
Alternative Targets for sPLA2 Activity: Role of Membrane-Enzyme Interactions


{kind=link}
{kind=link}
{kind=link}
{kind=link}
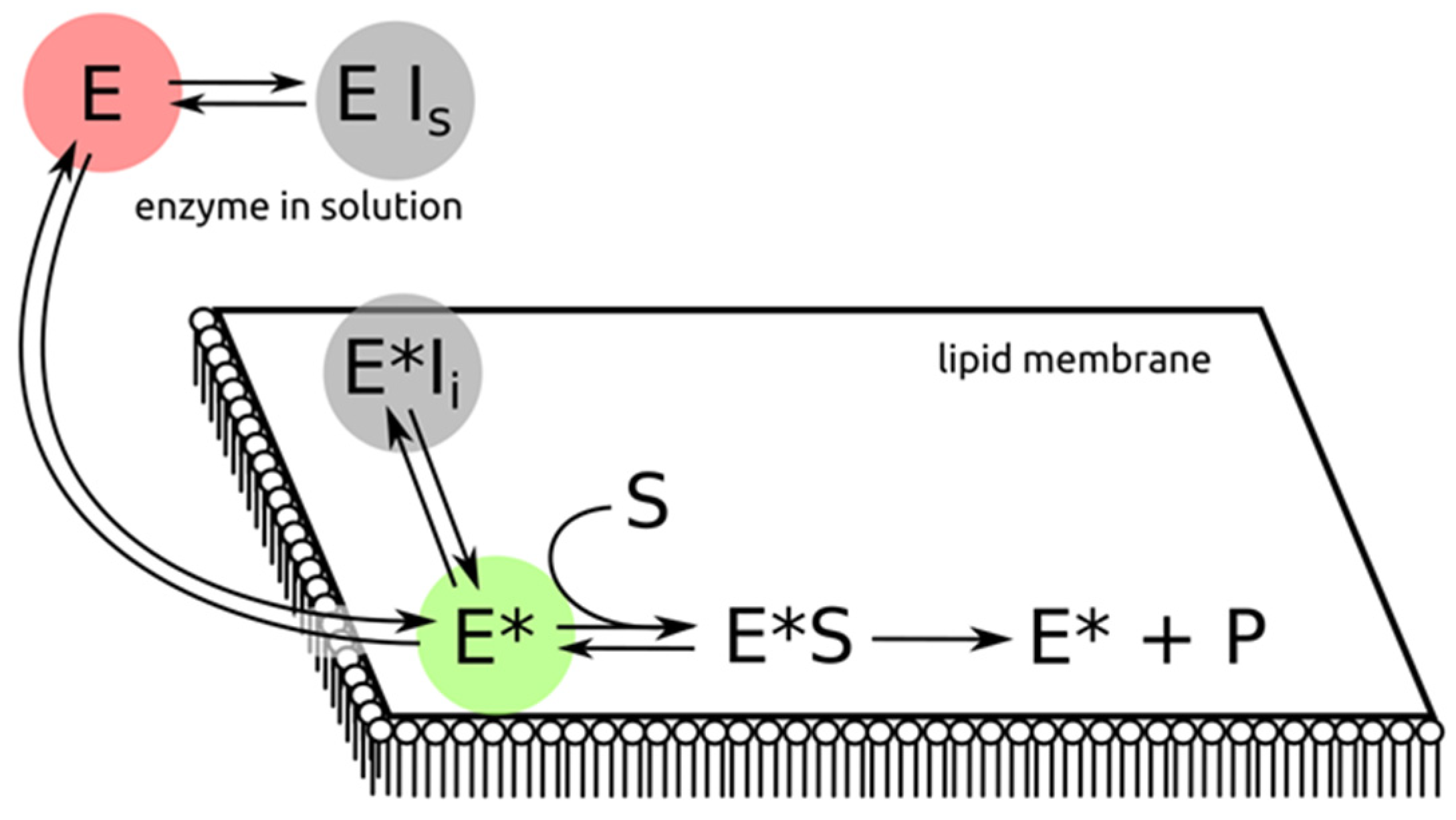
Abstract
:1. Introduction
2. Directions to Attack sPLA2 to Control its Enzymatic Activity
3. Preventing sPLA2 Binding to the Target Membrane
4. sPLA2-Binding Proteins with Inhibitory Properties
5. Blocking the Enzyme in the Wrong Orientation on the Membrane Surface
6. Blocking and Modulating Substrate Binding
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Dennis, E.A.; Cao, J.; Hsu, Y.-H.; Magrioti, V.; Kokotos, G. Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Ilies, M.A. The Phospholipase A2 Superfamily: Structure, Isozymes, Catalysis, Physiologic and Pathologic Roles. Int. J. Mol. Sci. 2023, 24, 1353. [Google Scholar] [CrossRef]
- Snider, J.M.; You, J.K.; Wang, X.; Snider, A.J.; Hallmark, B.; Zec, M.M.; Seeds, M.C.; Sergeant, S.; Johnstone, L.; Wang, Q.; et al. Group IIA secreted phospholipase A2 is associated with the pathobiology leading to COVID-19 mortality. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Taketomi, Y.; Girard, C.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: Lessons from transgenic and knockout mice. Biochimie 2010, 92, 561–582. [Google Scholar] [CrossRef]
- Murakami, M.; Sato, H.; Miki, Y.; Yamamoto, K.; Taketomi, Y. A new era of secreted phospholipase A2. J. Lipid Res. 2015, 56, 1248–1261. [Google Scholar] [CrossRef] [Green Version]
- Crowl, R.M.; Stoller, T.J.; Conroy, R.R.; Stoner, C.R. Induction of phospholipase A2 gene expression in human hepatoma cells by mediators of the acute phase response. J. Biol. Chem. 1991, 266, 2647–2651. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.R.; Chen, Z.; Mann, T.J.; Bastard, K.; Scott, K.F.; Church, W.B. Structural and Functional Aspects of Targeting the Secreted Human Group IIA Phospholipase A2. Molecules 2020, 25, 4459. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-Y.; Farooqui, T.; Kokotos, G.; Farooqui, A.A. Synthetic and Natural Inhibitors of Phospholipases A2: Their Importance for Understanding and Treatment of Neurological Disorders. ACS Chem. Neurosci. 2015, 6, 814–831. [Google Scholar] [CrossRef] [PubMed]
- Batsika, C.S.; Gerogiannopoulou, A.-D.D.; Mantzourani, C.; Vasilakaki, S.; Kokotos, G. The design and discovery of phospholipase A2 inhibitors for the treatment of inflammatory diseases. Expert Opin. Drug Discov. 2021, 16, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.L.; Nidorf, S.M.; Eikelboom, J. Targeting the Unstable Plaque in Acute Coronary Syndromes. Clin. Ther. 2013, 35, 1099–1107. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Elliott, M.; Stasiv, Y.; Hislop, C.; Imburgia, M.; Weiss, R.; Underwood, P.; Ende, D.; Brown, C.; Nadar, V.; et al. Randomized trial of an inhibitor of secretory phospholipase A2 on atherogenic lipoprotein subclasses in statin-treated patients with coronary heart disease. Eur. Hear. J. 2010, 32, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Rosenson, R.S.; Hislop, C.; McConnell, D.; Elliott, M.; Stasiv, Y.; Wang, N.; Waters, D.D. Effects of 1-H-indole-3-glyoxamide (A-002) on concentration of secretory phospholipase A2 (PLASMA study): A phase II double-blind, randomised, placebo-controlled trial. Lancet 2009, 373, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Fraser, H.; Trias, J.; Hislop, C. Varespladib methyl in cardiovascular disease. Expert Opin. Investig. Drugs 2010, 19, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Kastelein, J.J.P.; Schwartz, G.G.; Bash, D.; Rosenson, R.S.; Cavender, M.A.; Brennan, D.M.; Koenig, W.; Jukema, J.W.; Nambi, V.; et al. Varespladib and Cardiovascular Events in Patients with an Acute Coronary Syndrome. JAMA 2014, 311, 252–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewin, M.; Samuel, S.; Merkel, J.; Bickler, P. Varespladib (LY315920) Appears to Be a Potent, Broad-Spectrum, Inhibitor of Snake Venom Phospholipase A2 and a Possible Pre-Referral Treatment for Envenomation. Toxins 2016, 8, 248. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.J.; Faiz, M.A.; Abela-Ridder, B.; Ainsworth, S.; Bulfone, T.C.; Nickerson, A.D.; Habib, A.G.; Junghanss, T.; Fan, H.W.; Turner, M.; et al. Strategy for a globally coordinated response to a priority neglected tropical disease: Snakebite envenoming. PLOS Neglected Trop. Dis. 2019, 13, e0007059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikolaou, A.; Kokotou, M.G.; Vasilakaki, S.; Kokotos, G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1864, 941–956. [Google Scholar] [CrossRef]
- Winget, J.M.; Pan, Y.H.; Bahnson, B.J. The interfacial binding surface of phospholipase A2s. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2006, 1761, 1260–1269. [Google Scholar] [CrossRef]
- Kuzmina, N.; Volynsky, P.; Boldyrev, I.; Alekseeva, A. sPLA2 Wobbles on the Lipid Bilayer between Three Positions, Each Involved in the Hydrolysis Process. Toxins 2022, 14, 669. [Google Scholar] [CrossRef]
- Yu, B.-Z.; Ghomashchi, F.; Cajal, Y.; Annand, R.R.; Berg, O.G.; Gelb, M.H.; Jain, M.K. Use of an Imperfect Neutral Diluent and Outer Vesicle Layer Scooting Mode Hydrolysis to Analyze the Interfacial Kinetics, Inhibition, and Substrate Preferences of Bee Venom Phospholipase A2. Biochemistry 1997, 36, 3870–3881. [Google Scholar] [CrossRef]
- Jain, M.K.; Yu, B.Z.; Rogers, J.; Ranadive, G.N.; Berg, O.G. Interfacial catalysis by phospholipase A2: Dissociation constants for calcium, substrate, products, and competitive inhibitors. Biochemistry 1991, 30, 7306–7317. [Google Scholar] [CrossRef]
- Dennis, E.A. Kinetic dependence of phospholipase A2 activity on the detergent Triton X-100. J. Lipid Res. 1973, 14, 152–159. [Google Scholar] [CrossRef]
- Bayburt, T.; Yu, B.Z.; Lin, H.K.; Browning, J.; Jain, M.K.; Gelb, M.H. Human nonpancreatic secreted phospholipase A2: Interfacial parameters, substrate specificities, and competitive inhibitors. Biochemistry 1993, 32, 573–582. [Google Scholar] [CrossRef]
- Jain, M.K.; Jahagirdar, D. Action of phospholipase A2 on bilayers. Effect of inhibitors. Biochim. et Biophys. Acta (BBA)-Biomembr. 1985, 814, 319–326. [Google Scholar] [CrossRef]
