state, complete the NETGP approach.
NETGP has been highly successful at describing gas sorption [
15,
17,
18,
19] over a wide range of conditions. However, this formulation of NETGP is not well suited to describe sorption from complex liquid mixtures. First, the fugacity dependence of swelling is not necessarily linear. Second, Equation (10) does not account for competitive sorption effects. One can assume alternative non-linear fugacity dependence in Equation (10). However, it is difficult to utilize Equation (10) when different species may have swelling profiles with a different functional dependence on fugacity. While Equation (10) is sufficient to describe multi-component sorption from gases, it lacks the theoretical rigor to be applied for complex liquid mixtures.
Recently Marshall et al. [
12] proposed an alternative closure relation to NETGP called dry glass reference perturbation theory (DGRPT). DGRPT develops a closure relation for the non-equilibrium polymer chemical potential by treating guest species sorption as the perturbation to a dry polymer reference state.
is the chemical potential of the polymer (subscript p refers to the polymer, and superscript poly to the polymer phase) as a function of the polymer density (ρg) in the presence of the set of guest species densities (ρk) and temperature T. The term represents the dry reference polymer chemical potential in the absence of sorbed species, i.e., = 0. It is assumed that the dry glass density is known. Finally, the derivatives in the expansion are evaluated in the limit of infinite dilution of guest species.
Equations (9) and (11) summarize the DGRPT approach. It is an alternative realization of NETGP, in which the theoretical closure Equation (11) is used in place of the empirical closure Equation (10). The major benefit of Equation (11) over Equation (10) is in its application to dilating mixtures. Equation (11) provides a closed framework without the need to make additional assumptions. This allows for applications to complex liquid mixtures over a wide range of conditions.
In the applications considered thus far [
9,
12], the accuracy has been sufficient when truncating Equation (11) at first order
The dry glass density
has a rheological dependence on the temperature and pressure. This dependence is accounted for with the following relation [
9],
is the dry glass density at the temperature
To and atmospheric pressure. The dry polymer modulus
γ and thermal expansion coefficient α are dry pure polymer properties and are independent of any sorbed guest species. For bulk phase behavior calculations,
γ is best interpreted as a bulk modulus, while for membrane operations,
γ is interpreted as a dry Young’s modulus due to the uniaxial stress. Note, the actual modulus of the solvent swollen polymer is different from
γ, and is calculated self-consistently in DGRPT.
Finally, an equilibrium equation of state for the evaluation of all chemical potentials must be specified. As in previous applications of DGRPT [
9,
12], we employed the simplified [
21] polar [
22] PC-SAFT [
23] equation of state. In polar PC-SAFT, molecules are treated as chains of tangentially bonded spheres. The chain length
m, sphere diameter
σ, and sphere–sphere interactive energy
ε are parameters in the model. In addition, polar species are further described by the polar strength
αp =
mxpolμ2, which provides the magnitude of polar attractions. Here,
xpol is the fraction of segments in a molecule which are polar, and
μ is the dipole moment of a polar segment.
PC-SAFT parameters for fluid phase species are fit to pure component vapor pressures and liquid densities [
23]. For glassy polymers, equilibrium phase behavior does not exist to regress the
m,
σ,
ε of the polymer (
αp is set by the polymer molecular structure). Hence, these parameters are adjusted to the pure vapor sorption data of at least two vapor species with the polymer. In this work, we employed polymer parameters fit [
12] in this way to SBAD-1. Toluene and heptane pure vapor sorption data at 25 °C were employed to extract [
12] the polymer
m,
σ,
ε.
For all remaining fluid phase species, if sorption data of a given molecule with the polymer exist, a binary interaction parameter
kij between the fluid phase species and the polymer can be adjusted. The binary interaction parameter is used in the combining rule for the cross species
εij in terms of the pure component
εThe pure component PC-SAFT parameters of SBAD-1 and fluid phase species are given in
Table 2, while the binary interaction parameters between the fluid phase species and the polymer are given in
Table 3.
Table 3 also shows the fraction of aromatic carbon (
fa), fraction of saturated carbon (
fsat = 1 −
fa), and the fraction of carbon which is alkane branches (
fbr) in these nine molecules, which will be applied for building correlations between them and the interaction parameters, to be discussed later.
The dry modulus γ does not have an effect for the low pressure vapor isotherms. However, for elevated pressures in liquid membrane separations, γ becomes important. As in the previous publication [
9], we used
γ = 0.7 GPa. Further, since the dry thermal expansion coefficient of SBAD-1 is unknown, we simply set
α = 0. The dry glass density of SBAD-1 is
= 1.052 g/cc [
3].













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}