

Binding of Different Cyclosporin Variants to Micelles Evidenced by NMR and MD Simulations




Abstract
:1. Introduction
2. Materials and Methods
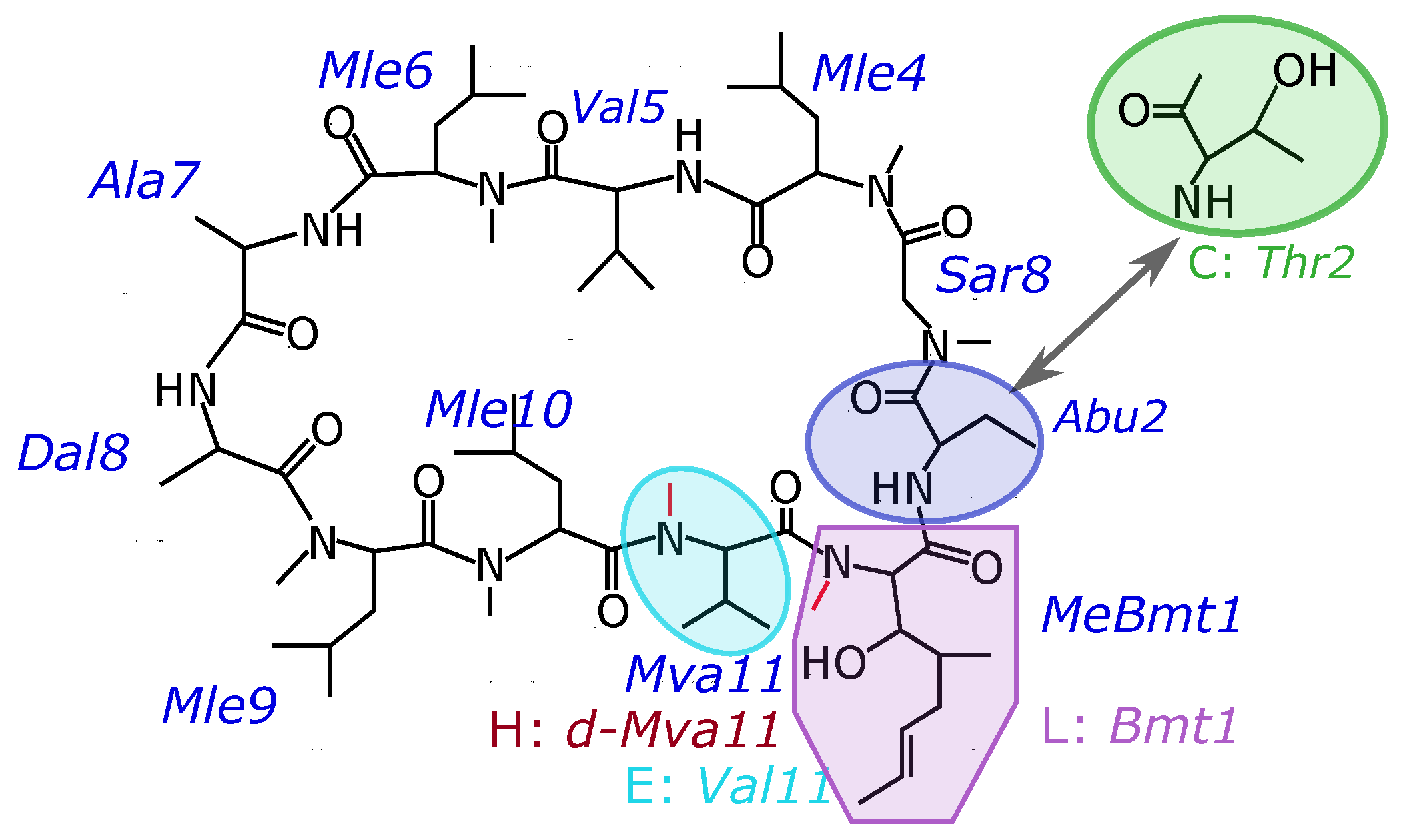
3. Results
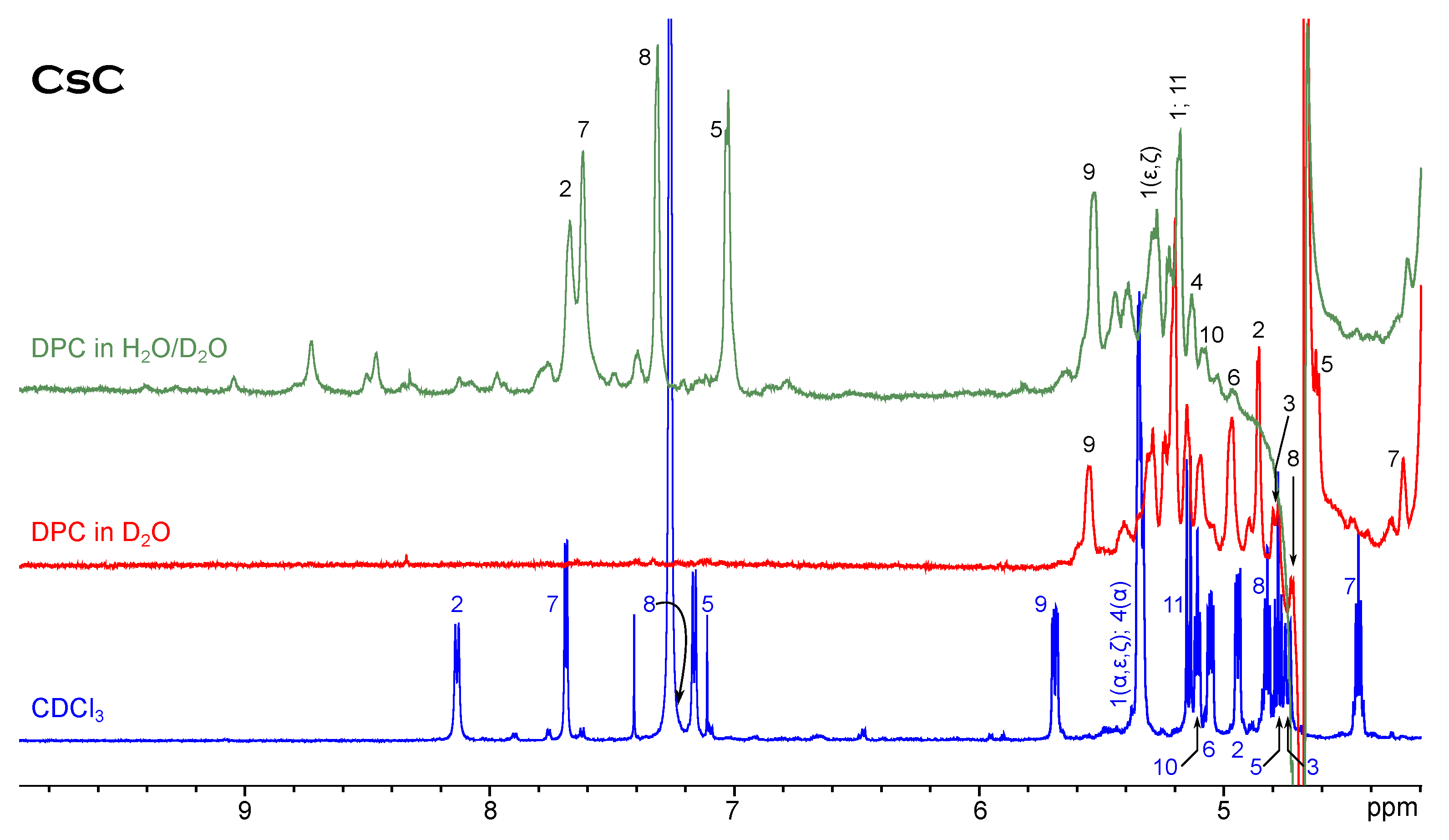
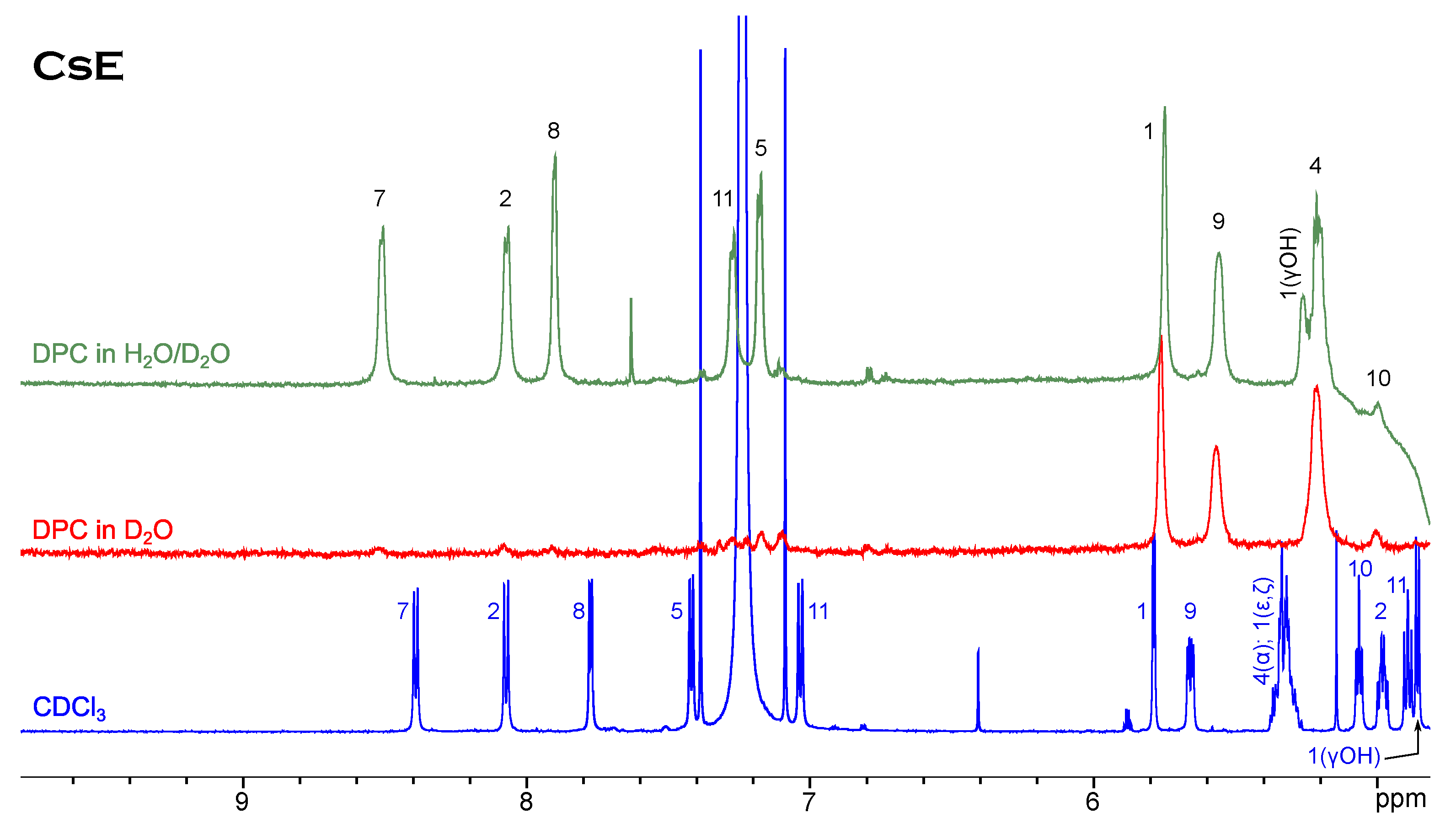
NMR Spectroscopy
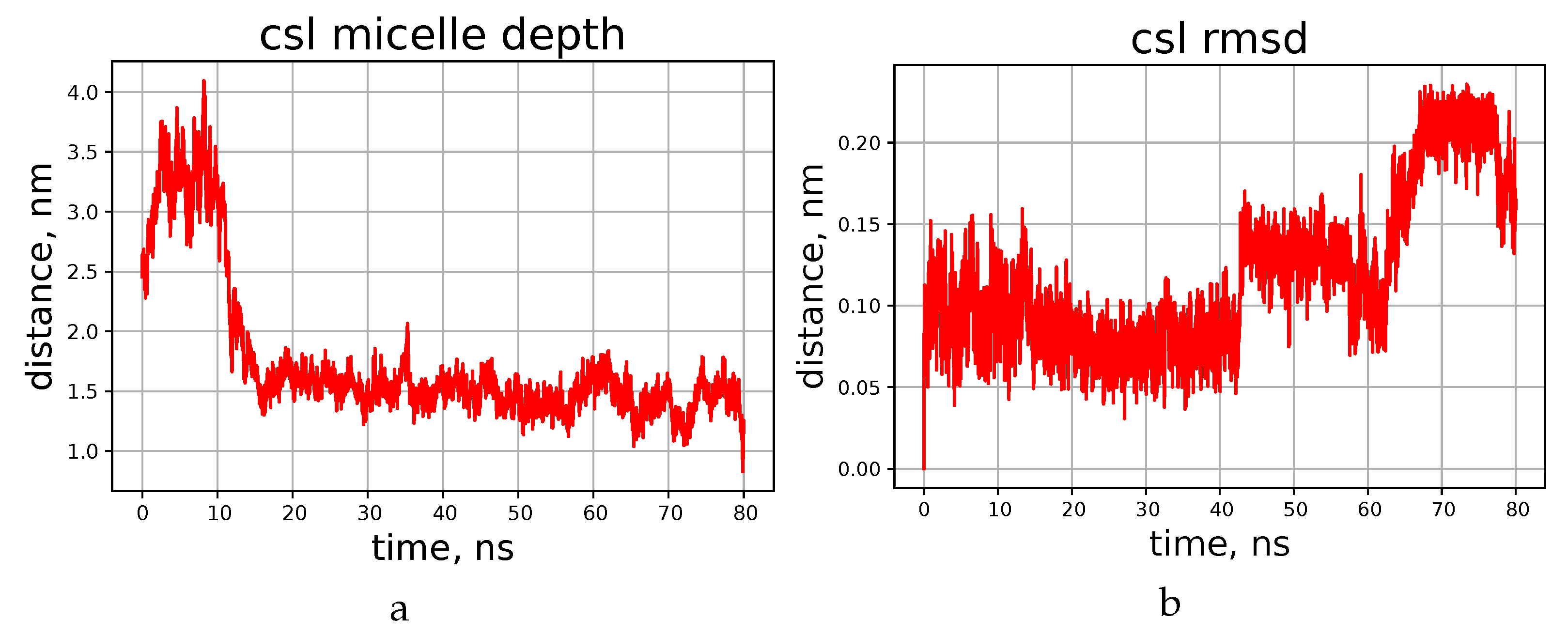
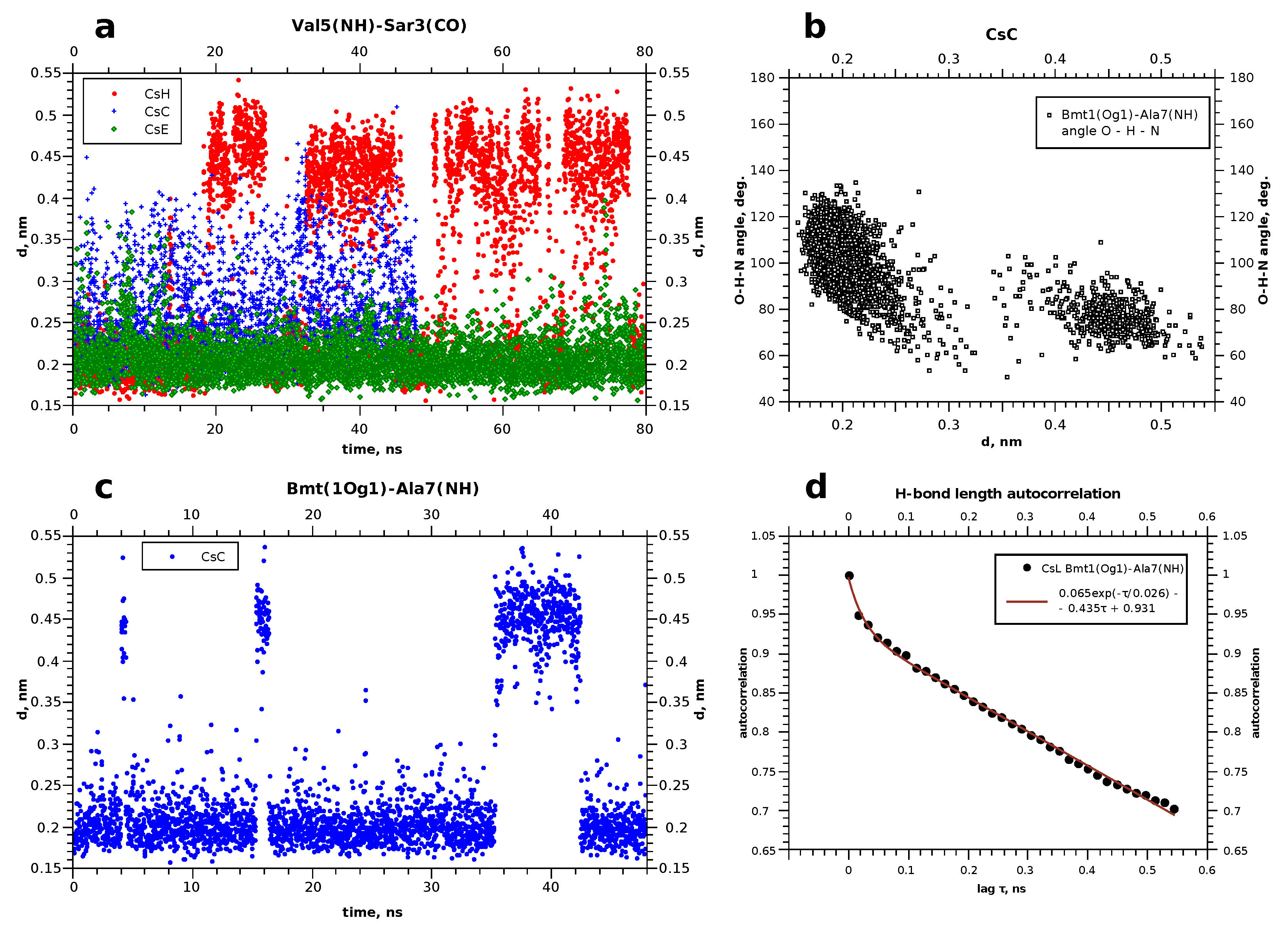
4. Molecular Dynamics
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NMR | Nuclear magnetic resonance |
| MD | Molecular dynamics |
| DPC | Dodecylphosphocholine |
| SDS | Sodium dodecylsulphate |
References
- Hamawy, M.M.; Knechtle, S.J. An Overview of the Actions of Cyclosporine and FK506. Transplant. Rev. 2003, 17, 165–171. [Google Scholar] [CrossRef]
- Myers, B.D.; Ross, J.; Newton, L.; Luetscher, J.; Perlroth, M. Cyclosporine-Associated Chronic Nephropathy. N. Engl. J. Med. 1984, 311, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Masri, M.A.; Naiem, M.; Pingle, S.; Daar, A.S. Cyclosporine A versus Cyclosporine G: A Comparative Study of Survival, Hepatotoxicity, Nephrotoxicity, and Splenic Atrophy in BALB/c Mice. Transplant Int. 1988, 1, 13–18. [Google Scholar] [CrossRef]
- Henry, M.L.; Elkhammas, E.A.; Davies, E.A.; Ferguson, R.M.A. Clinical Trial of Cyclosporine G in Cadaveric Renal Transplantation. Pediatr. Nephrol. 1995, 9, S49–S51. [Google Scholar] [CrossRef]