- Berg, O.G.; Gelb, M.H.; Tsai, M.-D.; Jain, M.K. Interfacial Enzymology: The Secreted Phospholipase A2-Paradigm. Chem. Rev. 2001, 101, 2613–2654. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Yao, L.; Lucena, A. Product inhibition of secreted phospholipase A2 may explain lysophosphatidylcholines' unexpected therapeutic properties. J. Inflamm. 2008, 5, 17. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, S.; Yuan, C. Active site competition is the mechanism for the inhibition of lipoprotein-associated phospholipase A2 by detergent micelles or lipoproteins and for the efficacy reduction of darapladib. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef]
- Ivanušec, A.; Šribar, J.; Križaj, I. Secreted Phospholipases A2—Not just Enzymes: Revisited. Int. J. Biol. Sci. 2022, 18, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Macchioni, L.; Corazzi, L.; Nardicchi, V.; Mannucci, R.; Arcuri, C.; Porcellati, S.; Sposini, T.; Donato, R.; Goracci, G. Rat Brain Cortex Mitochondria Release Group II Secretory Phospholipase A2 under Reduced Membrane Potential. J. Biol. Chem. 2004, 279, 37860–37869. [Google Scholar] [CrossRef] [Green Version]
- Alekseeva, A.S.; Volynsky, P.E.; Krylov, N.A.; Chernikov, V.P.; Vodovozova, E.L.; Boldyrev, I.A. Phospholipase A2 way to hydrolysis: Dint formation, hydrophobic mismatch, and lipid exclusion. Biochim. et Biophys. Acta (BBA)-Biomembr. 2020, 1863, 183481. [Google Scholar] [CrossRef] [PubMed]
- Fortes-Dias, C.L.; dos Santos, R.M.M.; Magro, A.J.; Fontes, M.R.D.M.; Chávez-Olórtegui, C.; Granier, C. Identification of continuous interaction sites in PLA2-based protein complexes by peptide arrays. Biochimie 2009, 91, 1482–1492. [Google Scholar] [CrossRef]
- Nicolas, J.-P.; Lambeau, G.; Lazdunski, M. Identification of the Binding Domain for Secretory Phospholipases A2 on Their M-type 180-kDa Membrane Receptor. J. Biol. Chem. 1995, 270, 28869–28873. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Cao, L.; Tang, H.; Shi, X.; He, Y. Structure of Human M-type Phospholipase A2 Receptor Revealed by Cryo-Electron Microscopy. J. Mol. Biol. 2017, 429, 3825–3835. [Google Scholar] [CrossRef]
- Higashino, K.-I.; Yokota, Y.; Ono, T.; Kamitani, S.; Arita, H.; Hanasaki, K. Identification of a Soluble Form Phospholipase A2Receptor as a Circulating Endogenous Inhibitor for Secretory Phospholipase A2. J. Biol. Chem. 2002, 277, 13583–13588. [Google Scholar] [CrossRef] [Green Version]
- Ancian, P.; Lambeau, G.; Mattéi, M.-G.; Lazdunski, M. The Human 180-kDa Receptor for Secretory Phospholipases A2. J. Biol. Chem. 1995, 270, 8963–8970. [Google Scholar] [CrossRef] [Green Version]
- Rouault, M.; Le Calvez, C.; Boilard, E.; Surrel, F.; Singer, A.; Ghomashchi, F.; Bezzine, S.; Scarzello, S.; Bollinger, J.; Gelb, M.H.; et al. Recombinant Production and Properties of Binding of the Full Set of Mouse Secreted Phospholipases A2 to the Mouse M-Type Receptor. Biochemistry 2007, 46, 1647–1662. [Google Scholar] [CrossRef]
- Hanasaki, K.; Arita, H. Phospholipase A2 receptor: A regulator of biological functions of secretory phospholipase A2. Prostaglandins Other Lipid Mediat. 2002, 68–69, 71–82. [Google Scholar] [CrossRef]
- Silliman, C.C.; Moore, E.E.; Zallen, G.; Gonzalez, R.; Johnson, J.L.; Elzi, D.J.; Meng, X.; Hanasaki, K.; Ishizaki, J.; Arita, H.; et al. Presence of the M-type sPLA2 receptor on neutrophils and its role in elastase release and adhesion. Am. J. Physiol. Physiol. 2002, 283, C1102–C1113. [Google Scholar] [CrossRef] [Green Version]
- Boilard, E.; Bourgoin, S.G.; Bernatchez, C.; Surette, M.E. Identification of an autoantigen on the surface of apoptotic human T cells as a new protein interacting with inflammatory group IIA phospholipase A2. Blood 2003, 102, 2901–2909. [Google Scholar] [CrossRef]
- Nomura, K.; Fujita, H.; Arita, H. Gene expression of pancreatic-type phospholipase-A2 in rat ovaries: Stimulatory action on progesterone release. Endocrinology 1994, 135, 603–609. [Google Scholar] [CrossRef]
- Ramanadham, S.; Ma, Z.; Arita, H.; Zhang, S.; Turk, J. Type IB secretory phospholipase A2 is contained in insulin secretory granules of pancreatic islet β-cells and is co-secreted with insulin from glucose-stimulated islets. Biochim. et Biophys. Acta (BBA)-Lipids Lipid Metab. 1998, 1390, 301–312. [Google Scholar] [CrossRef]
- Šribar, J.; Kovačič, L.; Oberčkal, J.; Ivanušec, A.; Petan, T.; Fox, J.W.; Križaj, I. The neurotoxic secreted phospholipase A2 from the Vipera a. ammodytes venom targets cytochrome c oxidase in neuronal mitochondria. Sci. Rep. 2019, 9, 283. [Google Scholar] [CrossRef] [Green Version]
- Mattiazzi, M.; Sun, Y.; Wolinski, H.; Bavdek, A.; Petan, T.; Anderluh, G.; Kohlwein, S.D.; Drubin, D.G.; Križaj, I.; Petrovič, U. A Neurotoxic Phospholipase A2 Impairs Yeast Amphiphysin Activity and Reduces Endocytosis. PLoS ONE 2012, 7, e40931. [Google Scholar] [CrossRef]
- Saegusa, J.; Akakura, N.; Wu, C.-Y.; Hoogland, C.; Ma, Z.; Lam, K.S.; Liu, F.-T.; Takada, Y.K.; Takada, Y. Pro-inflammatory Secretory Phospholipase A2 Type IIA Binds to Integrins αvβ3 and α4β1 and Induces Proliferation of Monocytic Cells in an Integrin-dependent Manner. J. Biol. Chem. 2008, 283, 26107–26115. [Google Scholar] [CrossRef] [Green Version]
- Beers, S.A.; Buckland, A.G.; Koduri, R.S.; Cho, W.; Gelb, M.H.; Wilton, D.C. The Antibacterial Properties of Secreted Phospholipases A2. J. Biol. Chem. 2002, 277, 1788–1793. [Google Scholar] [CrossRef] [Green Version]
- Faure, G.; Gowda, V.T.; Maroun, R.C. Characterization of a human coagulation factor Xa-binding site on Viperidae snake venom phospholipases A2 by affinity binding studies and molecular bioinformatics. BMC Struct. Biol. 2007, 7, 82. [Google Scholar] [CrossRef] [Green Version]