- Bell, A.; Monaghan, P.; Page, A.P. Peptidyl-Prolyl Cis-Trans Isomerases (Immunophilins) and Their Roles in Parasite Biochemistry, Host-Parasite Interaction and Antiparasitic Drug Action. Int. J. Parasitol. 2006, 36, 261–276. [Google Scholar] [CrossRef]
- Jiang, X.; Kelsey, S.; Wu, Y.; Newland, A. Circumvention of P-glycoprotein-mediated Drug Resistance in Human Leukaemic Cells by non-immunosuppressive Cyclosporin D Analogue, SDZ PSC 833. Br. J. Haematol. 1995, 90, 375–383. [Google Scholar] [CrossRef]
- Smith, S.; Peersen, O. Solid-State NMR Approaches for Studying Membrane Protein Structure. Annu. Rev. Biophys. Biomol. Struct. 1992, 21, 25–47. [Google Scholar] [CrossRef]
- Huster, D. Investigations of the structure and dynamics of membrane-associated peptides by magic angle spinning NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 79–107. [Google Scholar] [CrossRef]
- Henry, G.; Sykes, B. Methods to Study Membrane Protein Structure in Solution. Meth. Enzymol. 1994, 239, 515–535. [Google Scholar]
- Chill, J.; Naider, F. A solution NMR view of protein dynamics in the biological membrane. Curr. Opin. Struct. Biol. 2011, 21, 627–633. [Google Scholar] [CrossRef]
- Mäler, L. Solution NMR studies of cell-penetrating peptides in model membrane systems. Adv. Drug Deliv. Rev. 2013, 65, 1002–1011. [Google Scholar] [CrossRef]