- Zvaritch, E.; Lambeau, G.; Lazdunski, M. Endocytic Properties of the M-type 180-kDa Receptor for Secretory Phospholipases A2. J. Biol. Chem. 1996, 271, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Murakami, M.; Taketomi, Y.; Miki, Y.; Sato, H.; Yamamoto, K.; Lambeau, G. Emerging roles of secreted phospholipase A2 enzymes: The 3rd edition. Biochimie 2014, 107, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Chabot, S.; Koumanov, K.; Lambeau, G.; Gelb, M.H.; Balloy, V.; Chignard, M.; Whitsett, J.A.; Touqui, L. Inhibitory Effects of Surfactant Protein A on Surfactant Phospholipid Hydrolysis by Secreted Phospholipases A2. J. Immunol. 2003, 171, 995–1000. [Google Scholar] [CrossRef] [Green Version]
- Thwin, M.; Samy, R.P.; Satyanarayanajois, S.D.; Gopalakrishnakone, P. Venom neutralization by purified bioactive molecules: Synthetic peptide derivatives of the endogenous PLA2 inhibitory protein PIP (a mini-review). Toxicon 2010, 56, 1275–1283. [Google Scholar] [CrossRef]
- Sánchez, E.E.; Rodríguez-Acosta, A. Inhibitors of Snake Venoms and Development of New Therapeutics. Immunopharmacol. Immunotoxicol. 2008, 30, 647–678. [Google Scholar] [CrossRef] [PubMed]
- Santos-Filho, N.A.; Santos, C.T. Alpha-type phospholipase A2 inhibitors from snake blood. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Okumura, K.; Ohkura, N.; Inoue, S.; Ikeda, K.; Hayashi, K. A Novel Phospholipase A2 Inhibitor with Leucine-rich Repeats from the Blood Plasma of Agkistrodon blomhoffii siniticus. J. Biol. Chem. 1998, 273, 19469–19475. [Google Scholar] [CrossRef] [Green Version]
- Gelb, M.H.; Cho, W.; Wilton, D.C. Interfacial binding of secreted phospholipases A2: More than electrostatics and a major role for tryptophan. Curr. Opin. Struct. Biol. 1999, 9, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Cho, W. Differential Roles of Ionic, Aliphatic, and Aromatic Residues in Membrane−Protein Interactions: A Surface Plasmon Resonance Study on Phospholipases A2. Biochemistry 2001, 40, 4672–4678. [Google Scholar] [CrossRef]
- Bezzine, S.; Bollinger, J.G.; Singer, A.G.; Veatch, S.; Keller, S.; Gelb, M.H. On the Binding Preference of Human Groups IIA and X Phospholipases A2 for Membranes with Anionic Phospholipids. J. Biol. Chem. 2002, 277, 48523–48534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, S.F.; Othman, R.; Wilton, D.C. Tryptophan-Containing Mutant of Human (Group IIa) Secreted Phospholipase A2 Has a Dramatically Increased Ability to Hydrolyze Phosphatidylcholine Vesicles and Cell Membranes. Biochemistry 1998, 37, 13203–13211. [Google Scholar] [CrossRef]
- Beers, S.A.; Buckland, A.G.; Giles, N.; Gelb, M.H.; Wilton, D.C. Effect of Tryptophan Insertions on the Properties of the Human Group IIA Phospholipase A2: Mutagenesis Produces an Enzyme with Characteristics Similar to Those of the Human Group V Phospholipase A2. Biochemistry 2003, 42, 7326–7338. [Google Scholar] [CrossRef]
- Nasri, Z.; Memari, S.; Wenske, S.; Clemen, R.; Martens, U.; Delcea, M.; Bekeschus, S.; Weltmann, K.; von Woedtke, T.; Wende, K. Singlet-Oxygen-Induced Phospholipase A2 Inhibition: A Major Role for Interfacial Tryptophan Dioxidation. Chem. A Eur. J. 2021, 27, 14702–14710. [Google Scholar] [CrossRef]
- Bollinger, J.G.; Diraviyam, K.; Ghomashchi, F.; Murray, D.; Gelb, M.H. Interfacial Binding of Bee Venom Secreted Phospholipase A2 to Membranes Occurs Predominantly by a Nonelectrostatic Mechanism. Biochemistry 2004, 43, 13293–13304. [Google Scholar] [CrossRef]