- Shenkarev, Z.; Lyukmanova, E.; Paramonov, A.; Panteleev, P.; Balandin, S.; Shulepko, M.; Mineev, K.; Ovchinnikova, T.; Kirpichnikov, M.; Arseniev, A. Lipid–Protein Nanodiscs Offer New Perspectives for Structural and Functional Studies of Water-Soluble Membrane-Active Peptides. Acta Naturae 2014, 6, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Mineev, K.; Nadezhdin, K.; Goncharyuk, S.; Arseniev, A. Façade detergents as bicelle rim-forming agents for solution NMR spectroscopy. Nanotechnol. Rev. 2016, 6, 93–103. [Google Scholar] [CrossRef]
- Khodov, I.; Musabirova, G.; Klochkov, V.; Karataeva, F.; Huster, D.; Scheidt, H. Structural details on the interaction of fenamates with lipid membranes. J. Mol. Liq. 2022, 367, 120502. [Google Scholar] [CrossRef]
- Nikitina, L.; Pavelyev, R.; Startseva, V.; Kiselev, S.; Galiullina, L.; Aganova, O.; Timerova, A.; Boichuk, S.; Azizova, Z.; Klochkov, V.; et al. Structural details on the interaction of biologically active sulfur-containing monoterpenoids with lipid membranes. J. Mol. Liq. 2020, 301, 112366. [Google Scholar] [CrossRef]
- Jurczak, P.; Sikorska, E.; Czaplewska, P.; Rodziewicz-Motowidlo, S.; Zhukov, I.; Szymanska, A. The Influence of the Mixed DPC:SDS Micelle on the Structure and Oligomerization Process of the Human Cystatin C. Membranes 2021, 11, 17. [Google Scholar] [CrossRef]
- Usachev, K.; Efimov, S.; Kolosova, O.; Filippov, A.; Klochkov, V. High-resolution NMR structure of the antimicrobial peptide protegrin-2 in the presence of DPC micelles. J. Biomol. NMR 2015, 61, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, Ł.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the Mitochondrial Translocator Protein in Complex with a Diagnostic Ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef]
- Brown, L.; Braun, W.; Kumar, A.; Wüthrich, K. High resolution nuclear magnetic resonance studies of the conformation and orientation of melittin bound to a lipid-water interface. Biophys. J. 1982, 37, 319–328. [Google Scholar] [CrossRef]
- Irudayam, S.; Pobandt, T.; Berkowitz, M. Free Energy Barrier for Melittin Reorientation from a Membrane-Bound State to a Transmembrane State. J. Phys. Chem. B 2013, 117, 13457–13463. [Google Scholar] [CrossRef]
- Jamasbi, E.; Batinovic, S.; Sharples, R.; Sani, M.A.; Robins-Browne, R.; Wade, J.; Separovic, F.; Hossein, M. Melittin peptides exhibit different activity on different cells and model membranes. Amino Acids 2014, 46, 2759–2766. [Google Scholar] [CrossRef] [PubMed]
- Hyung, S.J.; Feng, X.; Che, Y.; Stroh, J.; Shapiro, M. Detection of conformation types of cyclosporin retaining intramolecular hydrogen bonds by mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 5785–5794. [Google Scholar] [CrossRef] [PubMed]