- Gaspar, D.; Lúcio, M.; Rocha, S.; Lima, J.C.; Reis, S. Changes in PLA2 activity after interacting with anti-inflammatory drugs and model membranes: Evidence for the involvement of tryptophan residues. Chem. Phys. Lipids 2011, 164, 292–299. [Google Scholar] [CrossRef]
- Alekseeva, A.S.; Volynsky, P.E.; Boldyrev, I.A. Estimation of the Phospholipase A2 Selectivity on POPC/POPG Membranes Using the Interaction Map. Biochem. (Moscow) Suppl. Ser. A Membr. Cell Biol. 2021, 15, 329–333. [Google Scholar] [CrossRef]
- Ghomashchi, F.; Lin, Y.; Hixon, M.S.; Yu, B.-Z.; Annand, R.; Jain, M.K.; Gelb, M.H. Interfacial Recognition by Bee Venom Phospholipase A2: Insights into Nonelectrostatic Molecular Determinants by Charge Reversal Mutagenesis. Biochemistry 1998, 37, 6697–6710. [Google Scholar] [CrossRef]
- Renetseder, R.; Brunie, S.; Dijkstra, B.W.; Drenth, J.; Sigler, P.B. A comparison of the crystal structures of phospholipase A2 from bovine pancreas and Crotalus atrox venom. J. Biol. Chem. 1985, 260, 11627–11634. [Google Scholar] [CrossRef]
- Ye, L.; Dickerson, T.; Kaur, H.; Takada, Y.K.; Fujita, M.; Liu, R.; Knapp, J.M.; Lam, K.S.; Schore, N.E.; Kurth, M.J.; et al. Identification of inhibitors against interaction between pro-inflammatory sPLA2-IIA protein and integrin αvβ3. Bioorganic Med. Chem. Lett. 2012, 23, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouchlis, V.D.; Chen, Y.; McCammon, J.A.; Dennis, E.A. Membrane Allostery and Unique Hydrophobic Sites Promote Enzyme Substrate Specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. [Google Scholar] [CrossRef] [Green Version]
- Woolford, A.J.-A.; Pero, J.E.; Aravapalli, S.; Berdini, V.; Coyle, J.E.; Day, P.J.; Dodson, A.M.; Grondin, P.; Holding, F.P.; Lee, L.Y.W.; et al. Exploitation of a Novel Binding Pocket in Human Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) Discovered through X-ray Fragment Screening. J. Med. Chem. 2016, 59, 5356–5367. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Hayashi, D.; Vasquez, A.M.; Cao, J.; McCammon, J.A.; Dennis, E.A. Lipoprotein-associated phospholipase A2: A paradigm for allosteric regulation by membranes. Proc. Natl. Acad. Sci. USA 2022, 119, e2102953118. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseeva, A.S.; Boldyrev, I.A. Alternative Targets for sPLA2 Activity: Role of Membrane-Enzyme Interactions. Membranes 2023, 13, 618. https://doi.org/10.3390/membranes13070618
Alekseeva AS, Boldyrev IA. Alternative Targets for sPLA2 Activity: Role of Membrane-Enzyme Interactions. Membranes. 2023; 13(7):618. https://doi.org/10.3390/membranes13070618
Chicago/Turabian StyleAlekseeva, Anna S., and Ivan A. Boldyrev. 2023. "Alternative Targets for sPLA2 Activity: Role of Membrane-Enzyme Interactions" Membranes 13, no. 7: 618. https://doi.org/10.3390/membranes13070618
APA StyleAlekseeva, A. S., & Boldyrev, I. A. (2023). Alternative Targets for sPLA2 Activity: Role of Membrane-Enzyme Interactions. Membranes, 13(7), 618. https://doi.org/10.3390/membranes13070618