- Bock, J.E.; Gavenonis, J.; Kritzer, J.A. Getting in Shape: Controlling Peptide Bioactivity and Bioavailability Using Conformational Constraints. ACS Chem. Biol. 2013, 8, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Loor, F.; Tiberghien, F.; Wenandy, T.; Didier, A.; Traber, R. Cyclosporins: Structure-Activity Relationships for the Inhibition of the Human MDR1 P-Glycoprotein ABC Transporter. J. Med. Chem. 2002, 45, 4598–4612. [Google Scholar] [CrossRef]
- Navia, M.A.; Chaturvedi, P.R. Design Principles for Orally Bioavailable Drugs. Drug Discov. Today 1996, 1, 179–189. [Google Scholar] [CrossRef]
- Witek, J.; Keller, B.G.; Blatter, M.; Meissner, A.; Wagner, T.; Riniker, S. Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model. 2016, 56, 1547–1562. [Google Scholar] [CrossRef]
- Tieleman, P. Available online: https://people.ucalgary.ca/tieleman/download.html (accessed on 1 January 2023).
- Berger, O.; Edholm, O.; Jähnig, F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997, 72, 2002–2013. [Google Scholar] [CrossRef]
- Pettersen, E.; Goddard, N.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Gfeller, D.; Michielin, O.; Zoete, V. SwissSidechain: A molecular and structural database of non-natural sidechains. Nucleic Acids Res. 2013, 41, D327–D332. [Google Scholar] [CrossRef]
- Langham, A.; Khandelia, H.; Kaznessis, Y. How Can a β-sheet Peptide Be Both a Potent Antimicrobial and Harmfully Toxic? Molecular Dynamics Simulations of Protegrin-1 in Micelles. Biopolym. Pept. Sci. 2006, 84, 219–231. [Google Scholar] [CrossRef]
- Abdel-Azeim, S. Revisiting OPLS-AA Force Field for Simulation of Anionic Surfactants in Concentrated Electrolyte Solutions. J. Chem. Theory Comput. 2020, 16, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Swedberg, J.; Harvey, P.; Kaas, Q.; Craik, D. Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J. Phys. Chem. B 2018, 122, 2261–2276. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.; Steren, C.; Haynes, I.; Bermejo, G.; Favretto, F.; Zweckstetter, M.; Do, T. Structural Flexibility of Cyclosporine A is Mediated by Amide Cis–Trans Isomerization and the Chameleonic Roles of Calcium. J. Phys. Chem. B 2021, 125, 1378–1391. [Google Scholar] [CrossRef] [PubMed]
- Efimov, S.; Dubinin, M.; Kobchikova, P.; Zgadzay, Y.; Khodov, I.; Belosludtsev, K.; Klochkov, V. Comparison of cyclosporin variants B–E based on their structural properties and activity in mitochondrial membranes. Biochem. Biophys. Res. Commun. 2020, 526, 1054–1060. [Google Scholar] [CrossRef]
- Minch, M. Orientational Dependence of Vicinal Proton-Proton NMR Coupling Constants: The Karplus Relationship. Concepts Magn. Reson. 1994, 6, 41–56. [Google Scholar] [CrossRef]
- Majerz, I. Directionality of Inter- and Intramolecular OHO Hydrogen Bonds: DFT Study Followed by AIM and NBO Analysis. J. Phys. Chem. A 2012, 116, 7992–8000. [Google Scholar] [CrossRef]
- Gao, X.; Wong, T. NMR Studies of Adrenocorticotropin Hormone Peptides in Sodium Dodecylsulfate and Dodecylphosphocholine Micelles: Proline Isomerism and Interactions of the Peptides with Micelles. Biopolymers 2001, 58, 20–32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cyclosporin | Amino Acid Sequence | Chemical Formula | CAS No. |
|---|---|---|---|
| CsC | cyclo[N(Me)Bmt(E)-Thr-Sar-Mle-Val-Mle-Ala-Dal-Mle-Mle-Mva] | CHNO | 59787-61-0 |
| CsE | cyclo[N(Me)Bmt(E)-Abu-Sar-Mle-Val-Mle-Ala-Dal-Mle-Mle-Val] | CHNO | 63798–73-2 |
| CsH | cyclo[N(Me)Bmt(E)-Abu-Sar-Mle-Val-Mle-Ala-Dal-Mle-Mle-N(Me)dVal] | CHNO | 83602-39-5 |
| CsL | cyclo[Bmt(E)-Abu-Sar-Mle-Val-Mle-Ala-Dal-Mle-Mle-Mva] | CHNO | 108027-39-0 |
| Bmt1 | Xxx2 | Val5 | Ala7 | Dal8 | Val11 | |
|---|---|---|---|---|---|---|
| CsC | 9.7 | 8.9 | 7.0 | 7.2 | ||
| 5.63 (1) | 8.49 (1) | 7.88 (1) | 7.07 (1) | |||
| CsE | 9.8 | 8.4 | 9.1 | 6.2 | 9.7 | |
| 7.94 (2) | 8.57 (2) | 8.18 (2) | 7.62 (2) | 8.25 (2) | ||
| CsH | 5.8 | 7.2; | n/d | 8.1 | ||
| 7.61 (1) | 6.45 (2) | 6.68 (1) | 6.70 (1) | |||
| CsL | 7.5 | 9.9 | 8.6 | 7.3 | 8.4 | |
| 10.0 | 8.7 | 6.9 | n/d | |||
| 8.72 (1) | 5.78 (2) | 8.02(2) | 7.61 (2) | 7.2 (1) |
| CsC | CsL | CsH | CsE | |
|---|---|---|---|---|
| Bmt1(H)-Mle10(O) | – | 100% | – | – |
| ine Bmt1(Og1)-Ala7(H) | 81% (14.2) | |||
| ine Bmt1(Hg1)-Bmt1(O) | 97% (18.7) | 91% (38) | 44% (12.5) | 19% (19.9) |
| ine Val5(H)-Sar3(O) | 96% (21.4) | 47% (13.4) | 100% | |
| ine Bmt1(Hg1)-Val5(O) | 13% (1.2) | 43% (8) | ||
| ine Thr2(Hg1)-Thr2(O) | 58% (2.4) | – | – | – |
| ine Thr2(H)-Mle6(O) | 88.8% (19) | |||
| ine Ala7(H)-Bmt1(Og1) | 50% (18) | |||
| ine Dal8(O)-Val11(H) | 51% (5.8) | |||
| ine Dal8(H)-Val11(O) | 76% | 58% (9.6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kobchikova, P.P.; Efimov, S.V.; Klochkov, V.V. Binding of Different Cyclosporin Variants to Micelles Evidenced by NMR and MD Simulations. Membranes 2023, 13, 196. https://doi.org/10.3390/membranes13020196
Kobchikova PP, Efimov SV, Klochkov VV. Binding of Different Cyclosporin Variants to Micelles Evidenced by NMR and MD Simulations. Membranes. 2023; 13(2):196. https://doi.org/10.3390/membranes13020196
Chicago/Turabian StyleKobchikova, Polina P., Sergey V. Efimov, and Vladimir V. Klochkov. 2023. "Binding of Different Cyclosporin Variants to Micelles Evidenced by NMR and MD Simulations" Membranes 13, no. 2: 196. https://doi.org/10.3390/membranes13020196
APA StyleKobchikova, P. P., Efimov, S. V., & Klochkov, V. V. (2023). Binding of Different Cyclosporin Variants to Micelles Evidenced by NMR and MD Simulations. Membranes, 13(2), 196. https://doi.org/10.3390/membranes13020196